

Abstract
Real time multiphoton imaging provides a great opportunity to study cell trafficking and cell-to-cell interactions in their physiological 3-dimensionnal environment. Biological activities of immune cells mainly rely on their motility capacities. Blood monocytes have short half-life in the bloodstream; they originate in the bone marrow and are constitutively released from it. In inflammatory condition, this process is enhanced, leading to blood monocytosis and subsequent infiltration of the peripheral inflammatory tissues. Identifying the biomechanical events controlling monocyte trafficking from the bone marrow towards the vascular network is an important step to understand monocyte physiopathological relevance. We performed in vivo time-lapse imaging by two-photon microscopy of the skull bone marrow of the Csf1r-Gal4VP16/UAS-ECFP (MacBlue) mouse. The MacBlue mouse expresses the fluorescent reporters enhanced cyan fluorescent protein (ECFP) under the control of a myeloid specific promoter 1, in combination with vascular network labelling. We describe how this approach enables the tracking of individual medullar monocytes in real time to further quantify the migratory behaviour within the bone marrow parenchyma and the vasculature, as well as cell-to-cell interactions. This approach provides novel insights into the biology of the bone marrow monocyte subsets and allows to further address how these cells can be influenced in specific pathological conditions.
Keywords: Immunology, Issue 96, Immunology, hematology, Intravital imaging, bone marrow, monocytes, cell trafficking.
Introduction
The bone marrow plays a central role in hematopoiesis and represents the main reservoir of monocytes that constitutively recirculate between the blood and the medullar parenchyma, renew the pool of circulating monocytes with a short life span 2,3 and participate in the reconstitution of the steady state tissue-macrophages and dendritic cells 4. During inflammation or after transient aplasia, monocytes are actively mobilized from either the bone marrow or the spleen 5, 6, 7 and colonize inflamed organs. Several chemoattractant axis have been involved in the process of myeloid cell mobilization from the bone marrow 8, 5, 6,9. Beyond the myeloid compartment the bone marrow is also an important site of T lymphocyte priming 10 and a niche of immunological memory 11,12. Thus, this tissue is central for numerous investigations in the field of hematology and immunology. Our knowledge on the structural organization of medullar myeloid cells mainly arises from the analysis of histological section of fixed tissues 13. This static view does not allow for a study of the cellular exchange dynamic between the different compartments of the bone marrow, which is the basis of its functional activity.
Intravital imaging constitutes an important biological input in the study of cell mobility, cell adherence and cell-to-cell interactions, which were previously described only from in vitro systems. Technical challenges for proper intravital imaging include the ability to reach the tissue of interest in an optical point of view, and to maximize its isolation from physiological (breath, muscle or peristaltic contractions) or mechanical drifts (tissue disruption and extension following surgery, and exposure to microscope objective as well as temperature and vascular/oxygenation perturbations). Microscopic drifts may limit the ability to keep the focus a long time and could introduce artifacts in the quantification of cell motility. One alternative, validated for several tissues to reduce these technical difficulties, is to work on explanted tissue incubated in a thermostated and oxygenated medium; however, complete disruption of the lymphatic and vascular circulation may be problematic. Intravital imaging of skull bone marrow has several advantages concerning these issues. Firstly, it requires minimal surgical action. Secondly, thickness of the bone in this region allows direct visualization of bone marrow niches without abrasion, thus reducing physiological perturbations. The medullar network can be imaged in the parasagittal region of the bone; however the sinusoids are more visible in the fronto-parietal area where the bone matrix is thinner12,14.
Intravital imaging relies on the availability of the most accurate fluorescent reporter tagging the population of interest. In vitro labelling of purified cell population before adoptive transfer led to important characterization of hematopoietic stem cell niches 15 or bone marrow endothelial microdomains favouring tumor engraftment 16, and provided several fundamental inputs on key concepts in immunology 17 . However, this approach usually requires hundreds of thousands of cells to get a chance to detect them afterwards in vivo. This could be explained by the high mortality rate following staining, the dilution in the whole body and the change in the activation state, which might lead to biased homing. Endogenous tagging from transgenic mouse system greatly overcomes these limitations and has allowed to image the behaviour of endogenous osteoblast 8, megakaryocytes 18 or myeloid-lineage subsets 6 . Nevertheless, one has to be cautious when considering the specificity of the fluorescent reporter among the studied subset.
The Csf1r-Gal4VP16/UAS-ECFP, called MacBlue mouse 1, is a valuable transgenic system to study medullar monocytes with real time imaging 6. Intravenous injection of high molecular weight rhodamin-dextran distinguishes the medullar parenchyma from the vascular sinusoid network of the bone marrow. Using this approach, it is possible to track the monocyte behaviour in the different medullar compartments in a specific physiopathological context of interest. Furthermore, we propose an additional strategy to compare monocyte dynamics with that of neutrophils through in vivo labelling using a specific antibody.
Protocol
NOTE: All experiment protocols were approved by the French Animal Experimentation and Ethics Committee and validated by the “Service Protection et Santé Animales, Environnement” with number A-75-2065. Sample sizes are chosen to ensure reproducibility of the experiments, and according to the 3R of animal ethic regulation.
1. Preparation of the Mouse
- Anaesthesia
- For short period of imaging (less than 1 hr), anaesthetize the mouse with an intraperitoneal injection of 200 µl of a saline solution containing Ketamine (100 mg/kg) and Xylazine (10 mg/kg).
- Alternatively, for longer period of imaging (up to 4 hr), anaesthetize the mouse with an inhalation of isoflurane 2.5% vaporized in a 70/30 mixture of O2/N2O, through an adapted mask.
- NOTE: This way provides for more stable unconsciousness and avoids serial intraperitoneal injection of drugs.
- Once the mouse is anesthetized, check for unconsciousness through stimulation of the foot pad with nippers.
- Vascular staining/ additional staining
- For vascular staining: Inject intravenously in the tail vein, 200 µl of Rhodamin-Dextran (2 MDa, 10 mg/ml in saline buffer) .
- For neutrophil staining: inject 2 µg of Ly6G-PE (clone 1A8) in 100 µl of saline solution intravenously in the tail vein.
- Immobilization
- For immobilization, use a custom made stereotactic holder adapted to the microscope and to the anaesthetic gas inhalator. NOTE: The sterotactic holder is a metal support with two metal plate brackets, one being fixed and the other removable and adaptable, to tighten the animal’s head.
- Install the head of the mouse between the retaining plates to ensure isolation from breathing movements. Tighten the ears between retaining plates and head to stretch the skin. Check for stability and breathing welfare of the animal.
- Remove the scalp
- Carefully use ethanol 70% to wet the hair of the scalp with a sterile applicator, avoiding contact with the eyes.
- Cut the skin with sterile scissors and nippers to fully remove the scalp from the backward of the parietal bone to the frontal bone. Keep away up to 3 mm from eyes and ears to prevent extensive bleeding. Remove the loose connective tissue covering the skull (periosteum). Wash out remaining hair with warm PBS-soaked paper towel.
- Immersion system set up
- Paste a rubber ring of 18mm diameter with surgical glue directly on the skull using a small gauge needle to control distribution. Glue the entire periphery of the rubber ring to prevent leakage (30 µl should be sufficient).
- Clean the skull under the ring one last time with PBS-soaked paper towel and then add 37 °C PBS for complete immersion of the skull. Add a drop of NaCl 0.9% solution or specific ophthalmic ointment on each eye of the mouse to avoid eye dryness. Drag the stereotactic holder under the objective.
- Approximately position the field using the microscope ocular through direct transmission.
2. Two-photon Imaging Acquisition
Use a multiphoton microscope coupled with a Ti:Sapphire crystal laser, which provides 140fs pulses of NIR light, selectively tunable from 680 to 1,080 nm, and an acousto-optic modulator for laser power control. Use a configuration including 3 external non-descanned detectors (NDD) (that enable simultaneous recording of 3 fluorescent channels) with a combination of 2 dichroic mirrors (565 nm and 690 nm), 565/610 and 500/550 bandpass filters, and a 485 short pass filter, with a plan apochromat ×20 (NA = 1) water immersion objective.
Set the heating chamber to allow good homeothermy of the anaesthetized mouse., NOTE: Temperature might be set 1 hr before imaging session for better stability. In our case, 32 °C has been chosen to get optimal body temperature of the anaesthetized mouse (36-37 °C) and is maintained up to 4 hr. This temperature may be adapted to the system used (digital probe can be used to check this point). In addition, the heating chamber used is dark/black and covers a part of the system (objectives and motorized plate) to avoid external light contamination.
- Turn on the system
- Turn on the Ti:Sapphire laser, then the whole microscope system. Launch the acquisition software. Set the laser wavelength range to 870 nm. Select appropriate NDD for recording. NOTE: In our case, second harmonic generation (SHG) 19and cyan fluorescent protein (ECFP) are detected by the NDD after the 485 short pass filter. ECFP is also detected by the NDD after the 500/550 bandpass filter and rhodamin-dextran is detected by the NDD after the 565/610 bandpass filter.
- Adjust acquisition settings
- Use the “live acquisition” imaging command of the software and using the microscope directional control, adjust the focus on a region with ECFP and Rhodamine signal. Set the laser power to the minimum to obtain best signal-to-noise ratio with minimal photo-damage. NOTE: The power setting used varies according to the depth of the region of interest. Typically the AOM settings are set around 20%, which represents a laser power beyond the objective around 15 mW. It is better to increase gain power on the PMTs for each NDD to reduce photo-bleaching and photo-damage. Typical acquisition settings are single scanning with a pixel dwell time of 1.58 µs, and a resolution of 512 x 512 pixels with a 1.5 magnification.
- Select regions of interest
- Once recording parameters are set, use again the “live” acquisition command, and survey the tissue to choose a desired (x,y,z) field to be monitored in real time. NOTE: Typically, we chose a field including both vascular and parenchymal areas.
- Register the field with the “position” software command. NOTE: This command memorizes x,y,z coordinates of different distant fields.
- Select and register additional fields of interest (up to 3) using the same procedure.
- Set a 3D z-stack with the “z-stack” software command on the first memorized position according to the required volume with chosen magnification. Memorize deeper position first, and then the highest position. Normalize laser power with the depth. NOTE: For multiple concomitant field imaging, we acquire 5 slices separated by 3 µm z-steps for each field to acquire a volume of 12 µm thickness. These settings might be adapted to the type of cell studied.
- Launch acquisition
- Select the “time series” software command and choose the duration of time interval and the number of cycles. Begin time-lapse imaging according to the time lapse and duration required by selecting the “start experiment” software command. NOTE: In our case, we acquire one volume each 30 sec during 60 cycles representing 30 min real time. Smaller time lapse can be performed to track fast cell rolling in the blood vessel. In this case thinner z-stack and no more than 2 different fields can be recorded at the same time.
- Regular monitoring of the set up.
- Always monitor the animal to make sure it is unconscious. NOTE: Shaking of whiskers or legs of the animal are indicators of revival. In case the animal’s breathing is jerky, the quantity of isoflurane can be slightly reduced. Strong tissue drift during acquisition is an indicator of mouse revival.
- Ensure the objective is properly immersed. Renew 37 °C PBS between two acquisitions (30 min). NOTE: Progressive disappearance of signal during acquisition is indicator of PBS leakage.
- End of procedure
- Once the required videos are recorded, euthanize the animal by cervical dislocation rapidly before resuscitation. NOTE: wound healing leads to the formation of a scab in 24 to 48 hr which limits the possibility to perform longitudinal imaging.
3. Data Analysis
- Drift correction
- Use Imaris Bitplane software for 3D automatic tracking.
- Open the 3D image sequence on Imaris. Select “spot” command, and generate manual tracking (mouse click+shift) for all time points for a minimum of 4 different anchor points, if possible homogeneously distributed in the field.
- Apply the drift correction using “correct drift” command in the “edit track” tab.
- Check for the quality of the correction in x, y and particularly in z to avoid artefactual wavering. NOTE: If the correction is not satisfactory, try other anchor points or exclude the video from the analysis.
- Cell tracking
- Use “add new spots” command and select “track spots over time” algorithm setting. Go to next step.
- Apply automatic cell tracking procedure either on the whole channel of interest using “source channel” command. Set objects (i.e., cells) diameter to 10 µm. Go to next step. NOTE: The choice will depend on the distinction between objects and specificity of the measured signal. In short, automatic tracking on the whole channel is possible only if detected signal is specific to the object of interest. Because both SHG and ECFP are detected by the NDD after the 485 short pass filter, tracking will be more specific and efficient using the ECFP signal recorded by the NDD after the 500/550 bandpass filters. Clustered or dense packed objects will require manual selection of individual objects similar to step 3.1.2.
- Select “quality” filter type, set the threshold to exclude tracking of unspecific objects. Go to next step.
- Choose “Brownian Algorithm Motion” with accurate parameters of “Max distance” and “Gap size” between two spots. Validate track generation.
- Once tracks are automatically calculated, select “track duration” filter type, set the threshold to eliminate tracks shorter than 120 sec (representing 4 time points).
- Mandatory: After automatic tracking, check the quality of the tracks.
- Unroll the time bar to check that the objects follow the track paths. If not, use the options of track point correction available in the “edit” tab: Delete false or improper tracks with “delete” command (delete any track point individually using “disconnect” command). If track path is discontinuous, add missing time points (mouse click+shift), select all points on a given track, and use “connect” command in the “edit track” tab.
- Quantification parameters
- Select the “Statistic” tab in “Preferences” command, choose the dynamic parameters of interest in the list. Select “Export all statistics to file” command and save the excel file generated. NOTE: Several dynamic parameters are directly obtained from the software such as track lengths, instantaneous velocity, mean velocity, and track straightness. Motility coefficient 6,20 can be determined but is not directly provided by the software.
- For each cell, calculate the mean of the displacements for each period of time (δt) (for a 30 sec time lapse movie, δt will be 30, 60, 90 sec, etc). Plot the mean of the square displacement as a function of the time interval. Obtain motility coefficient by calculating the slope of the linear part of the curve.
Representative Results
Mouse skull structure offers a good opportunity to study the bone marrow physiology by intravital imaging. The bone being thin around the front-parietal area, it is possible to get access to medullar niches without abrasion of the bone. Figure 1 represents a wide 2D field of the skull of a MacBlue transgenic mouse. The bone matrix is mainly composed of collagen I easily detectable by SHG 19. Injection of rhodamin-dextran stains the vascular network of the bone marrow and allows for the identification of parenchymal bone marrow niches between the bone matrix and the vessels 14. ECFP cells are distributed in the two compartments of the bone marrow, but are absent from the bone matrix. Monocytes are manually classified according to their location within the tissue. Vascular monocyte is defined when the whole cell is within the vasculature, disregarding the size of the vessel. Only big collecting venules are excluded from our analysis. Parenchymal monocyte is defined when the cell is located between the vasculature and the bone matrix.
Time-lapse imaging of specific region of interest (supplementary video 1 and 2) allows for the calculation of individual cell trajectory over time for either vascular (Figure 2A) or parenchymal (Figure 2B) monocytes. The track length shows that parenchymal monocytes display reduced displacement compared to vascular monocytes. Analysis of the displacement over time compares the mean velocity of monocytes in both compartments (Figure 2C). The mean square displacement as a function of time is a measure of the spatial extent of random motion. Motility coefficient is determined by the slope of the linear part of the curve and provides a better idea of the cell displacement (see discussion section). The motility coefficient of vascular monocytes (MC=71µm²/s) is higher than the motility coefficient of parenchymal monocytes (MC=4.4µm²/s).
The time resolution of acquisition must be adapted to the average cell velocity. Rolling vascular monocytes can make important displacement within a short time delay. For a more accurate cell tracking, reducing the time interval of acquisition is preferable. Figure 3 shows an illustration of monocyte tracking with two different time-resolutions. A 30 sec time-interval reduces the accuracy as well as the length of the pathways (in green dashed line) compared to 10 sec time-interval.
Finally, Figure 4 illustrates the possibility to analyze another cell subset concomitantly to monocytes. Ly6G is a specific marker of mature mouse neutrophils. The injection of anti-Ly6G combined with a fluorochrome (in this case Phyco-erythrin) 5 min before the imaging sequence stain neutrophils directly in vivo. This approach provides an additional dimension of analysis and the possibility to compare the behaviour of the different cell subsets. Such approach could be used to define subsets in the monocyte compartments.
 Figure 1. Wide field of skull bone marrow organization. In vivo two-photon laser scanning microscopy image of skull steady-state bone marrow from MacBlue mouse. Vasculature (in red) is stained by tail vain injection of 2MDa Rhodamin-Dextran 5 min before imaging session. The bone is visualized by second harmonic generation (blue). ECFP+ monocytes (Cyan) are located within parenchymal niches (dark areas between vasculature and bone) and within the vessels. Please click here to view a larger version of this figure.
Figure 1. Wide field of skull bone marrow organization. In vivo two-photon laser scanning microscopy image of skull steady-state bone marrow from MacBlue mouse. Vasculature (in red) is stained by tail vain injection of 2MDa Rhodamin-Dextran 5 min before imaging session. The bone is visualized by second harmonic generation (blue). ECFP+ monocytes (Cyan) are located within parenchymal niches (dark areas between vasculature and bone) and within the vessels. Please click here to view a larger version of this figure.
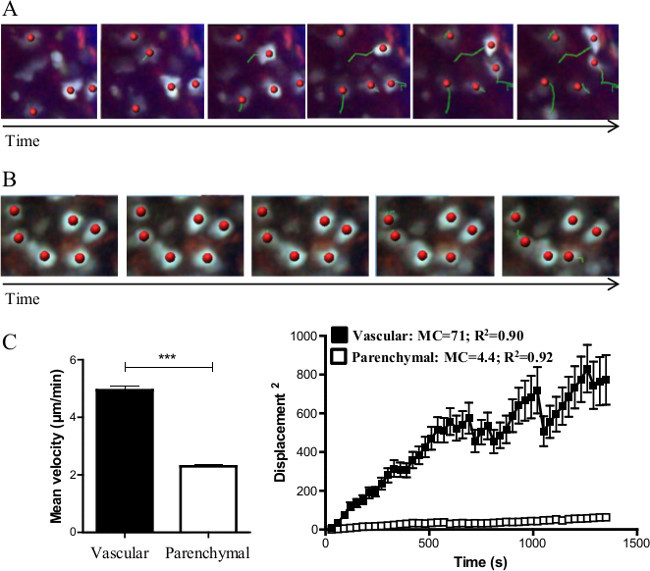 Figure 2. Real time cell tracking. Representative track paths along time of monocytes within (A) the vasculature and (B) the parenchyma. Tracks (green line) for each cell are automatically generated by the software. Red spots represent the centre of mass of tracked cells. (C) Mean velocity comparison between vascular and parenchymal monocytes. Mann-Whitney statistical analysis has been performed, *** represents p<0,001. (D) Mean squared displacement (msd) as a function of time is represented for vascular and parenchymal monocytes. Asymptotic curve indicates the constrained displacement of the cells over time, whereas linear curve indicates random walk in a non-constrained environment. Motility coefficient indicates the extent of cell displacement and is determined by the slope of the curve calculated by linear regression. Please click here to view a larger version of this figure.
Figure 2. Real time cell tracking. Representative track paths along time of monocytes within (A) the vasculature and (B) the parenchyma. Tracks (green line) for each cell are automatically generated by the software. Red spots represent the centre of mass of tracked cells. (C) Mean velocity comparison between vascular and parenchymal monocytes. Mann-Whitney statistical analysis has been performed, *** represents p<0,001. (D) Mean squared displacement (msd) as a function of time is represented for vascular and parenchymal monocytes. Asymptotic curve indicates the constrained displacement of the cells over time, whereas linear curve indicates random walk in a non-constrained environment. Motility coefficient indicates the extent of cell displacement and is determined by the slope of the curve calculated by linear regression. Please click here to view a larger version of this figure.
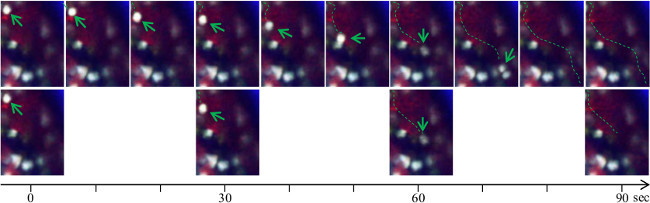 Figure 3. Time interval resolution in the study of rolling monocytes. Representative image sequences show that increased time resolution improves precision of the rolling cell trajectory. (Up) Image sequence with 10 sec time intervals. (Down) Image sequence with 30 sec time intervals. The quality of the track path is improved and the risk of considering two different cells for the same or the opposite is reduced. Please click here to view a larger version of this figure.
Figure 3. Time interval resolution in the study of rolling monocytes. Representative image sequences show that increased time resolution improves precision of the rolling cell trajectory. (Up) Image sequence with 10 sec time intervals. (Down) Image sequence with 30 sec time intervals. The quality of the track path is improved and the risk of considering two different cells for the same or the opposite is reduced. Please click here to view a larger version of this figure.
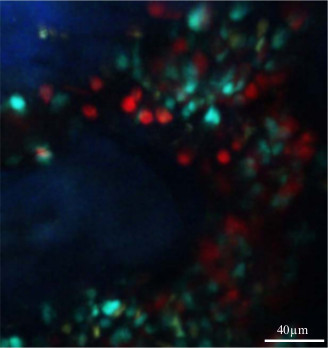 Figure 4. In vivo co-localization of medullar neutrophils and monocytes. This figure illustrates the possibility to detect another cell subset in vivo concomitantly with monocytes, by injecting specific monoclonal antibody combined with a fluorochrome. 2 µg of Ly6G-PE antibody has been injected intravenously 5 min before skull bone marrow imaging of the MacBlue mouse. Please click here to view a larger version of this figure.
Figure 4. In vivo co-localization of medullar neutrophils and monocytes. This figure illustrates the possibility to detect another cell subset in vivo concomitantly with monocytes, by injecting specific monoclonal antibody combined with a fluorochrome. 2 µg of Ly6G-PE antibody has been injected intravenously 5 min before skull bone marrow imaging of the MacBlue mouse. Please click here to view a larger version of this figure.
Supplementary video 1. Migratory behavior of parenchymal monocytes. In vivo 3D-imaging of the steady-state migratory activity of monocytes in the skull bone marrow parenchyma of a MacBlue mouse. The ECFP+ signal is in cyan. Migratory paths for each cell (red dot) appear in green.
Supplementary video 2. Migratory behavior of vascular monocytes. In vivo 3D-imaging of the steady-state migratory activity of monocytes in the skull bone marrow vasculature of a MacBlue mouse. The ECFP+ signal is in cyan. (2M Da) rhodamine-dextran was injected before the imaging session to distinguish the bone marrow vasculature (red). Migratory paths for each cell (red dot) appear in green.
Discussion
The critical points of in vivo imaging methodology are to ensure stability of the focus in order to maximize the duration of imaging and to minimize the risk of bacterial contamination and inflammation, which might impact the dynamics of the inflammatory cells. Imaging of the skull bone marrow follows these aims as the surgery performed to get access to the bone marrow is minimal. The use of sterile material and antiseptics is essential to limit the risk of infection that might induce perturbation in cell homeostasis.
The development of the stereotactic holder might be challenging and requires specific customization for each system (distance between objective and motorized stage of the microscope, use of gas inhalator for anaesthesia). It is important to get the head of the mouse as horizontal as possible for a wider imaging surface access. Once the animal is positioned, it is likely that several minutes can be required to obtain good stability of the focal plane. Therefore, it is recommended to check for tissue drift before launching the acquisition recording. Because this process together with the selection of regions of interest can last a while, it is also recommended to regularly check that the animal is unconscious if Ketamin /Xylazin have been used, and to check for proper immersion of the objective as leakage and evaporation can occur.
The good positioning of the rubber ring on the skull represents another critical step in the process. Cyanoacrylate glue provides good results in terms of stability and sealing, but one needs to prevent excessive loading of glue, which could cover the skull and block access to the region of interest of the tissue. The advantage of direct immersion of the skull without a glass coverslip is to get access to deeper regions of the bone. In addition, since the skull is not a plain surface, this will not limit the imaging area to the contact surface between the skull and the coverslip, allowing a wide imaging field, as displayed in Figure 1.
For all imaging technologies, the choice between acquisition speed and resolution quality is a compromise. Thus, the number and the quality of images per unit of time are limited, and the choice depends on the scientific question addressed. Typically, vascular monocytes are fast rolling cells, whereas parenchymal monocytes are slow motile or sessile. An accurate tracking of rolling cells requires higher time resolution of acquisition, as shown in Figure 2. High imaging rate for sessile cells is useless and high x,y,z resolution can be preferred to analyze protrusive activity of dendrites for example. Fast cells would require thicker volume to get a longer tracking of their displacement in z.
The mean square displacement as a function of time is not provided by the software Imaris. The principle of this representation based on the fact that, for an object travelling ballistically without any encounter or structural constriction, the distance it travelled would be proportional to the time interval (Figure 2D). Thus linearity of the slope indicates a random walk in a non-constrained environment. In contrast, asymptotic shape of the curve would reflect a constrained displacement over time of the cell population. The second advantage of this calculation is that velocity is sometimes overestimated by artefactual displacement of the centre of mass. This is particularly important for slow motile cell with protrusive activity. Thus, calculation of the motility coefficient by the slope of the curve calculated by linear regression indicates a real displacement of the cellover time 21.
Ly6G staining in vivo illustrates the possibility to add a new parameter of analysis during the imaging process (Figure 3). We have used this technique to stain vascular neutrophil infiltration into skin wound, and observed no defect in their mobility suggesting no functional alteration (Rodero et al in press). Although more than 98% of vascular neutrophils are properly labeled, the staining of bone marrow neutrophils is less efficient, and the mean fluorescence intensity is strongly reduced, suggesting reduced access of the antibody toward the bone marrow parenchyma (data not shown). Rhodamin dextran, quantum dots, or other specific dyes of apoptotic or necrotic cells such as Dapi, Propidium iodide, sytox 22, or fluorescent reporter for cell functions such as production of reactive oxygen species 23, can be added intravenously through the tail vein during imaging process and offer a good opportunity to quantify functional properties such as cell death, phagocytosis and neutrophil extracellular trap. Mazo et al12 nicely used two different vascular dyes with low and high molecular weight to better contrast parenchyma and vasculature. The choice of the fluorescent dye is critical to allow concomitant acquisition of the different fluorescent reporters. Indeed, the excitation wavelength will be chosen accordingly to get the best compromise between all used fluorescent dyes.
The protocol used herein makes it possible to analyze in vivo the biological activity of a cell compartment, study of which was difficult so far. Histological analysis of the bone marrow tissue usually requires a long preparation of bone decalcification to allow thin section, and it only provides a static view of the tissue. The dynamic view highlights the rate of cell exchange between the parenchyma and the bone marrow sinusoids. This rapid process is a crucial step to decipher in order to have a better understanding of cell mobilization mechanism upon inflammatory signal 5 or preconditioning myeloaplasive chemotherapy regimen 6. We have reported event of cell intravasation6 but this process is barely detectable due to the low frequency at steady state. However, it is likely to be enhanced upon inflammatory signaling. Chemokine receptors such as CCR2, CX3CR1 or CXCR4 are highly implicated in the process of intravasation, hence the use of genetic invalidation of these receptors should provide novel fundamental insights in the pathways of cell migration
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors wish to thank Anne Daron and Pierre Louis Loyher for editorial assistance, the Plateforme Imagerie Pitié-Salpêtrière (PICPS) for assistance with the two-photon microscope and the animal Facility “NAC” and Camille Baudesson for mice breeding assistance. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n° 304810 – RAIDs, and n°241440-Endostem, from Inserm, from Université Pierre et Marie Curie “Emergence”, from la “Ligue contre le cancer”, from “Association pour la Recherche sur le Cancer” and from “Agence Nationale de la Recherche” Programme Emergence 2012 (ANR-EMMA-050). P.H. was supported by la “Ligue contre le cancer”.
References
- Ovchinnikov DA, et al. Expression of Gal4-dependent transgenes in cells of the mononuclear phagocyte system labeled with enhanced cyan fluorescent protein using Csf1r-Gal4VP16/UAS-ECFP double-transgenic mice. J Leukoc Biol. 2008;83:430–433. doi: 10.1189/jlb.0807585. [DOI] [PubMed] [Google Scholar]
- Parihar A, Eubank TD, Doseff AI. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J Innate Immun. 2010;2:204–215. doi: 10.1159/000296507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Robbins CS, Swirski FK. The multiple roles of monocyte subsets in steady state and inflammation. Cell Mol Life Sci. 2010;67:2685–2693. doi: 10.1007/s00018-010-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquelin S, et al. CX3CR1 reduces Ly6Chigh-monocyte motility within and release from the bone marrow after chemotherapy in mice. Blood. 2013;122:674–683. doi: 10.1182/blood-2013-01-480749. [DOI] [PubMed] [Google Scholar]
- Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN, Miller MJ, Dustin ML, Nussenzweig MC. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat Rev Immunol. 2006;6:497–507. doi: 10.1038/nri1884. [DOI] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Milo I, et al. Dynamic imaging reveals promiscuous crosspresentation of blood-borne antigens to naive CD8+ T cells in the bone marrow. 2013;122:193–208. doi: 10.1182/blood-2012-01-401265. [DOI] [PubMed] [Google Scholar]
- Cavanagh LL, et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat Immunol. 2005;6:1029–1037. doi: 10.1038/ni1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazo IB, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity. 2005;22:259–270. doi: 10.1016/j.immuni.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Travlos GS. Normal structure, function, and histology of the bone marrow. Toxicol Pathol. 2006;34:548–565. doi: 10.1080/01926230600939856. [DOI] [PubMed] [Google Scholar]
- Mazo IB, et al. Hematopoietic progenitor cell rolling in bone marrow microvessels: parallel contributions by endothelial selectins and vascular cell adhesion molecule 1. J Exp Med. 1998;188:465–474. doi: 10.1084/jem.188.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidi NM, et al. In vivo time-lapse imaging of mouse bone marrow reveals differential niche engagement by quiescent and naturally activated hematopoietic stem cells. Blood. 2014. [DOI] [PMC free article] [PubMed]
- Sipkins DA, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariappa A, et al. Perisinusoidal B cells in the bone marrow participate in T-independent responses to blood-borne microbes. Immunity. 2005;23:397–407. doi: 10.1016/j.immuni.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Junt T, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317:1767–1770. doi: 10.1126/science.1146304. [DOI] [PubMed] [Google Scholar]
- Zoumi A, Yeh A, Tromberg BJ. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc Natl Acad Sci U S A. 2002;99:11014–11019. doi: 10.1073/pnas.172368799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- Cahalan MD, Parker I. Choreography of cell motility and interaction dynamics imaged by two-photon microscopy in lymphoid organs. Annu Rev Immunol. 2008;26:585–626. doi: 10.1146/annurev.immunol.24.021605.090620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost CC, et al. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood. 2009;113:6419–6427. doi: 10.1182/blood-2008-07-171629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi S, et al. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med. 2013;19:107–112. doi: 10.1038/nm.3024. [DOI] [PubMed] [Google Scholar]