

Abstract
Tumor cells secrete not only a variety of soluble factors, but also extracellular vesicles that are known to support the establishment of a favorable tumor niche by influencing the surrounding stroma cells. Here we show that tumor-derived microvesicles (T-MV) also directly influence the tumor cells by enhancing their invasion in a both autologous and heterologous manner. Neither the respective vesicle-free supernatant nor MV from benign mammary cells mediate invasion. Uptake of T-MV is essential for the proinvasive effect. We further identify the highly glycosylated form of the extracellular matrix metalloproteinase inducer (EMMPRIN) as a marker for proinvasive MV. EMMPRIN is also present at high levels on MV from metastatic breast cancer patients in vivo. Anti-EMMPRIN strategies, such as MV deglycosylation, gene knockdown, and specific blocking peptides, inhibit MV-induced invasion. Interestingly, the effect of EMMPRIN-bearing MV is not mediated by matrix metalloproteinases but by activation of the p38/MAPK signaling pathway in the tumor cells. In conclusion, T-MV stimulate cancer cell invasion via a direct feedback mechanism dependent on highly glycosylated EMMPRIN.
Keywords: breast cancer, microvesicles, invasion, EMMPRIN, glycosylation
Introduction
Intercellular communication is an essential aspect of malignant tumors that constitute complex tissues. It serves not only to maintain tissue homeostasis but also to mediate migration and subsequent invasion of tumor cells. While, in the past, soluble factors such as cytokines and growth factors have been considered the main mediators of these processes, the contribution of extracellular vesicles (EV) is now increasingly recognized (Thery et al., 2009; Kucharzewska and Belting, 2013).
Tumor cells constitutively produce distinct populations of extracellular vesicles that can be distinguished based on their biogenesis and physical properties. Among the most studied vesicles are exosomes (Exo, diameter: 50–100 nm) and microvesicles (MV, 100–1000 nm). While Exo are endosomal-derived and can be released upon fusion of multivesicular bodies (MVB) with the cell membrane, MV are actively shed from the plasma membrane (Raposo and Stoorvogel, 2013). Both particle populations carry a plethora of bioactive molecules including lipids, proteins, as well as different types of nucleic acids. Apart from the content of the vesicles, their uptake seems to be an important prerequisite for their function. Although the mechanism of MV uptake is still poorly understood, it was recently shown that low temperature as well as blocking the GTPase dynamin can reduce MV uptake and even abrogate MV function (Kawamoto et al., 2012), which points to an endocytosis-mediated process.
Tumor-derived MV (T-MV) are known to enhance progression of human tumors by modulating the surrounding stroma cells including fibroblasts, endothelial cells, and immune cells (Muralidharan-Chari et al., 2010). However, little is known about the autologous effects of T-MV on the tumor cells themselves. Some studies demonstrate that T-MV can enhance autologous tumor invasion by carrying matrix metalloproteinases (MMPs) or their regulators and thereby mediate degradation of the extracellular matrix (Muralidharan-Chari et al., 2010). The extracellular matrix metalloproteinase inducer (EMMPRIN) is known to stimulate MMP expression in tumor as well as stroma cells (Biswas et al., 1995). Overexpression of EMMPRIN has been described in many cancers and correlates with shortened survival (Nabeshima et al., 2006). EMMPRIN has three N-glycosylation sites, resulting in a highly glycosylated form (HG, 45–65 kDa) and a low glycoform (LG, ∼32 kDa) (Tang et al., 2004a). Among four splice variants, EMMPRIN-2 is the most abundant one (Liao et al., 2011). EMMPRIN can induce its own transcription (Tang et al., 2004b) by homophilic interaction of EMMPRIN on opposing cells (Yu et al., 2008), resulting in activation of p38-/mitogen-activated protein kinase (MAPK) (Lim et al., 1998).
Recently, we were able to demonstrate that T-MV induce a proinvasive phenotype in tumor-associated macrophages, which can be partly blocked by inhibition of MV uptake (Menck et al., 2013; Rietkotter et al., 2013). We thus hypothesized that T-MV not only influence the surrounding stroma cells, but also represent an important means of communication between neighboring breast cancer cells to facilitate their spontaneous invasion. Our aim was to specify their mode of action in relation to their uptake and to identify potential MV-related markers that mediate tumor invasion.
Results
Cancer cell-derived MV but not MV from nonmalignant cells induce invasion
EV from the breast cancer cell lines MCF-7 (T-EVM), SK-BR-3 (T-EVS), and MDA-MB231 (T-EVMDA), as well as the benign, immortalized mammary epithelial cell line hTERT-HME1 (hTERT-EV) were prepared by differential centrifugation without any previous stimulation. Upon transmission electron microscopy (TEM), the MV fraction consisted of a heterogeneous population of membrane-enclosed vesicles with a diameter of 100 up to 1000 nm, which clearly differed from the population of smaller, typically cup-shaped Exo (Figure 1A). There was no evidence of apoptotic bodies or cell debris.
Figure 1.

T-EV induce breast cancer invasion. (A) Electron microscopy (TEM) of T-MV and T-Exo. (B) Microinvasion assay of MCF-7 breast cancer cells stimulated with T-MV, T-Exo, and MV from normal epithelial cells (hTERT-MV) (all at 1 µg/ml), as well as the respective particle-free supernatants (sn) (mean ± SD, n = 3, *P < 0.001). Suffix M: vesicles/supernatant from MCF-7 cells; S: SK-BR-3; MDA: MDA-MB231; hTERT: hTERT-HME1. (C) Comparative analysis of cell invasion of MCF-7 and benign hTERT-HME1 cells stimulated with MV (10 µg/ml) (mean ± SD, n = 3, *P < 0.001).
Invasion of MCF-7 (Figure 1B) as well as SK-BR-3 cells (Supplementary Figure S1A) was enhanced by incubation with both autologous and heterologous T-MV. T-Exo also induced invasion, but to a significantly lesser extent. Therefore, we only focused on the MV population in further experiments. Incubation with the respective particle-free tumor cell supernatants (T-sn) or benign hTERT-MV had no effect, indicating that MV-induced invasion is a specific feature of distinct MV populations. Dilution experiments elucidated that T-MV at a concentration as low as 0.1 µg/ml were able to significantly increase tumor invasion (Supplementary Figure S1B), arguing against an unspecific, artificial effect. Moreover, T-MV did not influence cell proliferation (Supplementary Figure S1C–E). Since it has been described that T-MV can confer malignant characteristics to benign recipient cells, hTERT-HME1 cells were exposed to heterologous T-MVM and autologous hTERT-MV; however, they remained noninvasive (Figure 1C).
Incorporation is not specific, but necessary for proinvasive MV
We next asked whether MV are incorporated by tumor cells and whether this differs between MV populations explaining their differential features. Time course experiments revealed that a major part of T-MVM was taken up into MCF-7 cells after 24 h of stimulation (Supplementary Figure S2A). However, all of the investigated MV populations, regardless of their origin, were incorporated into both breast cancer cell lines at this time point (Figure 2A, Supplementary Figure S2B and C). Although the uptake of heterologous MV, including proinvasive T-MVS and non-proinvasive hTERT-MV, into MCF-7 cells seemed to be lower compared with the uptake of autologous T-MVM, the tumor cells still ingested a significant portion of all MV populations. This suggests that MV uptake per se does not differ between proinvasive and non-proinvasive MV. To investigate whether MV incorporation is nonetheless essential for MV function, we tried to antagonize T-MV uptake into breast cancer cells with known inhibitors of endocytosis at concentrations that did not interfere with cell viability (Supplementary Figure S2D and E). Neither inhibition of clathrin-mediated endocytosis with dansylcadaverine, nor interfering with caveolae-dependent endocytosis by cholesterol depletion in the recipient cells with filipin III decreased T-MV ingestion (Supplementary Figure S2F). Thus, T-MV incorporation occurs independent of either clathrin- or caveolin-mediated endocytosis. However, pharmacological inhibition of dynamin with dynasore led to a significant concentration-dependent reduction of T-MV uptake into MCF-7 (Figure 2B) as well as SK-BR-3 (Supplementary Figure S2F). Consistently, T-MV-induced invasion was significantly decreased after dynasore treatment (Figure 2C, Supplementary Figure S2G). Taken together, these data imply a dynamin-dependent uptake of T-MV as an important requirement for MV-triggered invasion. Furthermore, heating of T-MV significantly decreased their proinvasive phenotype (Figure 2D), indicating that their tumor-promoting effect is not mediated by transferring lipids, but by proteins most likely differentially expressed on T-MV and benign MV.
Figure 2.

MV uptake is important for their proinvasive function. (A) Fluorescence microscopy showing the uptake of MV (5 µg/ml) labeled with the red fluorescent dye PKH26 by MCF-7 cells after 24 h of stimulation (magnification, 10×; inserts: bright field). (B) Uptake of PKH26-labeled T-MV (5 µg/ml) into MCF-7 cells preincubated with the endocytosis inhibitor dynasore (Dyn) was quantified by flow cytometry (mean ± SD, n = 4, *P < 0.01, **P < 0.001; MFI, mean fluorescence intensity). (C) Microinvasion assay of MCF-7 cells pretreated with Dyn (12.5 µM) prior to stimulation with T-MV (mean ± SD, n = 3, *P < 0.001). (D) Microinvasion assay of MCF-7 cells exposed to heat-inactivated (5 min, 95°C) T-MV (mean ± SD, n = 3, *P < 0.001).
EMMPRIN is involved in the proinvasive phenotype of T-MV
Previously, we have shown that EMMPRIN expression in human breast cancer cells is involved in reprogramming tumor stroma cells toward a proinvasive, tumor-supporting phenotype (Hagemann et al., 2005). To investigate whether EMMPRIN is present on both EV populations, i.e. MV and Exo, of breast cancer cells, we analyzed its expression on the individual populations in comparison with the whole cell lysate. In fact, we found an enrichment of EMMPRIN on MV of both MCF-7 and SK-BR-3 cells (Figure 3A), whereas it was absent on the respective Exo (Figure 3B). To further clarify whether EMMPRIN is suitable for discriminating MV and Exo, we comparably analyzed various known markers on both vesicle populations (Supplementary Figure S3A). Interestingly, Flotillin-2, which is often used as a marker for Exo, was found mainly in MV preparations, while the MVB protein TSG101 was indeed specifically expressed on Exo. Tubulin was found predominantly on MV. Next, MV and Exo were loaded separately on sucrose gradients. Based on the distribution of the different markers, both populations were localized in comparable density fractions ranging from 1.13 to 1.22 g/ml (Supplementary Figure S3B). Again, EMMPRIN was present exclusively in the MV fraction, confirming that it can be used to visualize MV in sucrose gradient preparations and to distinguish them from Exo.
Figure 3.

EMMPRIN is expressed on T-MV in vitro and in vivo and contributes to their proinvasive phenotype. (A and B) Western blots showing the expression of highly (HG) and lowly glycosylated (LG) EMMPRIN (EMP) in whole cell lysates (C) and T-MV (A) as well as the corresponding T-Exo (B) of both breast cancer cell lines. (C) Western blots showing stable knockdown of EMMPRIN via shRNA (shEMP) in SK-BR-3 cells and MV (ns ctl, non-sense control). (D) Microinvasion assay of MCF-7 cells exposed to T-MVS (1 µg/ml) from EMMPRIN knockdown cells (mean ± SD, n = 3, *P < 0.01). (E) Total MV from peripheral blood of metastatic breast cancer (BC) patients and matched controls (ctl). The percentage of EMMPRIN-positive MV was quantified by flow cytometry. (F) Double staining for EMMPRIN and MUC1 of MV from a BC and a control patient (representative density plots).
To clarify whether the presence of EMMPRIN on T-MV is significant for their proinvasive feedback effect on tumor cells, we performed a stable knockdown of EMMPRIN via shRNA in SK-BR-3 cells (Figure 3C) that have the strongest EMMPRIN enrichment on the MV. This significantly diminished MV-induced autologous and heterologous tumor cell invasion (Figure 3D, Supplementary Figure S3C), confirming the role of EMMPRIN in the proinvasive MV phenotype. However, MV uptake into breast cancer cells was not affected by EMMPRIN knockdown (Supplementary Figure S3D and E).
EMMPRIN is expressed at high levels in patient-derived MV
To confirm that EMMPRIN expression on MV is not only involved in tumor invasion in vitro, but also plays a role in cancer progression in vivo, we isolated MV from peripheral blood of breast cancer patients with metastatic disease and analyzed the expression of EMMPRIN by flow cytometry (Supplementary Figure S4A). Indeed, the number of EMMPRIN-positive MV was significantly enhanced in patient-derived MV (% of EMMPRIN-positive MV: median 31.7 [IQR 21.35–37.35]) compared with matched controls (% of EMMPRIN-positive MV: median 19.5 [IQR 10.03–24.88], P = 0.0075, Wilcox one-sided rank test) (Figure 3E). Since blood-derived MV consist of different subpopulations, also containing MV from platelets, endothelial cells, and leukocytes, we performed staining for the tumor marker MUC1, which was found to be expressed on both in vitro T-MV and patient-derived MV in vivo (Supplementary Figure S4B and C). Flow cytometry revealed that the majority of EMMPRIN-positive MV also carried MUC1 (Figure 3F), indicating that these are in fact tumor cell-derived MV.
The glycosylation status of EMMPRIN on T-MV is important for its function
In order to test whether differential EMMPRIN expression could explain the proinvasive effect of T-MV in contrast to benign MV, we analyzed EMMPRIN expression in various cells and their MV. EMMPRIN was strongly enriched in T-MV compared with their parental cells, but only weakly expressed on platelet-derived MV (P-MV) (Figure 4A) that were previously identified as being not proinvasive (Menck et al., 2013). Surprisingly, the strongest EMMPRIN enrichment was demonstrated on MV from the benign hTERT-HME1. Analysis of the protein size revealed that all investigated MV contained a large form of EMMPRIN ranging from 45 to 65 kDa, indicating glycosylation at multiple sites. However, within this fraction, two different glycoforms can be distinguished: (i) a very highly glycosylated protein (HG-EMMPRIN, ∼50–65 kDa) characterizing all proinvasive MV, and (ii) a variant with intermediate glycosylation (IG-EMMPRIN, ∼45 kDa) in all non-proinvasive MV. The low glycosylation variant (LG-EMMPRIN, 32 kDa) was only very weakly detectable.
Figure 4.
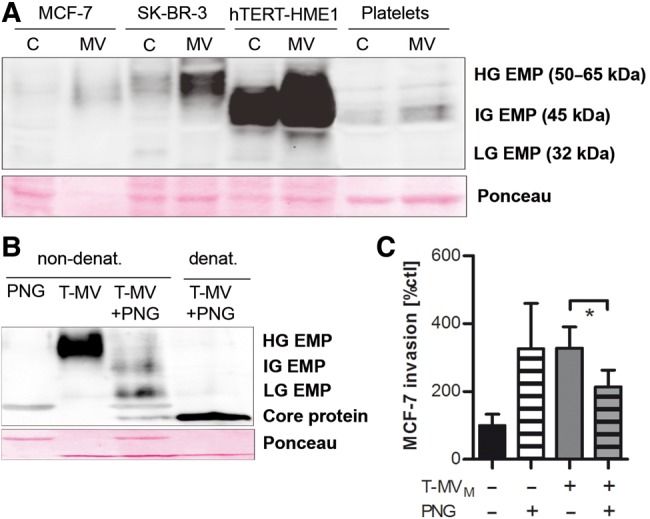
High glycosylation is essential for the proinvasive function of EMMPRIN. (A) The EMMPRIN glycosylation status on tumor and benign cells/MV was analyzed by western blotting (HG-EMP, highly glycosylated EMMPRIN; IG-EMP, intermediately glycosylated EMMPRIN; LG-EMP, lowly glycosylated EMMPRIN). (B) T-MVM were deglycosylated by PNGaseF (visible as an unspecific band in lanes 1 and 3) under denaturing or nondenaturing conditions and the EMMPRIN glycosylation status was assessed by western blotting. (C) Microinvasion assay of MCF-7 cells stimulated with deglycosylated T-MV (10 µg/ml, non-denat. conditions) (mean ± SD, n = 3, *P < 0.001).
To test whether only HG-, but not IG-EMMPRIN, is responsible for the proinvasive MV phenotype, we deglycosylated T-MVM with Peptide N-Glycosidase F (PNGaseF). Under nondenaturing experimental conditions, this led to a significant reduction of the HG fraction in favor of lesser glycosylated forms (Figure 4B). In contrast, complete deglycosylation down to the core protein (27 kDa) was only observed under denaturing conditions. T-MV deglycosylated under nondenaturing conditions induced significantly less tumor invasion than control MV (Figure 4C). To rule out possible anti-invasive effects of N-deglycosylation of other proteins involved in invasion on the recipient cells, cancer cells were also incubated with PNGaseF without T-MV addition. Exposing tumor cells to PNGase alone did not reduce, but rather enhanced invasion. This observation argues against a nonspecific anti-invasive effect of the enzyme's presence in the treated T-MV population.
Mass spectrometry analysis of T-MV confirms differential EMMPRIN glycosylation
To further analyze EMMPRIN glycosylation patterns on T-MV, T-MVM were digested with PNGaseF and all EMMPRIN bands detected by western blotting (see Figure 4B) were excised from one-dimensional SDS electrophoresis gels and subjected to LC-MS/MS. Alignment of the assigned peptides displayed the highest coverage for the isoforms EMMPRIN-2 and EMMPRIN-3, whereas peptides specific for EMMPRIN-1 and EMMPRIN-4 could not be identified (Supplementary Figure S5A). Spectral counting revealed that the N-terminal fragment of EMMPRIN-2 was present in a ratio of 10:1 compared with EMMPRIN-3. This is in line with an mRNA screening of three cell lines and the human brain metastases from eight patients with primary breast cancer, which showed that EMMPRIN-2 is expressed at very high levels in vitro and in vivo compared with the other two isoforms, EMMPRIN-3 and EMMPRIN-4 (Figure 5A). These results suggest that EMMPRIN-2 is the predominant isoform on T-MV. However, since it is also present on hTERT-MV, its expression per se is not sufficient for the proinvasive MV effect, which requires additional posttranslational modifications, in particular strong glycosylation.
Figure 5.

HG-EMMPRIN on T-MV is glycosylated at N160 and N268. (A) Expression of EMMPRIN (EMP) isoforms EMP-2, EMP-3, and EMP-4 in human breast (cancer) cells and in brain metastases from eight breast cancer patients (BC brain mets) (qRT–PCR, mean, n = 3). EMP-3 and EMP-4 were not detectable (n.d.) in hTERT-HME1 cells. (B) Schematic representation of EMP-2 with the three predicted N-glycosylation sites. (C) Mass spectrometry of the three predicted EMMPRIN N-glycosylation sites (see text for further details). (D) Microinvasion assay of MCF-7 cells preincubated with specific blocking peptides against the two EMMPRIN glycosylation sites (P-N160, P-N268) and stimulated with T-MV (mean ± SD, n = 3, *P < 0.05, **P < 0.01). Random control peptides (P-N160rd, P-N268rd) were used as controls.
In order to further confirm that HG- and IG-EMMPRIN differ in their glycosylation pattern, the three predicted EMMPRIN N-glycosylation sites (Figure 5B) were analyzed. While glycosylated peptides are too large for analysis, deglycosylation by PNGaseF treatment leads to deamidation of the glycan-carrying asparagine residues (N) to aspartic acid (D), which can then be detected due to an increase in peptide mass by 1 mass unit. In line with this, the peptides carrying the two glycosylation sites N160 and N268 were missing in HG-EMMPRIN, whereas they were present with deamidated N-residues in IG-EMMPRIN (Figure 5C, Supplementary Figure S5B–D). This confirms that HG- and IG-EMMPRIN are indeed differentially glycosylated on T-MV, since HG-EMMPRIN carries glycans at N160 and N268, which are not glycosylated in the non-proinvasive IG-EMMPRIN form. In addition, subsequent in-gel treatment of the HG-EMMPRIN spot with PNGaseF led to detection of the deamidated peptides (Supplementary Figure S5E). No consistent deamidation pattern was found for the third putative glycosylation site at N302.
Specific blocking peptides against the two identified glycosylation sites N160 and N268 (Sato et al., 2009) reduced T-MV-mediated tumor invasion, while random control peptides had no such effect (Figure 5D). This result confirms that these sites are indeed critical for the proinvasive function of EMMPRIN on T-MV. To identify other potential proinvasive factors on T-MV, we thoroughly characterized T-MVM by LC-MS/MS and all identified proteins were subjected to Ingenuity Pathway Analysis (Supplementary Table S1). Interestingly, the most probably putative upstream regulators were TP53, MYC, APP, PSEN1, and MAPT, which have been implicated in either EV genesis or the regulation of oncogenic pathways (Yu et al., 2006; Ikeda et al., 2010; Cho et al., 2011; Takagi et al., 2013).
EMMPRIN does not act via MMP induction, but through activation of p38/MAPK
Next, we investigated how EMMPRIN mediates MV-induced tumor invasion. Although a main function of EMMPRIN is to induce MMP transcription, there was no significant effect of EMMPRIN-positive T-MV on the expression of selected MMPs in the supernatant of MCF-7 or SK-BR-3 cells (Figure 6A, Supplementary Figure S6A–C). Moreover, neither MMP-2 nor MMP-9 could be detected on T-MV, although MV can function as carriers for MMPs as shown by the expression of MMP-9 on MV of human macrophages and platelets (Figure 6B). This suggests that EMMPRIN acts MMP-independently in this context and that the conveyance of MMPs is not the critical discriminator between proinvasive and non-proinvasive MV.
Figure 6.

p38 signal transduction partially mediates MV-induced cancer cell invasion. (A) MMP mRNA expression in MCF-7 cells stimulated for 24 h with MV (25 µg/ml) (qRT–PCR, mean ± SD, n = 3). (B) Zymography for MMP-9 in tumor and benign cells (C), as well as their respective MV (SK, SK-BR-3; MΦ, human macrophages; P, platelets). (C) Western blots showing the phosphorylation of p38 and c-jun in MCF-7 cells stimulated with T-MV for the indicated time periods. (D) Analysis of p38 phosphorylation in MCF-7 cells incubated for 1 h with T-MV from EMMPRIN knockdown (shEMP) or non-sense control (ns) cells. (E) Microinvasion assay of MCF-7 cells pretreated with the p38 inhibitor SB-203580 (SB) for 2 h followed by stimulation with T-MV (1 µg/ml) (mean ± SD, n = 3, *P < 0.001). (F) Western blots showing p38 phosphorylation in MCF-7 cells pretreated with two EMMPRIN blocking peptides P-N160 and P-N268, respectively, for 2 h prior to stimulation with T-MV.
The expression of the proinvasive factors VEGF, TNFα, and M-CSF, which have been described as EMMPRIN target genes (Tang et al., 2005; Schmidt et al., 2008; Seizer et al., 2010), remained unchanged in MCF-7 and SK-BR-3 upon T-MV stimulation (Supplementary Figure S6D). Furthermore, no positive feedback loop could be observed for the EMMPRIN expression itself. We then investigated activation of p38/MAPK as a potential downstream signal. Upon incubation with T-MV, p38 was rapidly phosphorylated in both breast cancer cell lines (Figure 6C, Supplementary Figure S6E). However, this phosphorylation was absent when stimulating with T-MVS from EMMPRIN knockdown cells (Figure 6D), confirming that p38 activation is indeed specifically induced by EMMPRIN on T-MV. Consistently, T-MV-induced invasion was significantly reduced by the p38 inhibitor SB-203580 (Figure 6E, Supplementary Figure S6F) at concentrations not interfering with cell viability (Supplementary Figure S6G). To test whether p38 phosphorylation depends specifically on the presence of HG-EMMPRIN, MCF-7 cells were preincubated with the two blocking peptides P-N160 and P-N268 prior to T-MV stimulation. This resulted in a significant reduction of phosphorylated p38 (Figure 6F). In line with this finding, stimulation of the tumor cells with PNGaseF-treated, deglycosylated T-MV also resulted in a reduction in phosphorylated p38 protein (Supplementary Figure S6H).
T-MV incubation also induced rapid phosphorylation of c-jun, which, however, was accompanied by a parallel increase of total c-jun (Figure 6C). While there was no evidence for de novo synthesis of c-jun mRNA (Supplementary Figure S7A), T-MV carry considerable amounts of c-jun protein (Supplementary Figure S7B). Thus, the early increase in phosphorylated c-jun may be due to the transfer via T-MV containing the c-jun protein. Inhibition of c-jun phosphorylation by application of the c-Jun N-terminal kinase (JNK) inhibitor-II did not antagonize T-MV-induced invasion (Supplementary Figure S7C), arguing for a c-jun-independent effect.
Discussion
Recently, MV have been increasingly appreciated as important mediators of cancer progression. Their role in malignant processes, such as invasion and metastasis, has mostly been attributed to either a horizontal interaction with the surrounding tumor stroma or direct extracellular matrix degradation. Our data show that T-MV also directly increase cancer invasion via an autologous and heterologous loop. This effect has not been described so far for MV, although similar observations have been reported for Exo (Graves et al., 2004; Higginbotham et al., 2011). However, the recipient cell type has to be predisposed towards an invasive state, as T-MV alone are not sufficient to induce invasion in benign mammary epithelial cells. Another important prerequisite for MV function is the uptake into their target cell. The mechanism by which extracellular vesicles are taken up is controversially discussed. While some studies observed blocking of Exo uptake by common inhibitors of clathrin- or caveolin-mediated endocytosis (Escrevente et al., 2011; Svensson et al., 2013), others argue that Exo are taken up by membrane fusion (Parolini et al., 2009). However, in our study, MV were ingested by endocytosis that occurred independent of clathrin OR caveolin, but was reduced by 50% through inhibition of dynamin, suggesting that alternative endocytotic routes are involved in this process.
Interestingly, heating of T-MV significantly reduced their proinvasive potential, showing that MV function is influenced by tumor-promoting proteins expressed on the vesicles. Contribution of lipids cannot be completely ruled out, since MV remained partly invasive after heating. In search for molecules that mediate the T-MV phenotype, we focused on EMMPRIN, which we had described earlier as indispensable for breast cancer invasion induced by macrophages (Hagemann et al., 2005). EMMPRIN is present on T-MV where it is strongly enriched in comparison with the parent cells. To our surprise, we did not find EMMPRIN on the Exo of our investigated cell lines, which contradicts recent data (Redzic et al., 2013). Since our isolation protocol clearly discriminates between Exo and MV, this discrepancy may be due to different isolation methods and contamination of Exo preparations with EMMPRIN-carrying MV or differential expression patterns in distinct cell lines. Next to EMMPRIN, IPA of the T-MV protein content revealed that more than a third of the identified proteins are involved in cell growth, assembly, and proliferation. These include many members of the integrin or mTOR signaling pathways, both known to play a role in cancer progression. Since the employed anti-EMMPRIN strategies did not completely abrogate the proinvasive function of T-MV, it is very likely that they harbor other tumor-promoting factors that might be regulated by the identified upstream regulators MYC, APP, PSEN1, or MAPT.
In line with our in vitro finding of EMMPRIN enrichment on proinvasive T-MV, we detected a significantly increased number of EMMPRIN-positive MV in vivo in the blood of breast cancer patients with metastatic disease compared with matched controls. This is in accordance with a study from Baran et al. (2010) that EMMPRIN levels are elevated in plasma samples from gastric cancer patients, whereas EMMPRIN could no longer be detected after centrifugation of the samples at 50000 g. However, we also detected EMMPRIN on MV from tumor-free control patients. This is not surprising, since EMMPRIN expression has also been described for nonmalignant blood cells, i.e. platelets and lymphocytes (Nabeshima et al., 2004; Schmidt et al., 2008). In contrast to the controls, EMMPRIN on patient-derived MV colocalized with the tumor marker MUC1 (also known as CA 15-3), which is used as prognostic serum marker in breast cancer (Duffy et al., 2000) and is frequently overexpressed on cancer cells. This clearly shows that EMMPRIN is present predominantly on tumor cell-derived MV.
P-MV, which were demonstrated to be non-proinvasive (Menck et al., 2013), carry only low amounts of EMMPRIN. Nevertheless, the different MV effects cannot be attributed to varying EMMPRIN levels alone, since EMMPRIN expression is strongest on hTERT-MV that do not confer invasion. A clear influence on EMMPRIN function has been demonstrated for its glycosylation status. Strong glycosylation is necessary from self-aggregation and MMP induction (Weidle et al., 2010). The HG form is also critical for adhesion to integrins and invasion of hepatoma cells (Dai et al., 2009). We show that EMMPRIN is glycosylated to a maximum extent in all proinvasive T-MV, whereas P- and hTERT-MV are characterized by a less glycosylated form of EMMPRIN, which we termed intermediately glycosylated (IG) EMMPRIN. Both stable knockdown of EMMPRIN and MV deglycosylation down to the IG form counteract MV-induced invasion. This indicates that not only the presence of EMMPRIN per se, but particularly the expression of the HG form is necessary for this proinvasive effect. A nonspecific action through deglycosylation of other glycoproteins involved in invasion can be ruled out, since addition of PNGaseF alone does not inhibit invasiveness.
Through mass spectrometry, we demonstrated that the difference between HG- and the newly described IG-EMMPRIN is due to N-glycosylation at the residues N160 and N268, which are only glycosylated in the HG form. Specific peptides covering these two sites led to a significant reduction of T-MV-induced tumor invasion. Interestingly, others have already shown that inhibition of the first immunoglobulin loop domain of EMMPRIN via synthetic peptides covering N160, results in decreased induction of MMP in fibroblasts by malignant cells of different origins (Sato et al., 2009; Koga et al., 2011). Moreover, targeting the second N-glycosylation site with synthetic peptides directed against the second immunoglobulin loop regulates cell migration of ovarian cancer cells (Sato et al., 2012), showing that both domains are involved in the tumor-promoting effects of EMMPRIN.
Although the role of EMMPRIN in MV-mediated stromal MMP induction is well known (Muralidharan-Chari et al., 2010), we did not observe increased activation or expression of MMPs in the tumor cells. While we can confirm that MV act as carriers of MMPs, this cannot be the critical determinant of proinvasive MV, since we found that MMPs are present only on benign MV, but not on the investigated T-MV. Equally, both the expression of EMMPRIN itself and known tumor-promoting EMMPRIN target genes, i.e. M-CSF, TNFα, and VEGF (Tang et al., 2005; Schmidt et al., 2008; Seizer et al., 2010), remained unchanged. This prompted us to look for upstream signaling events in MV-stimulated tumor cells instead. A potential candidate is p38 that has been described to mediate EMMPRIN signaling in 16-Lu lung fibroblasts (Lim et al., 1998). We demonstrate that exposure to T-MV leads to phosphorylation of p38 in the tumor cells, which is specifically mediated by HG-EMMPRIN. Inhibition of p38 phosphorylation, in contrast, antagonizes MV-induced invasion.
In conclusion, we show that T-MV significantly contribute to tumor progression by enhancing invasion of surrounding tumor cells in an auto- and heterologous intercellular communication loop. We identified EMMPRIN that is expressed at high levels on T-MV in vitro and in vivo as one of the proinvasive factors that mediate this effect. Its function is dependent on the glycosylation status. Glycosylation at N160 and N268 is indispensable for the tumor-promoting effects. Surprisingly, EMMPRIN-bearing T-MV confer their proinvasive potential through activation of the p38/MAPK signaling cascade without affecting MMPs. Both the presence of HG-EMMPRIN on MV and the uptake of these vesicles by the receiving cells are essential for MV-induced tumor invasion. Since EMMPRIN knockdown and deglycosylation only partially counteract the proinvasive effect, we propose a two-step model: (i) MV-bound HG-EMMPRIN interacts with and activates proinvasive factors on the tumor cell surface, and (ii) MV horizontally deliver tumor-promoting proteins or nucleic acids that exert their effects after ingestion by the tumor cells.
Materials and methods
Full methods including standard cell culture techniques, peptide synthesis, and mass spectrometry can be found in Supplementary Materials and methods.
Isolation of MV and Exo, sucrose gradient
EV were isolated as described previously (Menck et al., 2013). Briefly, tumor cells or macrophages were cultured for 48 h in medium with particle-free FCS (ultracentrifuged overnight at 100000 g, 4°C). The supernatant was collected and centrifuged for 5 min at 750 g, followed by 15 min at 1500 g to remove residual cells and debris. MV were precipitated from the supernatant (14000 g, 35 min, 4°C), washed once with PBS, and resuspended in PBS or RIPA lysis buffer. To isolate Exo, the supernatant after ultracentrifugation at 14000 g was filtered (0.22 µm, Sarstedt) and Exo pelleted at 100000 g for 2 h at 4°C. The pellet was washed once in PBS and resuspended in PBS or RIPA lysis buffer. The particle-free supernatant was used as negative control for microinvasion assays. EV from human platelets were isolated from outdated (<2 days) platelet concentrates from healthy blood donors. MV and Exo protein counts were quantified using the Lowry assay (Bio-Rad). For sucrose gradients, MV or Exo preparations were loaded onto a sucrose step gradient (0.25–2.25 M) and centrifuged for 16 h at 100000 g, 4°C. Eight fractions of 2 ml were collected and protein content was enriched by precipitation with ice-cold acetone (1:10, v/v) overnight at −20°C.
Quantitative real-time PCR
Total RNA from cells and MV was extracted using the High Pure RNA isolation kit (Roche). RNA from brain metastases samples was prepared using a modified Trizol (Invitrogen) method including a DNase I (Roche) digestion step. Reverse transcription was carried out from 1 µg of RNA using the iScript cDNA synthesis kit (Bio-Rad) and quantitative real-time PCR (qRT–PCR) was carried out from 10 ng cDNA with SYBR green detection (7900 HT system, Applied Biosystems). The primer sequences are listed in Supplementary Table S2. Relative mRNA expression was determined with SDS version 2.4 (Applied Biosystems) after normalization on the expression of the housekeeping genes HPRT1 and GNB2L1 and calculated as fold change versus control.
MV uptake studies
To evaluate MV uptake into MCF-7 cells, MV were labeled with the red-fluorescent dye PKH26 (Sigma-Aldrich) according to the manufacturer's instructions. Tumor cells were preincubated with the indicated concentrations of endocytosis inhibitors for 2 h followed by 24 h of stimulation with the red-labeled MV (5 µg/ml). Cells were washed once with PBS and their red mean fluorescence intensity (MFI) was measured by flow cytometry.
Microinvasion and proliferation assays
Cell invasion upon stimulation with MV was measured in a modified Boyden chamber assay as described previously (Hagemann et al., 2004). Briefly, 1 × 105 cells were plated in triplicates in particle-free medium onto a polycarbonate membrane (10 µm pore diameter, Nucleopore) that was coated with Matrigel (R&D systems). Cells were stimulated for 48 h up to 96 h with particle-free supernatant (diluted 1/3) or MV at the indicated concentrations. Cell invasion was quantified by relating the number of invasive cells in the lower wells to the unstimulated controls. To assess changes in cell proliferation, MCF-7 (1 × 104/well) and SK-BR-3 (5 × 103/well) cells were seeded in E-Plates 16 in the xCELLigence RTCA DP system (Roche). MV (10 µg/ml) were added and changes in morphology and proliferation were recorded in quadruplets for 96 h.
Western blotting and zymography
Cells or vesicles were homogenized in RIPA lysis buffer (150 mM NaCl, 0.1% SDS, 0.5% Na-deoxycholate, 1% Triton X-100, 50 mM Tris, pH 7.2). Up to 50 µg of protein were separated by SDS–PAGE (8% gels) and transferred onto a nitrocellulose membrane (75 min at 10 V). The blocked membrane was incubated with primary antibodies specific to EMMPRIN (#sc-13976), MUC-1 (#sc-7313), TSG101 (#sc-7964, all three from Santa Cruz), Flotillin-2 (#610383, BD), Tubulin (#05-829, Millipore), p38 MAPK total protein (#9212), phospho-p38 MAPK (#9211), c-jun total protein (#9165), and phospho-c-jun (#9261, all four from cell signaling). Signals were detected after incubation with anti-rabbit (#sc-2004) or anti-mouse (#sc-2005, both from Santa Cruz) HRP-labeled secondary antibodies using ECL™ Prime (Amersham Biosciences). Ponceau S staining was routinely used as loading control. Zymography was performed as previously described (Hagemann et al., 2004). Briefly, cell lysates (35 µg), supernatants (12 µl), or MV (35 µg) were loaded onto SDS–PAGE gels (8%) supplemented with 1 mg/ml gelatine. After electrophoresis, gels were incubated overnight in renaturation buffer (200 mM NaCl, 5 mM CaCl2, 0.02% Brij-35, pH 7.5) and stained with Coomassie brilliant blue.
Patient samples and flow cytometry
Cerebral metastasis samples were collected from eight patients who were previously diagnosed with primary breast cancer and underwent medically indicated neurosurgical procedures. Patient-derived MV were isolated from EDTA-anticoagulated peripheral blood (<30 min after blood withdrawal) of 15 breast cancer patients who had metastatic disease and did not receive chemotherapy at the time of sample acquisition. Sixteen matched tumor-free patients were used as negative controls. The median patient age at the time of blood withdrawal was 63 (interquartile range (IQR): 59.5–69.5) years and the median age of the controls was 62.5 (IQR: 56.5–71.5) years with a P-value of 0.8432 (Wilcox two-sided rank test). All samples were obtained under an informed consent approved by the local ethics committee. Samples were centrifuged for 15 min at 1200 g and serum from blood cells was further separated through the application of a serum filter (Sarstedt). The serum was used for isolation of MV as described above. MV (5 µg) were stained with PE-conjugated mouse anti-MUC1 (#355603) and/or anti-EMMPRIN conjugated to Alexa Fluor 488 (#306207, both BioLegend). Irrelevant IgG1 antibodies were used as respective isotype-matched negative controls (#400114, #400132, BioLegend). Fluorescence was measured by a FACSCanto II flow cytometer (BD Biosciences) with the FACSDiva software (version 6.1.3).
Protein deglycosylation
For analysis of the glycosylation status of EMMPRIN, T-MVM were incubated under nondenaturing conditions with 1000 U Peptide N-Glycosidase F (PNGaseF, New England Biolabs) for 1 h at 37°C and directly subjected to microinvasion assays or lysed in RIPA buffer for western blotting. As a deglycosylation control, T-MVM were incubated with 20 U PNGaseF under denaturing conditions according to the manufacturer's instructions.
Mass spectrometry
In order to investigate the total protein content, T-MVM were subjected to 1D SDS–PAGE and individual gel lanes were excised into equally sized gel slices prior to mass spectrometry. For the identification of the EMMPRIN N-glycosylation sites on T-MV, T-MVM before and after treatment with PNGaseF under nondenaturing conditions were loaded onto a 1D gel. All EMMPRIN-corresponding Coomassie-stained gel bands were manually excised and digested overnight (37°C) with trypsin (Promega) in a ratio of 1:25. Digestion was stopped with 1% acetic acid and the peptide solution was desalted with C-18 ZipTip (Millipore) following the manufacturer's instructions. In parallel, one half of the HG-EMMPRIN 1D-gel band was in-gel digested with PNGaseF (denaturing conditions) to achieve complete deglycosylation. LC–MS/MS and subsequent data analysis are described in Supplementary Materials and methods.
Statistical analysis
Data were plotted as mean ± SD using GraphPad Prism for Windows (version 5.04, GraphPad software). Statistical significance was calculated with a two-tailed Student's t-test unless indicated otherwise, P-values <0.05 were considered significant.
Supplementary material
Supplementary material is available at Journal of Molecular Cell Biology online.
Funding
This work was supported by grants no. 109615 (Deutsche Krebshilfe), BI 703/3-2 (DFG), and eBIO MetastaSys (BMBF).
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We would like to thank Lena Ries, Meike Schaffrinski, and Matthias Schulz (University Medical Center Göttingen, Germany) for their excellent technical assistance. We greatly appreciate Kathrin Darm (University Medical Center Greifswald, Germany) for her support in the proteomic studies.
References
- Baran J., Baj-Krzyworzeka M., Weglarczyk K., et al. (2010). Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol. Immunother. 59, 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas C., Zhang Y., DeCastro R., et al. (1995). The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 55, 434–439. [PubMed] [Google Scholar]
- Cho S., Lu M., He X., et al. (2011). Notch1 regulates the expression of the multidrug resistance gene ABCC1/MRP1 in cultured cancer cells. Proc. Natl Acad. Sci. USA 108, 20778–20783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J.Y., Dou K.F., Wang C.H., et al. (2009). The interaction of HAb18G/CD147 with integrin alpha6beta1 and its implications for the invasion potential of human hepatoma cells. BMC Cancer 9, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy M.J., Shering S., Sherry F., et al. (2000). CA 15-3: a prognostic marker in breast cancer. Int. J. Biol. Markers 15, 330–333. [DOI] [PubMed] [Google Scholar]
- Escrevente C., Keller S., Altevogt P., et al. (2011). Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 11, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves L.E., Ariztia E.V., Navari J.R., et al. (2004). Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res. 64, 7045–7049. [DOI] [PubMed] [Google Scholar]
- Hagemann T., Robinson S.C., Schulz M., et al. (2004). Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-alpha dependent up-regulation of matrix metalloproteases. Carcinogenesis 25, 1543–1549. [DOI] [PubMed] [Google Scholar]
- Hagemann T., Wilson J., Kulbe H., et al. (2005). Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J. Immunol. 175, 1197–1205. [DOI] [PubMed] [Google Scholar]
- Higginbotham J.N., Demory Beckler M., Gephart J.D., et al. (2011). Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. 21, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H., Taira N., Hara F., et al. (2010). The estrogen receptor influences microtubule-associated protein tau (MAPT) expression and the selective estrogen receptor inhibitor fulvestrant downregulates MAPT and increases the sensitivity to taxane in breast cancer cells. Breast Cancer Res. 12, R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto T., Ohga N., Akiyama K., et al. (2012). Tumor-derived microvesicles induce proangiogenic phenotype in endothelial cells via endocytosis. PLoS One 7, e34045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K., Aoki M., Sameshima T., et al. (2011). Synthetic emmprin peptides inhibit tumor cell-fibroblast interaction-stimulated upregulation of MMP-2 and tumor cell invasion. Int. J. Oncol. 39, 657–664. [DOI] [PubMed] [Google Scholar]
- Kucharzewska P., Belting M. (2013). Emerging roles of extracellular vesicles in the adaptive response of tumour cells to microenvironmental stress. J. Extracell. Vesicles 2, 20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C.G., Kong L.M., Song F., et al. (2011). Characterization of basigin isoforms and the inhibitory function of basigin-3 in human hepatocellular carcinoma proliferation and invasion. Mol. Cell. Biol. 31, 2591–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim M., Martinez T., Jablons D., et al. (1998). Tumor-derived EMMPRIN (extracellular matrix metalloproteinase inducer) stimulates collagenase transcription through MAPK p38. FEBS Lett. 441, 88–92. [DOI] [PubMed] [Google Scholar]
- Menck K., Klemm F., Gross J.C., et al. (2013). Induction and transport of Wnt 5a during macrophage-induced malignant invasion is mediated by two types of extracellular vesicles. Oncotarget 4, 2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan-Chari V., Clancy J.W., Sedgwick A., et al. (2010). Microvesicles: mediators of extracellular communication during cancer progression. J. Cell Sci. 123, 1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima K., Suzumiya J., Nagano M., et al. (2004). Emmprin, a cell surface inducer of matrix metalloproteinases (MMPs), is expressed in T-cell lymphomas. J. Pathol. 202, 341–351. [DOI] [PubMed] [Google Scholar]
- Nabeshima K., Iwasaki H., Koga K., et al. (2006). Emmprin (basigin/CD147): matrix metalloproteinase modulator and multifunctional cell recognition molecule that plays a critical role in cancer progression. Pathol. Int. 56, 359–367. [DOI] [PubMed] [Google Scholar]
- Parolini I., Federici C., Raggi C., et al. (2009). Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 284, 34211–34222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G., Stoorvogel W. (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redzic J.S., Kendrick A.A., Bahmed K., et al. (2013). Extracellular vesicles secreted from cancer cell lines stimulate secretion of MMP-9, IL-6, TGF-beta1 and EMMPRIN. PLoS One 8, e71225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietkotter E., Menck K., Bleckmann A., et al. (2013). Zoledronic acid inhibits macrophage/microglia-assisted breast cancer cell invasion. Oncotarget 4, 1449–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Ota T., Watanabe M., et al. (2009). Identification of an active site of EMMPRIN for the augmentation of matrix metalloproteinase-1 and -3 expression in a co-culture of human uterine cervical carcinoma cells and fibroblasts. Gynecol. Oncol. 114, 337–342. [DOI] [PubMed] [Google Scholar]
- Sato T., Watanabe M., Hashimoto K., et al. (2012). A novel functional site of extracellular matrix metalloproteinase inducer (EMMPRIN) that limits the migration of human uterine cervical carcinoma cells. Int. J. Oncol. 40, 236–242. [DOI] [PubMed] [Google Scholar]
- Schmidt R., Bultmann A., Fischel S., et al. (2008). Extracellular matrix metalloproteinase inducer (CD147) is a novel receptor on platelets, activates platelets, and augments nuclear factor kappaB-dependent inflammation in monocytes. Circ. Res. 102, 302–309. [DOI] [PubMed] [Google Scholar]
- Seizer P., Schonberger T., Schott M., et al. (2010). EMMPRIN and its ligand cyclophilin A regulate MT1-MMP, MMP-9 and M-CSF during foam cell formation. Atherosclerosis 209, 51–57. [DOI] [PubMed] [Google Scholar]
- Svensson K.J., Christianson H.C., Wittrup A., et al. (2013). Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 288, 17713–17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi K., Ito S., Miyazaki T., et al. (2013). Amyloid precursor protein in human breast cancer: an androgen-induced gene associated with cell proliferation. Cancer Sci. 104, 1532–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W., Chang S.B., Hemler M.E. (2004a). Links between CD147 function, glycosylation, and caveolin-1. Mol. Biol. Cell 15, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Kesavan P., Nakada M.T., et al. (2004b). Tumor-stroma interaction: positive feedback regulation of extracellular matrix metalloproteinase inducer (EMMPRIN) expression and matrix metalloproteinase-dependent generation of soluble EMMPRIN. Mol. Cancer Res. 2, 73–80. [PubMed] [Google Scholar]
- Tang Y., Nakada M.T., Kesavan P., et al. (2005). Extracellular matrix metalloproteinase inducer stimulates tumor angiogenesis by elevating vascular endothelial cell growth factor and matrix metalloproteinases. Cancer Res. 65, 3193–3199. [DOI] [PubMed] [Google Scholar]
- Thery C., Ostrowski M., Segura E. (2009). Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 9, 581–593. [DOI] [PubMed] [Google Scholar]
- Weidle U.H., Scheuer W., Eggle D., et al. (2010). Cancer-related issues of CD147. Cancer Genomics Proteomics 7, 157–169. [PubMed] [Google Scholar]
- Yu X., Harris S.L., Levine A.J. (2006). The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 66, 4795–4801. [DOI] [PubMed] [Google Scholar]
- Yu X.L., Hu T., Du J.M., et al. (2008). Crystal structure of HAb18G/CD147: implications for immunoglobulin superfamily homophilic adhesion. J. Biol. Chem. 283, 18056–18065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.