

Abstract
Recent data suggest that endothelial progenitor cells (EPCs) are involved in recanalizing venous thrombi. We examined the impact of a fibrin network, and particularly of adsorbed thrombin, on EPCs derived from cord blood CD34+ cells. Fibrin networks generated in microplates by adding CaCl2 to platelet-depleted plasma retained adsorbed thrombin at the average concentration of 4.2 nM per well. EPCs expressed high levels of endothelial cell protein C receptor and thrombomodulin, allowing the generation of activated protein C on the fibrin matrix in the presence of exogenous human protein C. The fibrin matrix induced significant EPC proliferation and, when placed in the lower chamber of a Boyden device, strongly enhanced EPC migration. These effects were partly inhibited by hirudin by 41% and 66%, respectively), which suggests that fibrin-adsorbed thrombin interacts with EPCs via the thrombin receptor PAR-1. Finally, spontaneous lysis of the fibrin network, studied by measuring D-dimer release into the supernatant, was inhibited by EPCs but not by control mononuclear cells. Such an effect was associated with a 10-fold increase in plasminogen activator inhibitor-1 (PAI-1) secretion by EPCs cultivated in fibrin matrix. Overall, our data show that EPCs, in addition to their angiogenic potential, have both anticoagulant and antifibrinolytic properties. Thrombin may modulate these properties and contribute to thrombus recanalization by EPCs.
Keywords: endothelial progenitor cells, fibrin matrix, thrombin, PAR-1, fibrinolysis, D-dimers, PAI-1
Introduction
Histologically, thrombus resolution closely resembles wound healing [1, 2]. New vessels appear within the body of the thrombus and contribute to restoring a patent vein lumen [3]. This is associated with an increase in local expression of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor, which are thought to drive neovascularization within the thrombus [4].
Endothelial progenitor cells (EPCs) appear to play an important role in adult neovascularization [5–8]. EPCs support a strong intrinsic urokinase-type plasminogen activator (u-PA)/u-PA receptor (u-PAR) dependent proteolytic activity that could contribute to their invasive and angiogenic properties [9]. Recent data suggest that EPCs are also involved in thrombus recanalization and resolution [3, 10]. Indeed, thrombi fail to resolve in u-PA knockout mice, while wild-type bone marrow–derived cells, potentially rich in progenitor cells, rescue thrombus re-organization and resolution in uPA−/− mice [11].
Thrombin was initially implicated in haemostasis and fibrin clot formation [12], and has been also involved in cell biology since the discovery of its major receptor, the protease-activated receptor-1 (PAR-1). PAR-1 is a protease-activated G protein-coupled receptor specifically cleaved by thrombin at its extracellular N-terminus. The amino-terminal sequence thereby unmasked acts as a tethered ligand, triggering a rapid response that can be reproduced by a hexapeptide (SFLLRN). In vascular biology, PAR-1 is the major mediator of thrombin signalling and is involved in platelet activation, smooth muscle cells migration and proliferation. PAR-1 activation also regulates many aspects of endothelial cell biology and has been involved in vascular development [12]. In previous work, we found that EPCs express the thrombin receptor PAR-1, and that specific activation of this receptor enhances EPC proangiogenic properties [13, 14]. Here we screened EPC for receptors other than PAR-1 that may influence thrombin activity and/or PAR-1 activation, such as thrombomodulin (TM or CD141) that enhances generation of activated protein C (APC) [15], and endothelial protein C receptor (EPCR or CD201) that facilitates APC generation and permits PAR-1 cleavage [16]. The presence of the two cofactors of protein C activation –i.e. TM and EPCR – could confer anticoagulant properties to EPCs.
Therapeutic use of EPCs is now the subject of intensive investigations, but possible interactions between these cells and fibrin clot have not yet been examined in experimental studies.
The aim of this study was to examine the behaviour of EPCs cultured on a fibrin network, and to determine whether the hypothetical interaction of EPCs with haemostasis –i.e. anticoagulant and fibrinolytic properties – are modulated by fibrin-adsorbed thrombin. In particular, we have studied the role of EPCs on fibrin lysis. Proteolysis of a fibrin clot is mediated by the serine protease plasmin, generated upon activation of the zymogen plasminogen by tissue-plasminogen activator (t-PA) and u-PA.
Plasmin generation is modulated by the major plas-minogen activator inhibitor, PAI-1. Fibrin degradation upon plasmin can be monitored by the release of specific fibrin degradation products, the D-dimers.
Methods
Late-EPC culture and characterization
Mononuclear cells (MNCs) were isolated from human cord blood by density gradient centrifugation with Histopaque-1077 (Sigma-Aldrich, Saint-Quentin Fallavier, France). Plastic non-adherent cells were enriched in CD34+ cells by magnetic activated cell sorting on MiniMacs columns (Miltenyi Biotec, Paris, France) following the manufacturer's instructions. Cells thus recovered were plated on 0.2% gelatin-coated 24-well plastic culture dishes at a density of 5 × 105/ml and maintained in endothelial growth medium-2 (EGM-2, Lonza, Saint-Beauzire, France) as previously described [13]. After 2 to 3 weeks of expansion, EPCs were characterized by means of flow cytometry with monoclonal antibodies (mAb) from Beckman Coulter (Villepinte, France); we also used a mAb against CD141 (thrombomodulin), that was a kind gift from Diagnostica Stago (Asnières, France), and a mAb against EPCR, that was a kind gift from Dr CT Esmon. For immunofluorescence analysis of EPCR expression, cells were seeded on gelatin-coated glass cover slips in 24-well plates, then incubated at room temperature with endothelial cell basal medium 2 (EBM2, Lonza), BSA 0.1% containing mAb against EPCR. Cells were fixed with 4% paraformaldehyde then incubated with goat secondary antibodies coupled to AlexaFluor 488 (Molecular Probes, Invitrogen, Cergy Pontoise, France). The cover slips were mounted with Mowiol containing ToPro-3 nuclear stain and observed with a Leica TCS SP2 confocal microscope (Leica Microsystems, Rueil-Malmaison, France).
Fibrin network preparation
A fibrin network was generated in microplates by adding 0.025 M CaCl2 to platelet-depleted plasma, obtained by centrifuging citrated blood for 15 min. at 2300 ×g, and holding it at 37°C for 1 hr. We used 24-well and 6-well microplates containing 600 μl and 1000 μl of plasma, respectively, for migration and proliferation assays. For proliferation assays, EPCs were plated at a density of 5 × 104/well in EBM-2. After 24 hrs of culture on the fibrin matrix, DNA synthesis was determined by measuring the incorporation of 5′-[3H]-thymidine ([3H]-Tdr, Amersham, Orsay, France) in EBM-2 medium for 4 hrs with a Betamatic counter (1900 CA Packard). EPC migration was measured by using modified Boyden chambers (Costar, Avon, France) with 8-μm pore size filters. EPCs were seeded at a density of 5 × 104/well in 200 μl of EBM-2/1% FBS medium and then allowed to migrate for 5 hrs at 37°C. A fibrin matrix was prepared in the lower chamber of the Boyden chamber. Thrombin activity on the matrix surface was quantified by measuring the hydrolysis of the thrombin-selective chromogenic substrate S2238 (Biogénic, Mauguio, France) at 405 nm at 60-sec. intervals. A calibration curve was constructed with serial dilutions of purified human thrombin, prepared as previously described [17].
Purification of human protein C
Vitamin K-dependent proteins were concentrated by BaCl2 precipitation. Protein C (PC) was then immunopurified with Sepharose-immobilized anti-PC mAb 2E2A8D2 coupled to CNBr–Sepharose 4B (Pharmacia, Piscataway, NJ, USA). This purification procedure was followed by anion-exchange chromatography (Mono Q) using a CaCl2 gradient to elute PC.
Assay of activated PC generation
Fibrin networks were pre-treated with 40 mM benzamidine, 100 nM hirudin (lepirudine, Refludan®, Pharmion Development, Paris, France) or tris buffer saline (TBS: 50 mM Tris, 150 mM NaCl2) for 30 min at 37°C. EPCs grown on gelatin were then detached and placed on the fibrin network at a density of 5 × 104/well and cultured for 24 hrs.
Then, 50 μl of TBS supplemented with 5 mM calcium, 0.2% bovine serum albumin and 640 nM human PC was added. The plates were incubated with 5% CO2 at 37°C for 0 to 180 min. After incubation, 20 μl of culture supernatant was diluted in 80 μl of 100 nM hirudin solution to stop APC gen-eration. APC was quantified in 96-well plates by adding 500 μM S-2366 substrate (Biogénic) and the initial rate of chromogenic substrate cleavage was assayed at 405 nm.
Real-time quantitative reverse-transcription polymerase chain reaction (RT-PCR)
The theoretical and practical aspects of real-time quantitative RT-PCR on the ABI Prism 7700 Sequence Detection System (Perkin-Elmer Applied Biosystems, Courtaboeuf, France) are described in detail elsewhere [18]. We quantified transcripts of the TBP gene which encodes the TATA box-binding protein (a component of the DNA-binding protein complex TFIID) as the endogenous RNA control, and each sample was normalized on the basis of its TBP content. Results, expressed as N-fold differences in target gene expression relative to the TBP gene, and termed Ntarget, were determined with the formula: Ntarget= 2ΔCtsample, where the ΔCt value of the sample was determined by subtracting the Ct value of the target gene from the Ct value of the TBP gene. The Ntarget values of the samples were subsequently normalized such that the untreated control Ntarget values were 1. Primers for TBP and five target genes were chosen with the assistance of the Oligo 5.0 computer program (National Biosciences, Plymouth, MN, USA). The primer sequences are shown in Table 1. To avoid amplifying contaminating genomic DNA, one of the two primers was placed at the junction between two exons. The thermal cycling conditions comprised an initial denaturation step at 95°C for 10 min. and 50 cycles at 95°C for 15 sec and 65°C for 1 min.
1.
Oligonucleotide primers used for real-time quantitative RT-PCR
| Genes | Oligonucleotide | Sequence | PCR product size (bp) | ||
|---|---|---|---|---|---|
| u-PA/PLAU | Upper primer | 5′– AAA ATG CTG TGT GCT GCT GAC C – 3′ | 87 | ||
| Lower primer | 5′– GCC TTG GAG GGA ACA GAC GA – 3′ | ||||
| u-PAR/PLAUR | Upper primer | 5′– ACA CCT GCG TCC CAG CCT CT – 3′ | 78 | ||
| Lower primer | 5′– CGC ACT CTT CCA CAC GGC A – 3′ | ||||
| PAI1/SERPINE1 | Upper primer | 5′– CAC AAA TCA GAC GGC AGC ACT – 3′ | 85 | ||
| Lower primer | 5′– CAT CGG GCG TGG TGA ACT C – 3′ | ||||
| PAI2/SERPINB2 | Upper primer | 5′– ACC CCC ATG ACT CCA GAG AAC T – 3′ | 124 | ||
| Lower primer | 5′– GAG AGC GGA AGG ATG AAT GGA T – 3′ | ||||
| t-PA/PLAT | Upper primer | 5′– AGC AGG CCC TGT ACT TCT CAG ATT – 3′ | 87 | ||
| Lower primer | 5′– ACG TGG CCC TGG TAT CTA TTT CA – 3′ | ||||
| TBP | Upper primer | 5′– TGC ACA GGA GCC AAG AGT GAA – 3′ | 132 | ||
| Lower primer | 5′– CAC ATC ACA GCT CCC CAC CA– 3′ | ||||
ELISA methods
EPCs were incubated for 24 hrs in EBM-2 at 37°C with 75 μM SFLLRN or 10 nM human thrombin (Diagnostica Stago) and the supernatants were then collected. The cells were non enzymatically detached with EDTA (Sigma Aldrich), counted and lysed on ice in 100 mM Tris-HCl (pH 8.1), 0.5% Triton X-100 buffer supplemented with a complete protease inhibitor mixture (Roche Diagnostic, Meylan, France). The u-PA and u-PAR antigen concentrations were determined in conditioned media and in cell lysates by using ELISA methods as recommended by the manufacturer (894 and 893 Imubind Elisa kits, American Diagnostica, Stamford, CT, USA). Results were expressed as nanograms of u-PA or u-PAR per 105 cells. T-PA was measured with the Asserachrom kit (Diagnostica Stago), and PAI-1 with the TintElize kit (Biopool International, Trinity Biotech, Wicklow, Ireland). Fibrin degradation products (D-dimers) released into the fibrin network supernatant were quantified with the Asserachrom D-Di kit (Diagnostica Stago).
Statistical analysis
Data are reported as means ± SEM. Significant differences were identified by ANOVA followed by Fisher's protected least-significant difference test. All statistical tests were performed with the StatView® software package (SAS, Cary, NC, USA). Differences with P values <0.05 were considered significant.
Results
EPCs express PAR-1, thrombomodulin and EPCR
Sorted human cord blood CD34+ cells were cultured with specific endothelial growth factors (EGM-2 medium), yielding small colonies of so-called late EPCs after 7 to 14 days [19]. At confluence, EPCs exhibited a cobblestone morphology (a growth pattern typical of the endothelial lineage) and expressed CD146 (S-Endo1) and CD31 (Fig. 1A). EPCs expressed CD34 antigen and, as expected, were negative for the haematopoietic stem cell antigen CD133 and for the leucocyte markers CD14 and CD45. Flow cytometry also showed that, respectively, 94% and 89% of EPCs expressed the thrombin receptors PAR-1 and thrombomodulin (CD141). EPCR was expressed by 91% of EPCs. This expression was homogeneously distributed over the cell surface, as shown by confocal microscopy (Fig. 1B).
1.

Characterization of EPCs derived from human cord blood (A). Representative flow cytometric histograms of detached EPCs after immunolabelling with a control antibody (black line) and specific antibodies (blue line) to endothelial markers (CD146, CD31), haematopoietic markers (CD34 and CD133), leucocyte markers (CD14 and CD45), thrombin receptors (PAR-1 and CD141 = thrombomodulin) and endothelial protein C receptor (EPCR). (B). Immunofluorescence was performed on unpermeabilized EPCs, with anti-EPCR antibodies. Bar = 50 μm (magnification × 40)
EPCs generate activated protein C on a fibrin network
EPCs express the two cofactors of PC activation on their surface, implying that they might have anticoagulant functions. To determine whether EPCs express anticoagulant properties when adherent to fibrin, we studied APC generation on the surface of EPCs cultured on gelatin for 30 to 40 days then placed on a fibrin matrix prepared by recalcification of human plasma (see Methods). Part of the thrombin formed during the clotting process is adsorbed to the fibrin network. Fibrin-bound thrombin activity, measured by selective chromogenic substrate S2238 hydrolysis, averaged 4.2 nM per well. APC generation was then quantified in the presence of EPCs and exogenous immunopurified human PC, by determining the kinetics of hydrolysis of the chromogenic substrate S2366 (Fig. 2). In our experimental conditions, APC generation reached a plateau at 90 nM within 1 hr. APC formation was inhibited when the fibrin surface was pretreated with hirudin or benzamidine, inferring that thrombin adsorbed to the fibrin network plays a major role in APC generation on the EPC surface.
2.
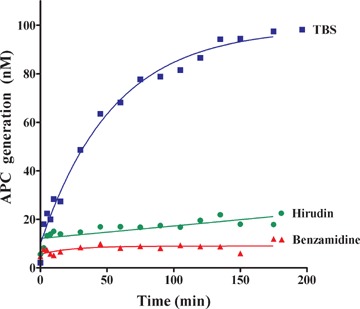
Protein C activation by EPCs on a fibrin clot, in the presence of exogenous protein C. Monolayer cultures of EPCs grown to confluence on a fibrin network in 96-well microtitre plates were overlaid with 50 μl of 50 mM Tris/150 mM NaCl2 (TBS) containing 5 mM calcium and 0.2% bovine serum albumin. Then, 64 nM human protein C (PC) was added with 30 mM benzamidine, 100 nM hirudin, or TBS, for 0 to 180 min. at 37°C. Twenty microlitres of supernatant was added to 80 μl of 100 nM hirudin solution to stop activated protein C (APC) generation, and APC was quantified by measuring the kinetics of hydrolysis of 500 μM S-2366 substrate at 405 nm.
Fibrin-induced EPC proliferation and migration is dependent on bound thrombin
When placed on the fibrin matrix, EPCs strongly adhered and proliferated, without the need for exogenous angiogenic growth factors (EBM2 medium). During the first hours of culture on the fibrin matrix, EPCs formed pseudo-tubes, but these rapidly disorganized owing to the rapid cell proliferation (data not shown). EPC proliferation was significantly reduced by hirudin, as measured by 3H thymidine incorporation (77,634 ± 15,644 versus 45,352 ± 12,822 counts per minute (cpm) for EPC proliferation on fibrin network in the absence or in the presence of hirudin, respectively, Fig. 3A), suggesting that thrombin bound to a fibrin clot is able to activate PAR-1 and to induce cell prolif-eration. We also examined whether EPCs were able to migrate towards a fibrin network in a modified Boyden chamber. Fibrin attracted EPCs through the chamber membrane, and this effect was inhibited by fibrin pretreatment with hirudin (100%versus 58 ± 5% for EPC migration towards fibrin in the absence or in the presence of hirudin, respectively, Fig. 3B). The partial inhibition of EPC proliferation and migration by hirudin suggests the involvement of mechanisms other than PAR-1 activation by thrombin bound to fibrin.
3.
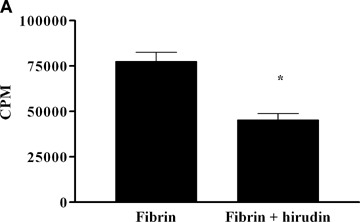
Impact of thrombin bound to a fibrin clot on EPC proliferation and migration (A) EPCs plated at a density of 5 × 104/well were cultured for 24 hrs on a fibrin network (pre-treated or not with hirudin for 30 min. in unsupple-mented EBM-2 medium. The cells were then washed twice in ice-cold PBS and lysed overnight at 4°C in 1 N NaOH. DNA synthesis was determined by measuring the incorporation of 5′-[3H]-thymidine for 4 hrs with a Betamatic counter. Results are expressed as mean ± SEM of at least three separate experiments.*P < 0.05.(B) EPC migration was measured in modified Boyden chambers with 8-μm pore-size filters. EPCs were seeded at a density of 5 × 104 per well in 200 μl of EBM-2 medium/1% SVF and were allowed to migrate for 5 hrs at 37°C toward a fibrin matrix placed in the lower chamber. Fibrin networks were pre-treated with 100 nM hirudin for 30 min. Results are expressed as mean ± SEM of at least three separate experiments.*P < 0.05
EPCs interfere with fibrin lysis
EPCs have strong proteolytic potential and express high levels of u-PA and u-PAR, two factors involved in the angiogenic properties of EPC in vitro[9]. Assuming that EPCs can promote fibrin solubilization, we examined the fibrin network lysis by measuring D-dimer release. In the absence of EPCs, or in the presence of MNCs, spontaneous fibrin lysis occurred within 72 hrs at 37°C. Surprisingly, fibrin lysis was inhibited by EPCs, as shown by a significant decrease in D-dimer generation (487 ± 8 versus 84 ± 10 ng/ml in the absence or in the presence of EPCs after 72 hrs of incubation, respectively). Such an inhibition of fibrin lysis by EPCs was unaffected by pre-treatment with hirudin (84 ± 10 versus 92 ± 3 ng/ml in the absence or in the presence of pre-treatment with hirudin, after 72 hrs of incubation).
In an attempt to explain these data, we first evaluated the expression level of genes encoding the main fibrinolytic system actors before and after PAR-1 activation. We used real-time quantitative RT-PCR to measure the expression of u-PA, u-PAR, t-PA, PAI-1 and PAI-2. All these genes were expressed at high levels by EPCs and were over-expressed 4 hrs after PAR-1 activation by thrombin or by the SFLLRN peptide (Table 2). The corresponding protein levels were determined in EPC lysates and supernatants by using ELISA. After thrombin and SFLLRN peptide treatment, u-PAR antigen levels in cell lysates increased by 283% and 331%, respectively (Fig. 4A). The increase in u-PAR membrane expression was confirmed by flow cytometry (data not shown). The level of secreted u-PA increased by 537% and 258%, respectively (Table 2). The level of PAI-1, a potent t-PA and u-PA inhibitor, also significantly increased after PAR-1 activation (Table 2). These results suggest that, in EPCs, PAR-1 activation is associated with modification of fibrinolytic balance.
2.
Effect of SFLLRN peptide and thrombin on mRNA and protein levels of t-PA, u-PA, u-PAR, PAI-1 and PAI-2
| SFLLRN 75 μM | Thrombin 10 nM | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mRNA fold-ratio (%± SD) | Protein fold-ratio (%± SD) | mRNA fold-ratio (%± SD) | Protein fold-ratio (%± SD) | ||||||||||
| u-PA | 420 ± 10 | 214 ± 71 | 795 ± 95 | 273 ± 26 | |||||||||
| secreted: 537 ± 362 | secreted: 258 ± 16 | ||||||||||||
| u-PAR | 400 ± 110 | 283 ± 70 | 275 ± 45 | 331 ± 104 | |||||||||
| t-PA | 230 ± 11 | 263 ± 36 | 370 ± 170 | 442 ± 309 | |||||||||
| PAI-1 | 235 ± 55 | 129 ± 17 | 220 ± 20 | 132 ± 26 | |||||||||
| PAI-2 | 415 ± 55 | Not detectable | 150 ± 24 | Not detectable | |||||||||
4.
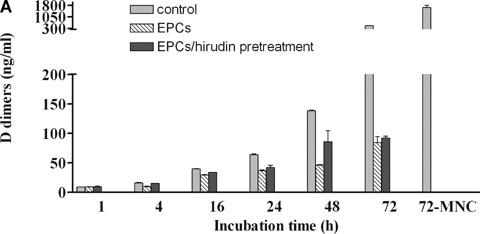
Fibrin network lysis by EPCs.(A) D-dimer release by EPCs and mononuclear cells (MNCs) cultured on fibrin matrices for 72 hrs. The control corresponds to spontaneous clot lysis in the absence of cells (EBM-2 medium). Closed histograms show the results of fibrin network pre-treatment with 100 nM hirudin for 30 min. before adding EPCs. Results are expressed as mean ± SEM of at least three separate experiments.*P < 0.05.(B) PAI-1 levels in supernatants of EPCs and MNCs placed on fibrin clots for 72 hrs. The control corresponds to basal levels of PAI-1 in the supernatant of the clot in the absence of cells (EBM-2 medium). When indicated, thrombin was inhibited by pre-treating the fibrin network with 100 nM hirudin for 30 min. Results are expressed as mean ±SEM of at least three separate experiments.*P < 0.05.
To understand the observed inhibition of fibrin lysis by EPCs, we measured fibrinolytic molecules in the supernatant of the fibrin network. No t-PA was detected in the supernatant of the fibrin network. The presence of EPCs (compared to MNCs) resulted in a 10-fold increase in the PAI-1 concentration in conditioned medium after 24 hrs (7.5 ± 2 versus 78 ± 3 ng of PAI-1 antigen/105 cells in fibrin network supernatants in the absence or in the presence of EPCs, respectively). Hirudin pre-treatment of the fibrin matrix partially inhibited PAI-1 secretion by EPCs that stay at very high levels in the supernatant (Fig. 4B) (78 ± 3 versus 57 ± 7 ng/105 cells in fibrin network supernatants in the absence or in the presence of pre-treatment of EPCs with hirudin, respectively, P= 0.03). All together, these data infer that EPCs locally express fibrinolytic enzymes to invade fibrin network, a phenomenon magnified by PAR-1 activation by adsorbed thrombin. However, high levels of secreted PAI-1 result in a global antifibrinolytic activity of EPCs within the fibrin network.
Discussion
Since their discovery in 1997, EPCs have been implicated in neovascularization [20, 21]. Recently, fibrin and activated platelets have been shown to guide stem cells to sites of vessel damage and to promote differentiation towards an endothelial cell phenotype [22]. Moreover, the presence of bone marrow–derived EPCs has been shown in resolving thrombi [3, 10]. However, EPCs are scarce in blood and their role in clot recanalization in vivo is unclear. Likewise, EPC transfer could interfere with fibrin clot dissolution and thrombosis resolution.
We have previously detected the main thrombin receptor, PAR-1, on EPCs. The present study provides the first experimental evidence of EPCR expression on EPCs and confirms that EPCs express thrombomodulin (CD141) [23, 24]. Recently, high-level EPCR expression was observed on haematopoietic stem cells [25]. EPCR facilitates PC activation by the thrombin-thrombomodulin complexes. Our finding that EPCs express the two cofactors required for PC activation on their surface suggests that EPCs may have anticoagulant properties when interacting with thrombin adsorbed on the fibrin clots. Indeed, fibrin matrix prepared to cultivate EPCs retains active thrombin. After adding purified human PC, we observed APC generation on the fibrin matrix in the presence of EPCs. Our results suggest that EPC recruitment or injection could modify thrombus structure by exerting a local anticoagulant effect.
During the clotting process, thrombin binds to the fibrin clot and retains its catalytic activity. To examine the impact of such an adsorption on EPCs, we prepared a human fibrin matrix. Thrombin adsorbed to the fibrin network triggered EPC chemotaxis and proliferation, suggesting PAR-1 activation by bound thrombin. In the fibrin matrix model used here, involvement of growth factors other than thrombin is likely to explain why hirudin only partially inhibited EPC expansion and migration. Large amounts of VEGF have been found at the site of haemostatic plug formation in vivo, with a possible role in thrombus recanalization [4]. In our experimental conditions, VEGF contained in the clot could explain part of the angiogenic effect of the fibrin network [26]. Indeed, fibrin incorporates a variety of proangiogenic factors, such as VEGF, fibroblast growth factor and interleukin-8 that may generate an angiogenic environment within the thrombus [4, 27]. Moreover, inte-grins appear to play a fundamental role in EPC biology [28–30]. Binding of fibrin RGD sequence with EPC integrins could thus increase proangiogenic properties of EPCs on the fibrin network.
In vivo, fibrin clots can induce EPC homing and differentiation from peripheral blood MNCs to ischemic sites [10]. The important role of fibrin deposition and degradation in neovessel formation highlights the crucial function of the haemostasis system in the regulation of angiogenesis and tumour growth [31]. The dense cross-linked structure of fibrin represents a major obstacle for EPC locomotion, and EPCs thus use u-PA and tPA fibrinolytic activities and matrix metalloproteinases (MMP) proteolytic systems to invade fibrin networks [32, 33]. In order to identify factors involved in the control of EPC-mediated fibrinolysis by thrombin, we studied the effect of PAR-1 activation on EPC synthesis of fibrinolytic proteins. As shown in Table 2, thrombin and SFL-LRN, a PAR-1-activating peptide, both significantly increased mRNA levels of tPA, u-PA and its receptor u-PAR, as determined by quantitative real-time PCR and ELISA. Expression of u-PAR, that plays an important role in the angiogenic properties of EPCs, was also increased at the cell surface after 24 hrs. This up-regulation of u-PA, t-PA and u-PAR in EPCs upon SFLLRN treatment points to the involvement of PAR-1. Transfer of bone marrow progenitors from wild-type mice restores thrombus resolution in u-PA−/–animals, suggesting that the fibrinolytic potential of bone marrow-derived cells plays an important role in this process. Thus, the increase in u-PA synthesis upon PAR-1 activation by EPCs entering a fibrin clot might contribute to thrombus resolution and subsequent recanalization. Finally, we studied EPC lysis of fibrin clots by measuring D-dimer release, and found that EPCs stabilize fibrin clot by inhibiting its degradation. Thrombolysis is regulated by the local balance between proteases and their inhibitors. PAI-1 is the key inhibitor of the fibrinolytic system, by inhibiting t-PA and u-PA, subsequently PAI-1 inhibits fibrin clot lysis [34, 35]. In our model, important PAI-1 secretion could be responsive of thrombus lysis inhibition induced by EPCs. It may counterbalance the local degradation of fibrin induced by the overexpression of u-PA and u-PAR by EPCs under the activation by thrombin. In humans, increased PAI-1 levels have been found associated with higher cardiovascular risk [36]. Both metabolic and genetic factors determine circulating PAI-1 levels [37]. Nine different polymorphisms have been detected in the PAI-1 gene, the most extensively studied being the 4G/5G polymorphism in the promoter [38]. Recently, the endothelial 4G/5G PAI-1 genotype has been found to influence PAI-1 secretion by endothelial cells [39]. Such a variable level of PAI-1 secretion by EPCs might modify their interaction with thrombus.
The role of fibrin on EPC migration and subsequent activation upon adsorbed thrombin suggests potential clinical implications. Indeed, PAR-1 specific antagonists have been developed [40, 41] and tested for platelet inhibition [42] in various diseases such as thrombosis, intestinal inflammatory disorders and experimental liver fibrosis [42–45]. These compounds have been found to inhibit angiogenesis in vitro[46]. Our findings are a supplementary argument justifying the development of PAR-1 antagonists in cancer and other angiogenesis-related diseases. Moreover, cardiovascular disease correlates with a decrease in EPC numbers [47], that might result in a delayed thrombus recanalization by EPCs.
Finally, the fibrin matrix could be used as an autologous matrix to expand EPCs ex vivo. However, the secretion of high levels of the fibrinolysis inhibitor PAI-1 could interfere with their angiogenic potential –i.e. proliferation, migration, differentiation or adhesive properties. Indeed, PAI-1 has been shown to inhibit adhesive properties of endothelial cells [48] or to interfere with MMP activation.
The major limitation of our study is that a fibrin clot prepared in vitro differs from an in vivo thrombus that is a complex biomaterial, not limited to thrombin and fibrin, that trap red blood cells and leucocytes releasing protease activities and binding plasminogen, u-PA and t-PA. These activities probably impair the ability of the thrombus to be colonized by EPCs. Moreover, the contribution of these cells to a thrombus in the early stages, such as platelets and neutrophils, and in the later stages, such as monocytes/macrophages, are likely to modify the balance between procoagulant and fibrinolytic properties and also the matrix composition. Indeed, cells within the thrombus can interact with plasma components trapped in the matrix, such as fibronectin [49]. Plasma fibronectin is a substrate for activated factor XIII and is thereby incorporated into fibrin clots. Other sources of fibronectin than plasma are available in vivo for potent interaction with EPCs, such as cellular fibronectin secreted from platelet-granules and other cells near an injury site, and tissue fibronectin present in the vascular basement membrane. Aside from altering the structure of the fibrin network, a possible role of fibronectin in blood clots is to mediate interactions between cells or platelets and fibrin. Thus, fibronectin as well as fibrin may interact with EPC integrins during clot formation or recanalization. Finally, platelets activated during the clot formation secrete other adhesive proteins including fibrinogen, thrombospondin, vitronectin, and von Willebrand factor. Another limitation of our study is the absence of blood flow, which role on EPC activation and on their ability to recanalize the thrombus is still unknown. Indeed, some studies have shown that shear stress induces differentiation of embryonic stem cells in endothelial phenotype [50, 51] and also modulates thrombogenic potential of human EPCs [52, 53].
In conclusion, thrombin adsorbed to fibrin clots may contribute to EPC recruitment. The anticoagulant properties of EPCs, including APC generation, could limit thrombus extension. This property is counterbalanced by strong PAI-1 secretion by EPCs, which might prevent premature thrombolysis. This latter property might hinder therapeutic use of EPCs for thrombus recanalization.
Acknowledgments
We thank Dr. CT. Esmon for kindly providing antibodies against EPCR. This study was supported by research grants from Inserm (‘Réseau de Recherche sur les Cellules Souches’), Programme Hospitalier de Recherche Clinique OPTIPEC (Ministère Chargé de la Santé, PHRC AOM 03 034, sponsor: AP-HP), and Leducq Transatlantic Network of Excellence on Atherothrombosis Research (Grant 04CVD01).
References
- 1.McGuinness CL, Humphries J, Waltham M, Burnand KG, Collins M, Smith A. Recruitment of labelled monocytes by experimental venous thrombi. Thromb Haemost. 2001;6:1018–24. [PubMed] [Google Scholar]
- 2.Wakefield TW, Linn MJ, Henke PK, Kadell AM, Wilke CA, Wrobleski SK, Sarkar M, Burdick MD, Myers DD, Strieter RM. Neovascularization during venous thrombosis organization:a preliminary study. J Vasc Surg. 1999;5:885–92. doi: 10.1016/s0741-5214(99)70013-3. [DOI] [PubMed] [Google Scholar]
- 3.Modarai B, Burnand KG, Sawyer B, Smith A. Endothelial progenitor cells are recruited into resolving venous thrombi. Circulation. 2005;20:2645–53. doi: 10.1161/CIRCULATIONAHA.104.492678. [DOI] [PubMed] [Google Scholar]
- 4.Waltham M, Burnand KG, Collins M, McGuinness CL, Singh I, Smith A. Vascular endothelial growth factor enhances venous thrombus recanalisation and organisation. Thromb Haemost. 2003;1:169–76. [PubMed] [Google Scholar]
- 5.Smadja DM, Cornet A, Emmerich J, Aiach M, Gaussem P. Endothelial progenitor cells: Characterization, in vitro expansion, and prospects for autologous cell therapy. Cell Biol Toxicol. 2007;4:223–39. doi: 10.1007/s10565-007-0177-6. [DOI] [PubMed] [Google Scholar]
- 6.Werner N, Nickenig G. Clinical and therapeutical implications of EPC biology in atherosclerosis. J Cell Mol Med. 2006;2:318–32. doi: 10.1111/j.1582-4934.2006.tb00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts N, Jahangiri M, Xu Q. Progenitor cells in vascular disease. J Cell Mol Med. 2005;3:583–91. doi: 10.1111/j.1582-4934.2005.tb00490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sata M, Fukuda D, Tanaka K, Kaneda Y, Yashiro H, Shirakawa I. The role of circulating precursors in vascular repair and lesion formation. J Cell Mol Med. 2005;3:557–68. doi: 10.1111/j.1582-4934.2005.tb00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basire A, Sabatier F, Ravet S, Lamy E, Mialhe A, Zabouo G, Paul P, Gurewich V, Sampol J, Dignat-George F. High urokinase expression contributes to the angiogenic properties of endothelial cells derived from circulating progenitors. Thromb Haemost. 2006;4:678–88. [PubMed] [Google Scholar]
- 10.Moldovan NI, Asahara T. Role of blood mononuclear cells in recanalization and vascularization of thrombi: past, present, and future. Trends Cardiovasc Med. 2003;7:265–9. doi: 10.1016/s1050-1738(03)00108-7. [DOI] [PubMed] [Google Scholar]
- 11.Singh I, Burnand KG, Collins M, Luttun A, Collen D, Boelhouwer B, Smith A. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice:rescue by normal bone marrow-derived cells. Circulation. 2003;6:869–75. doi: 10.1161/01.cir.0000050149.22928.39. [DOI] [PubMed] [Google Scholar]
- 12.Major CD, Santulli RJ, Derian CK, Andrade-Gordon P. Extracellular mediators in atherosclerosis and thrombosis: lessons from thrombin receptor knockout mice. Arterioscler Thromb Vasc Biol. 2003;6:931–9. doi: 10.1161/01.ATV.0000070100.47907.26. [DOI] [PubMed] [Google Scholar]
- 13.Smadja DM, Bieche I, Uzan G, Bompais H, Muller L, Boisson-Vidal C, Vidaud M, Aiach M, Gaussem P. PAR-1 activation on human late endothelial progenitor cells enhances angiogenesis in vitro with upregulation of the SDF-1/CXCR4 system. Arterioscler Thromb Vasc Biol. 2005;11:2321–7. doi: 10.1161/01.ATV.0000184762.63888.bd. [DOI] [PubMed] [Google Scholar]
- 14.Smadja DM, Laurendeau I, Avignon C, Vidaud M, Aiach M, Gaussem P. The angiopoietin pathway is modulated by PAR-1 activation on human endothelial progenitor cells. J Thromb Haemost. 2006;9:2051–8. doi: 10.1111/j.1538-7836.2006.02101.x. [DOI] [PubMed] [Google Scholar]
- 15.Olivot JM, Estebanell E, Lafay M, Brohard B, Aiach M, Rendu F. Thrombomodulin prolongs thrombin-induced extracellular signal-regulated kinase phosphorylation and nuclear retention in endothelial cells. Circ Res. 2001;7:681–7. doi: 10.1161/hh0701.088769. [DOI] [PubMed] [Google Scholar]
- 16.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;5574:1880–2. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 17.Picard V, Marque PE, Paolucci F, Aiach M, Le Bonniec BF. Topology of the stable serpin-protease complexes revealed by an autoantibody that fails to react with the monomeric conformers of antithrombin. J Biol Chem. 1999;8:4586–93. doi: 10.1074/jbc.274.8.4586. [DOI] [PubMed] [Google Scholar]
- 18.Bieche I, Onody P, Laurendeau I, Olivi M, Vidaud D, Lidereau R, Vidaud M. Real-time reverse transcription-PCR assay for future management of ERBB2-based clinical applications. Clin Chem. 1999:1148–56. [PubMed] [Google Scholar]
- 19.Hur J, Yoon CH, Kim HS, Choi JH, Kang HJ, Hwang KK, Oh BH, Lee MM, Park YB. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;2:288–93. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, Magner M, Isner JM, Asahara T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;4:434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 21.Kawamoto A, Tkebuchava T, Yamaguchi J, Nishimura H, Yoon YS, Milliken C, Uchida S, Masuo O, Iwaguro H, Ma H, Hanley A, Silver M, Kearney M, Losordo DW, Isner JM, Asahara T. Intramyocardial transplantation of autologous endothelial progenitor cells for therapeutic neovascu-larization of myocardial ischemia. Circulation. 2003;3:461–8. doi: 10.1161/01.cir.0000046450.89986.50. [DOI] [PubMed] [Google Scholar]
- 22.De Boer HC, Verseyden C, Ulfman LH, Zwaginga JJ, Bot I, Biessen EA, Rabelink TJ, Van Zonneveld AJ. Fibrin and activated platelets cooperatively guide stem cells to a vascular injury and promote differentiation towards an endothelial cell phenotype. Arterioscler Thromb Vasc Biol. 2006;7:1653–9. doi: 10.1161/01.ATV.0000222982.55731.f1. [DOI] [PubMed] [Google Scholar]
- 23.Bagley RG, Walter-Yohrling J, Cao X, Weber W, Simons B, Cook BP, Chartrand SD, Wang C, Madden SL, Teicher BA. Endothelial precursor cells as a model of tumor endothelium: characterization and comparison with mature endothelial cells. Cancer Res. 2003;18:5866–73. [PubMed] [Google Scholar]
- 24.Hatzopoulos AK, Folkman J, Vasile E, Eiselen GK, Rosenberg RD. Isolation and characterization of endothelial progenitor cells from mouse embryos. Development. 1998;8:1457–68. doi: 10.1242/dev.125.8.1457. [DOI] [PubMed] [Google Scholar]
- 25.Balazs AB, Fabian AJ, Esmon CT, Mulligan RC. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood. 2006;6:2317–21. doi: 10.1182/blood-2005-06-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;4:777–94. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henke PK, Wakefield TW, Kadell AM, Linn MJ, Varma MR, Sarkar M, Hawley A, Fowlkes JB, Strieter RM. Interleukin-8 administration enhances venous thrombosis resolution in a rat model. J Surg Res. 2001;1:84–91. doi: 10.1006/jsre.2001.6122. [DOI] [PubMed] [Google Scholar]
- 28.Chavakis E, Aicher A, Heeschen C, Sasaki K, Kaiser R, El Makhfi N, Urbich C, Peters T, Scharffetter-Kochanek K, Zeiher AM, Chavakis T, Dimmeler S. Role of beta2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J Exp Med. 2005;1:63–72. doi: 10.1084/jem.20041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin G, Ii M, Silver M, Wecker A, Bord E, Ma H, Gavin M, Goukassian DA, Yoon YS, Papayannopoulou T, Asahara T, Kearney M, Thorne T, Curry C, Eaton L, Heyd L, Dinesh D, Kishore R, Zhu Y, Losordo DW. Functional disruption of alpha4 integrin mobilizes bone marrow-derived endothelial progenitors and augments ischemic neovascularization. J Exp Med. 2006;1:153–63. doi: 10.1084/jem.20050459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smadja DM, Bieche I, Helley D, Laurendeau I, Simonin G, Muller L, Aiach M, Gaussem P. Increased VEGFR2 expression during human late endothelial progenitor cells expansion enhances in vitro angiogenesis with upregulation of integrin a6. J Cell Mol Med. 2007;11:1149–61. doi: 10.1111/j.1582-4934.2007.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rickles FR, Falanga A. Molecular basis for the relationship between thrombosis and cancer. Thromb Res. 2001;6:V215–24. doi: 10.1016/s0049-3848(01)00285-7. [DOI] [PubMed] [Google Scholar]
- 32.Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;7:1104–17. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 33.Kroon ME, Koolwijk P, Van Goor H, Weidle UH, Collen A, Van Der Pluijm G, Van Hinsbergh VW. Role and localization of urokinase receptor in the formation of new microvascular structures in fibrin matrices. Am J Pathol. 1999;6:1731–42. doi: 10.1016/S0002-9440(10)65429-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robbie LA, Booth NA, Croll AM, Bennett B. The roles of alpha 2-antiplasmin and plasminogen activator inhibitor 1 (PAI-1) in the inhibition of clot lysis. Thromb Haemost. 1993;2:301–6. [PubMed] [Google Scholar]
- 35.Mutch NJ, Thomas L, Moore NR, Lisiak KM, Booth NA. TAFIa, PAI-1 and alpha-antiplasmin: complementary roles in regulating lysis of thrombi and plasma clots. J Thromb Haemost. 2007;4:812–7. doi: 10.1111/j.1538-7836.2007.02430.x. [DOI] [PubMed] [Google Scholar]
- 36.Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;24:1792–801. doi: 10.1056/NEJM200006153422406. [DOI] [PubMed] [Google Scholar]
- 37.Humphries SE, Panahloo A, Montgomery HE, Green F, Yudkin J. Gene-environment interaction in the determination of levels of haemostatic variables involved in thrombosis and fibrinolysis. Thromb Haemost. 1997;1:457–61. [PubMed] [Google Scholar]
- 38.Eriksson P, Kallin B, Van 't Hooft FM, Bavenholm P, Hamsten A. Allele-specific increase in basal transcription of the plasminogen-activator inhibitor 1 gene is associated with myocardial infarction. Proc Natl Acad Sci USA. 1995;6:1851–5. doi: 10.1073/pnas.92.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roncal C, Orbe J, Belzunce M, Rodriguez JA, Paramo JA. The 4G/5G PAI-1 polymorphism influences the endothelial response to IL-1 and the modulatory effect of pravastatin. J Thromb Haemost. 2006;8:1798–803. doi: 10.1111/j.1538-7836.2006.02031.x. [DOI] [PubMed] [Google Scholar]
- 40.Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, Eckardt AJ, Hoekstra WJ, McComsey DF, Oksenberg D, Reynolds EE, Santulli RJ, Scarborough RM, Smith CE, White KB. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci USA. 1999;22:12257–62. doi: 10.1073/pnas.96.22.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;10:1161–5. doi: 10.1038/nm760. [DOI] [PubMed] [Google Scholar]
- 42.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;9:1244–54. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- 43.Derian CK, Damiano BP, Addo MF, Darrow AL, D'Andrea MR, Nedelman M, Zhang HC, Maryanoff BE, Andrade-Gordon P. Blockade of the thrombin receptor protease-activated receptor-1 with a small-molecule antagonist prevents thrombus formation and vascular occlusion in nonhuman primates. J Pharmacol Exp Ther. 2003;2:855–61. doi: 10.1124/jpet.102.042663. [DOI] [PubMed] [Google Scholar]
- 44.Vergnolle N, Cellars L, Mencarelli A, Rizzo G, Swaminathan S, Beck P, Steinhoff M, Andrade-Gordon P, Bunnett NW, Hollenberg MD, Wallace JL, Cirino G, Fiorucci S. A role for proteinase-activated receptor-1 in inflammatory bowel diseases. J Clin Invest. 2004;10:1444–56. doi: 10.1172/JCI21689. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Fiorucci S, Antonelli E, Distrutti E, Severino B, Fiorentina R, Baldoni M, Caliendo G, Santagada V, Morelli A, Cirino G. PAR1 antagonism protects against experimental liver fibrosis. Role of proteinase receptors in stellate cell activation. Hepatology. 2004;2:365–75. doi: 10.1002/hep.20054. [DOI] [PubMed] [Google Scholar]
- 46.Zania P, Kritikou S, Flordellis CS, Maragoudakis ME, Tsopanoglou NE. Blockade of angiogenesis by small molecule antagonists to protease-activated receptor-1 (PAR-1): Association with endothelial cell growth suppression and induction of apoptosis. J Pharmacol Exp Ther. 2006;318:246–54. doi: 10.1124/jpet.105.099069. [DOI] [PubMed] [Google Scholar]
- 47.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;7:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 48.Al-Fakhri N, Chavakis T, Schmidt-Woll T, Huang B, Cherian SM, Bobryshev YV, Lord RS, Katz N, Preissner KT. Induction of apoptosis in vascular cells by plasminogen activator inhibitor-1 and high molecular weight kininogen correlates with their anti-adhesive properties. Biol Chem. 2003;3:423–35. doi: 10.1515/BC.2003.048. [DOI] [PubMed] [Google Scholar]
- 49.Cho J, Mosher DF. Role of fibronectin assembly in platelet thrombus formation. J Thromb Haemost. 2006;7:1461–9. doi: 10.1111/j.1538-7836.2006.01943.x. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto K, Sokabe T, Watabe T, Miyazono K, Yamashita JK, Obi S, Ohura N, Matsushita A, Kamiya A, Ando J. Fluid shear stress induces differentiation of Flk-1-positive embryonic stem cells into vascular endothelial cells in vitro. Am J Physiol Heart Circ Physiol. 2005;4:H1915–24. doi: 10.1152/ajpheart.00956.2004. [DOI] [PubMed] [Google Scholar]
- 51.Wang H, Riha GM, Yan S, Li M, Chai H, Yang H, Yao Q, Chen C. Shear stress induces endothelial differentiation from a murine embryonic mesenchymal progenitor cell line. Arterioscler Thromb Vasc Biol. 2005;9:1817–23. doi: 10.1161/01.ATV.0000175840.90510.a8. [DOI] [PubMed] [Google Scholar]
- 52.Yang Z, Wang JM, Wang LC, Chen L, Tu C, Luo CF, Tang AL, Wang SM, Tao J. In vitro shear stress modulates antithrombogenic potentials of human endothelial progenitor cells. J Thromb Thrombolysis. 2007;2:121–7. doi: 10.1007/s11239-006-9045-0. [DOI] [PubMed] [Google Scholar]
- 53.Yang Z, Tao J, Wang JM, Tu C, Xu MG, Wang Y, Pan SR. Shear stress contributes to t-PA mRNA expression in human endothelial progenitor cells and nonthrombogenic potential of small diameter artificial vessels. Biochem Biophys Res Commun. 2006;2:577–84. doi: 10.1016/j.bbrc.2006.01.172. [DOI] [PubMed] [Google Scholar]