

Abstract
Phosphoinositide-3-kinase enhancers (PIKE) are GTP-binding proteins that posses anti-apoptotic functions. The PIKE family includes three members, PIKE-L, PIKE-S and PIKE-A, which are originated from a single gene (CENTG1) through alternative splicing or differential transcription initiation. Both PIKE-S and PIKE-L bind to phosphoinositide-3-kinase (PI3K) and enhance its activity. PIKE-A does not interplay with PI3K. Instead, it interacts with the downstream effector Akt and promotes its activity. These actions are mediated by their GTPase activity. Because both PI3K and Akt are important effectors in the growth factor-mediated signaling which triggers cellular growth and acts against apoptosis, PIKEs therefore serve as the molecular switch that their activation are crucial for growth factors to exert their physiological functions. In this review, the current understanding of different PIKE isoforms in growth factors-induced anti-apoptotic function will be discussed. Moreover, the role of PIKE in the survival and invasion activity of cancer cells will also be introduced.
Keywords: Akt, apoptosis, GTPase, PI3K, PIKE
Introduction
Nerve growth factor (NGF) is a protein secreted by various neuron targets during development in both peripheral tissues and central nervous system. It is an important factor that controls the proliferation, differentiation, migration, survival and death of neuronal cells [1]. This protein exerts its physiological function through binding to its specific receptor (Trk A) which is a single-transmembrane receptor tyrosine kinase [2]. Numerous signal transduction path ways are triggered upon activation of Trk A by NGF and could be roughly classified into two categories: the small-GTPase pathway and the phospholipids pathway.
Among all the pathways triggered by activated Trk A, the phosphoinositide-3-kinases (PI3K)/Akt pathway is critical for NGF to maintain neuronal cell survival. PI3Ks are lipid kinases that phosphorylate phosphatidylinositol (PI) and its derivatives phosphatidylinositol-4-phosphate (PI-4-P) and phosphatidylinositol-4,5-biphosphate (PI-4,5-P) to give phosphatidylinositol-3-phosphate (PI-3-P), phosphatidylinositol-3,4-biphosphate (PI-3,4-P2) and phosphatidylinositol-3,4,5-triphosphate (PI-3,4,5-P3), respectively. They are heterodimers composed of a catalytic subunit (p110) and a regulatory subunit (p85). The kinases are grouped into three classes (classes I, II and III) based on their sequence homologies and substrate preferences [3]. Upon Trk A activation, classes IA PI3K is recruited to the intracellular tail of Trk A via adaptor proteins like Shc and Gab1 [4,5] where they phosphorylate the PI into various inositol phospholipids. The conversion of PI to PI-3,4,5-P3 by class I PI3Ks have been studied most extensively because the PI-3,4,5-P3 is an important intracellular messenger that binds to the pleckstrin homology (PH) domain of a great variety of proteins and activates downstream effector molecules for further signal conduction. Nowadays, more than hundreds of proteins are found to posses the PH domains. Akt (protein kinase B) is one of the wellknown examples of such PH-containing proteins. Akt is a serine/threonine kinase which belongs to the cAMP-dependant protein kinase A/protein kinase G/protein kinase C (AGC) superfamily [6]. The major function of Akt is to promote growth factor-mediated cell survival and to block apoptosis viaphosphorylating a great variety of proteins. For example, Akt phosphorylates proteins in the Bcl-2 and caspases families, suppressing apoptosis initiation [7, 8]. It has also been reported that transcription factors like Forkhead (FoxO) family members and nuclear factor-κB (NF-κB) were substrates of Akt which suggested that Akt could regulate cell survival through interacting with the transcription factors that are responsible for apoptotic gene transcription [9,10].
Because the PI3Ks were found predominately in the cytosol, most attentions were drawn to delineate the signal transduction pathway involving the cytosolic PI3Ks. Since the first report by Neri et al. [11] it is now obvious that the translocation of PI3K from cytoplasm to nucleus is a common phenomenon in several cell types [12–16]. The generation of PI-3,4,5-P3 by PI3K in the nucleus promotes cell survival in a transcription-independent manner which inhibits the caspase-3-activated DNA fragmentation factor/Caspase-activated DNase (DFF40/CAD) [17]. It is also found that nuclear PI-3,4,5-P3 activates nucle-ophosmin/B23 which in turn prevents the apoptotic DNA fragmentation [18]. In fact, nuclear phiospho-inositid lipids are important in regulating cell prolifer-ation and differentiation [19, 20]. Nevertheless, the regulatory mechanisms of PI3K in the nucleus are not obvious. The identification of phosphoinositide-3-kinase enhancers (PIKE), therefore, represents a breakthrough in this area. In this review, characteristics of the three PIKE isoforms as well as their roles in preventing apoptosis will be discussed. Moreover, the role of PIKE in the survival and invasion of glioblastomas will also be introduced.
The structure, tissue distribution and cellular localization of PIKEs
PIKE proteins are GTP-binding proteins which contain three members, PIKE-S, PIKE-L and PIKE-A (Fig. 1). They are originated from a single gene, CENTG1, by alternative splicing (PIKE-S and PIKE-L) or differential transcription start site usage (PIKE-A) [21, 22]. PIKE-S contains three praline-rich domains (PRD) at the N-terminus, followed by a GTP-binding motif (the GTPase region) and a PH domain. PIKE-L is the longer isoform of PIKE-S which possesses an extra C-terminal extension that includes a domain showing sequence homology to ARF/GAP protein (Arf-GAP) and two ankyrin repeats (ANK). PIKE-A shares identical structure with PIKE-L at the C-terminal but differ at the N-terminal that PIKE-A has no PRD.
1.
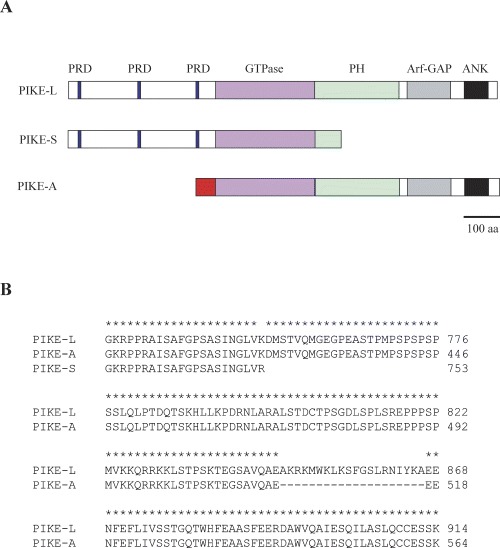
Structure of PIKE proteins. Schematic representations of PIKE proteins (A) and partial alignment of the PH domains of rat PIKEs amino acids (B) were shown. The structure which is different from that of other isoforms in PIKE-A is shown in red. Identical amino acids were marked with an asterisk and the corresponding position of the amino acid in each protein was numbered.
Although the PIKEs are generated from the same gene, their tissue distribution and cellular localization are different. Northern blot analysis and immunoblotting using a specific antibody against the N-terminal showed that PIKE-S was found prominently in brain [23]. Moreover, in situ hybridization showed that PIKE-S was expressed exclusively in the neurons of cortex, hippocampus and cerebellum. The highest level was found in both CA1 region of hippocampus and the dentate gyrus. Low intensity signal was detected in the molecular layer of cerebellum, but enriched in granule and Purkinje cells [23]. Further studies using immunofluorescent staining and cellular fractionation techniques revealed that PIKE-S was nuclear specific [23].
Like PIKE-S, PIKE-L is also brain specific. Although PIKE-S and PIKE-L could be found in the cortex, hippocampus and olfactory bulb, no PIKE-L was found in cerebellum as shown in the Western blot analysis [21]. Using an identical biochemical fractionation techniques as that used in PIKE-S studies, PIKE-L was detected primarily in the nuclear fraction but substantial level was also found in the cytoplasmic fraction of rat brain homogenates. Furthermore, immunohistochemical studies revealed that PIKE-L located in the cell body and the neuronal processes of hippocampal cultures [21]. These results suggest that, unlike PIKE-S which resides exclusively in the nucleus, PIKE-L presents both in the nucleus and the cytoplasm.
PIKE-A shows a distinct tissue distribution from the other PIKE isoforms. Using Northern blot analysis, high level of PIKE-A mRNA was detected in brain and small amount in liver, lung, skeletal muscle, spleen thymus, small intestine and periphery blood leucocytes [24–26]. Within the brain region, PIKE-A was detected in cerebellum, cerebral cortex, occipital pole, frontal lobe, temporal lobe and putamen [26]. Similar to PIKE-L, PIKE-A was found both in the cytoplasm and the nucleus of in transfected COS-7 cells [26]. In our evaluation of PIKE amplification in the human glioblastoma multiform, we found that PIKE-A but not -L or -S isoform is selectively over-expressed. Interestingly, the selectively over-expressed PIKE-A provokes cancer cell survival and invasion [22, 27]. Most recently, we found that PIKE-A act as a proto-oncogene, eliciting NIH3T3 cell transformation [28]. Thus, PIKE-A might contribute to tumourgenesis.
How different PIKE isoforms are shuttled and confined to specific cellular compartment is unknown.
Since the molecular mass of all PIKE isoforms exceed the size limit that could freely pass through the nuclear envelope by simple diffusion, their nuclear targeting must rely on protein-facilitated transportation. Sequence analysis of rat PIKE-L revealed two putative nuclear localization signals (NLS) in the PH domain [29]. Proteins that carry a NLS could be recognized by importin/karyopherins which are subsequently transported into the nucleus via the nuclear pore complex [30] The existence of the NLS in PIKE-L suggests that such importin/karyopherins-facilitated nuclear transportation might be the potential mechanism for PIKE localization in cell. However, the possibility that cytoplasm-nuclear transportation of PIKE via interaction with other proteins could not be excluded since the nuclear specific PIKE-S contains only a truncated PH domain [23]. The role of NLS in PIKE-L nuclear targeting will be further discussed in other section of this article.
Mitogenic PIKE-S signaling in the nucleus
Like Ras GTPase, PIKE-S showed a specific binding to GTP and hydrolyzed bound GTP into GDP. This GTP-loading was further increased in the presence of NGF [23]. These results suggest that PIKE-S is a functional GTP-binding protein in which its activity could be regulated by its intrinsic GTPase cycle: in the inactive state, GDP was bound to the GTP-binding domain whereas upon upstream signal stimulation, e.g. NGF stimulation, the GDP is replaced with GTP leading to the conformational change of the effector-binding region so that the region interacts with the downstream effectors. After activation, the GTP is hydrolyzed into GDP by its GTPase activity and thus returns to the resting state when the bound effector is released [31]. Further characterizations of PIKE-S indicated that it also bound to both p85 and p110 sub-units of PI3K in which the interactions consequently enhanced its activity.This augmentation of PI3K activity was GTPase dependant as interaction of a PIKE-S/K413AS414N) mutant, a mutation that abolished the GTP binding activity of PIKE-S, failed to activate PI3K. Moreover, the PIKE-S-stimulated PI3K activity was further enhanced by NGF stimulation. The temporal profile of PIKE-S activation, as shown by GTP-loading assay, correlated well with the enhanced PI3K activity after NGF treatment in which maximal activities of both proteins occurred 30 min after NGF stimulation and returned to basal level after 4 hrs [23]. Because PIKE-S is a nuclear specific protein, these observations, together with the fact that PIKE-S is an NGF-activated GTPase, reveal that PIKE-S is a regulator of PI3K activity in the nucleus (Fig. 2).
2.
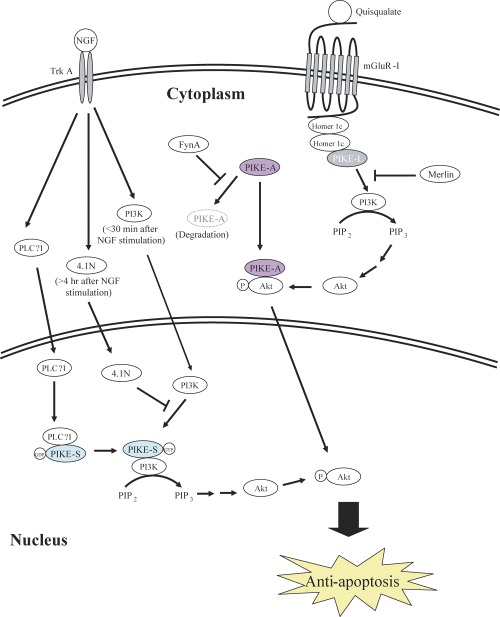
A summary of PIKE signaling pathways.
PIKE-S is firstly identified as the interaction partner of protein 4.1N. Protein 4.1N is a neuronal specific isoform of protein 4.1R which are members of 4.1 family which possess multiple functions including cytoskeleton networking, RNA processing, assembly of mitotic spindle, etc. [32–34]. In PC12 cells, 4.1N served as an important factor of NGF-mediated arrest of cell division in which NGF triggered 4.1N nuclear translocation and NuMA interaction [35]. By using yeast two hybrid screening, it was found that PIKE-S specifically bound to the C-terminal portion of 4.1N [23]. NGF treatment of PC12 cells also stimulated translocation of 4.1N from cytoplasm to nucleus where it bound to PIKE-S [23]. Within the nucleus, 4.1N serves as the negative regulator of PIKE-S in which its PI3K activity enhancer function is abolished. It was shown that the binding of PIKE-S and PI3K in PC12 cells was abolished in the presence of 4.1N as both 4.1N and PI3K interacted with PIKE-S on the same N-terminal site [23]. It was also found that the interaction of PIKE-S and 4.1N in PC12 cells increased after NGF treatment [23]. Taken into consideration that the temporal profile of this NGF-stimulated interaction between PIKE-S and 4.1N lags behind that of PIKE-S and PI3K (24 hrs versus 30 min), the negative regulatory role of 4.1N is therefore definite. Within minutes following NGF treatment, cytosolic PI3K is transported into the nucleus where it is activated by PIKE-S. Hours later, 4.1N is translocated into the nucleus where it binds to PIKE-S and diminishes the PI3K activities by displacing the PI3K from PIKE-S (Fig. 2, Table 1). To turn off the activity of nuclear PI3K, protein 4.1N might ensure the NGF-treated cells to be primed for cell-cycle arrest and trigger PC12 differentiation, since persistent PI3K activity has been shown to block PC12 differentiation.
1.
Functions of PIKE interacting proteins
| PIKE isoform | Interacting proteins | Interacting proteins | ||
|---|---|---|---|---|
| PIKE-L | Homer 1c | Adaptor protein which links PIKE and mGluR | ||
| Merlin | Preventing PI3K and PIKE-L interaction | |||
| PIKE-S | 4.1N | Preventing PI3K and PIKE-S interaction | ||
| PLCγ1 | Guanine nucleotide exchange factor of PIKE-S | |||
| PIKE-S | Akt | Kinase activity is enhanced when interacting with PIKE-A | ||
| Fyn | Phosphorylating PIKE-A and preventing its degradation | |||
It is well known that exchange of GDP to GTP in the GTP-binding site of a GTPase requires the association of a guanine nucleotide exchange factor (GEF) [31]. PRD typically binds to SH3 domains of other protein [36, 37]. Thus the PRD in PIKE-S might serve as an interacting domain with other regulatory protein to PIKE-S function. Using an in vitro binding assay, it was found that the PRD of PIKE-S bound to the SH3 domains of a variety of proteins including Fyn, Grb2 and phospholipase Cγ1 (PLC-γ1) [38]. However, only PLC-γ1 but not Fyn or Grb2 enhanced the GTP-binding or GDP dissociation of PIKE-S. This interaction was strongly enhanced in PC12 cells after stimulated by NGF, indicating that the PLC-γ1 played an important role in NGF mediated PIKE-S activation. Indeed, a functional PLC-γ1 was essential for PIKE-S signaling since the expression of a SH3 domain-deleted PLC-γ1 in PC12 cells greatly diminished the NGF triggered PIKE-S activation. Notably, the activation of PIKE-S by PLC-γ1 was independent of its lipase activity since the lipase-inactive mutant of PLC-1 displayed similar effect as its wild-type in PIKE-S activation.These results provide a mechanistic explanation to the mitogenic activity of PLC-γ1 which is independent of its lipase activity but requires its SH3 domain [39, 40]. PLC-γ1 is a well-characterized substrate of many growth factor receptor kinases to perform the proliferation and differentiation [41, 42]. Similar to PI3K, the cytosolic action of PLC-γ1 is extensively studied while its action in the nucleus remains obscure.The findings that NGF treatment causes the translocation of PLC-γ1 into the nucleus where it interacts with PIKE-S and its subsequent PI3K pathway thus provide a molecular mechanism in accounting for its mitogenic function in the nucleus. Similar findings were reported by Klein et al. [43] that a specific 120kDa PLC-1 fragment is activated and associated with PIKE-S in the nucleus after EGF treatment in vivo. In the studies, the authors also demonstrated that the interaction was triggered by nuclear EGF receptor but not a consequence of plasma membrane-located EGF receptor stimulation. Indeed, the nucleus possess sufficient machinery for production of PI-3,4,5-P3, Ca2+ stores and signaling apparatus, thus generating its own Ca2+ signaling that is independent to its cytosolic counterpart [44, 45]. Therefore, it is apparent that PIKE-S is a component of this unique system in regulating the nuclear lipid metabolism. Therefore, PIKE-S is a novel component of the unique system in regulating the lipid metabolism in the nucleus.
EGF enhances PC12 cell proliferation and NGF decreases its growth and arrests cell cycle, although both growth factors trigger numerous similar signaling cascades including PI3K and PLC-γ1 [46]. Recently, it has been reported that EGF provokes nuclear PLC-γ1 association with PIKE in mice liver in vivo [43]. Thus, PLC-γ1/PIKE/PI3K signaling cascade is conserved in both EGF and NGF provoked signaling pathways. Presumably, NGF but not EGF incurs 4.1N nuclear translocation to block PIKE's stimulatory effect in nuclear PI3K.
Anti-apoptotic function of PIKE-L in neurons
During the database search for sequences that resemble PIKE-S, several EST clones showing homology sequences to PIKE-S were identified [21]. Using the techniques of cDNA library screening, a longer isoform of PIKE-S, which designated as PIKE-L, was isolated from the rat brain library. Sequence analysis of the novel protein revealed a consensus sequence (PKPF) that interacted with the EVH1 domain of Homer [47] was located in the N-terminal (amino acid 187–190). This hypothetical interaction of Homer and PIKE-L was later confirmed based on the results of immunoprecipitation and in vitro binding assay [21]. It was found that the Homer 1c/PIKE-L interaction depends on the Homer EVH1 (ena/vasodilator-stimulated phosphoprotein homology) domain and the amino acid 180–225 (the PXXF motif where X represents any amino acid) of PIKE-L. Homer are adaptor proteins that locate at the postsynaptic densities where they interact with a great variety of proteins including group I metabotropic glutamate receptors (mGluR-I) [48], inositol-1,4,5-triphosphate receptors (IP3-R) [49], ryanodine receptors [50], transient receptor potential canonical-1 (TRPC1) [51], scaffold protein Shank [52], etc. Homer exists as a homodimer in which the coiled-coil motif at the C-terminal of two individual homer proteins binds together. Since the EVH1 motif of the Homer proteins are involved in protein–protein interactions with other proteins, the dimerized Homer proteins thus serve as an adaptor that links two individual proteins [53]. In fact, it has been reported that the Homer linked mGluR-I and IP3-R together which facilitates the mGluR-I mediated Ca2 response [49].
Because Homer 1c is known to be an interacting partner of mGluR-I in the neuron [48, 49], the demonstrations that PIKE-L associates with Homer 1c both in vitro and in vivo raised a possibility that the three proteins might form a functional complex. Immunoprecipiation studies from hippocampal neurons indicated that PIKE-L associated with mGluR-I in basal condition and this interaction was enhanced after quisqualate, an agonist of mGluR-I, stimulation [21]. In fact, this interaction of mGluR-I and PIKE-L via the Homer adaptor provides a novel linkage between mGluR-I and PI3K. In HKE293 cells transfected with mGluR5, Homer 1c and PIKE-L, quisqualate markedly increased the PI3K activity. This augmentation of PI3K activity was diminished if either one of the proteins were absence which indicated that an intact complex containing the three components were necessary for the quisqualate-triggered PI3K activation. Blockage of the interaction between mGluR-I and Homer, PIKE-L and PI3K or PIKE-L and Homer by using various mutated construct in the transfection also failed to increase the PI3K activity which provided further evidence to the crucial role of mGluR-I(Homer/PIKE-L complex. It was also found that the GTP-binding function of PIKE-L was essential to the quisqualate-stimulated PI3K activity as mutating the GTP-binding activity of PIKE-L greatly reduced the PI3K activation. Similarly, quisqualate markedly increased the PI3K activity in hippocampal neurons over-expressing the PIKE-L, but not those expressing the PIKE-L mutants (PIKE-L-KS) that failed to interact with PI3K. These results also demonstrated that, like PIKE-S, PIKE-L was able to associate with PI3K and regulated its activity accordingly (Fig. 2).
Previous studies reported that mGluR-I activation by its agonist prevent neuronal apoptosis via an unknown mechanism [54, 55]. The activation of PI3K by quisqualate-stimulated mGluR-I via PIKE-L therefore provides an explanation to this phenomenon. In hippoacmpal cultures, quisqualate stimulation prevented staurosporine-induced apoptosis [21]. In contrast, hippocampal neurons that over-expressing PIKE-L reduced the staurosporine elicited apoptosis which demonstrated the anti-apoptotic action of PIKE-L. This anti-apoptotic action of PIKE-L depended on its ability to activate the PI3K as no protective action was seem in neurons over-expressing the PIKE-L mutant PIKE-L-KS. When the neurons are infected with PIKE-L(P187L), a mutant that abolishes the interaction between Homer and PIKE-L which has no effect on the PI3K activation, the anti-apoptotic function of quisqualate was abolished. Furthermore, pretreatment of neurons with penetratin 1 conjugated peptide that disrupted the binding of PIKE-L and Homer abrogated the anti-apoptotic function of quisqualate. All these experiments suggested that mGluR-I stimulation of PI3K and prevention of apoptosis are mediated by interactions of mGluR-I, Homer, PIKE-L and PI3K. In addition to promoting neuronal survival, PIKE-L/Homer/PI3K signaling might also implicate a variety of mGluR-mediated cellular activities including synaptic plasticity, learning and memory formation. However, whether PIKE-L exerts its neuroprotective effects against neuronal excitotoxicities such as glutamate stress in vivo remain unknown. We strongly believe our ongoing PIKE knockout mice studies will elucidate all these activities.
Role of PIKE-L in merlin inhibited growth suppression
Using affinity chromatography purification method, another partner binding of PIKE family was found [56]. This protein, called merlin, is a tumour suppressor which shows structure homology to the proteins of the ezrin-eadxin-moesin (ERM) family which belong to the 4.1 protein superfamily [57]. Genetic studies indicated that merlin is encoded by the NF2 gene in which loss or mutation of the gene was responsible for the type 2 neurofibromatosis with schwannomas developed on or around the eighth cranial nerves [58]. It is now recognized that the mutation on NF2 gene is not restricted to schwannomas but also extended to thyroid carcinomas, hepatocellular carcinomas and perineurial tumours [59–62]. In genetically manipulated rat schwannoma cells RT4-D6P2T which contained inducible expression of merlin, the N-terminal of PIKE-L bind specifically to the N-terminal domain of merlin [56]. Interestingly, both merlin and Homer 1c shared the same binding site in the N-terminal of PIKE-L. On the other hand, the patient-derived missense mutation (L64P) within the N-terminal of merlin failed to interact with PIKE-L. The binding of merlin to PIKE-L leads to a diminished PIKE-L-enhanced PI3K activity, which was a result of binding site competition. In fact, this PI3K displacement from PIKE-L accounts for the anti-proliferative activity of merlin. In merlin over-expressed RT4-D6P2T cells, infection with adenovirus expressing PIKE-L reduced the anti-proliferation effect induced by merlin. This effect, however, could not be detected in cells infected with mutated PIKE-L (P187L) that showed no interaction with merlin. Over-expression of merlin also enhanced the caspase-3 activity which indicated that merlin expression could increase cell death. It has been reported that transfection of merlin into human schwannoma cells decreased the cell growth by inducing apoptosis [63]. Although the studies in merlin deficient cells revealed its important role in suppressing cell growth [64], the mechanism by which merlin acts as a tumour suppressor is not obvious. Recent work suggests that its function might come from the negative regulation of PAK1 or Rho/Rac GTPase [65, 66]. Since PI3K pathway plays an essential role in promoting cell survival, the negative regulatory effect of merlin in PIKE-L enhanced PI3K activity provides a novel mechanistic insight into how merlin regulated cell growth. Conceivably, selective inhibition of PIKE-L/PI3K signaling in neurofibromatosis might be conducive for NF2 patients since inhibition of PI3K/Akt signaling has been recognized as a potential approach to treat human malignancies [67].
Phosphoinositol lipids as a feedback regulator to PIKE-L activation and translocation
PH domain is a common structure motif found in numerous signaling molecules. Its function could be roughly categorized as a protein–protein interaction domain (e.g. β-ARK that catalyses β-adrenergic receptors phosphorylation [68]) or phosphoinositides binding site (e.g. Akt [69]). Because the PH domain presents in all the PIKE isoforms, it is conceivable that the domain should play some functional roles to PIKE activities. Studies by Hu et al. [29] suggested that the PH domain in PIKE-L acted as a regulatory domain in which the binding of phosphoinositol changed its cellular localization as well as the GTPase activity. In vitro binding assay using agarose-conjugated phosphoinositols revealed that the PH domain of PIKE-L bound preferentially to PI-3,4,5-P3 followed by PI-3,5-P3, PI-3,4-P3[29].The PH domains in PIKE-S and PIKE-A showed different phosolipid binding patterns. PH domain of PIKE-S did not bind to any phosphoinositol whereas PH domain of PIKE-A strongly bound to all phosphoinositols tested. This distinct binding affinity in different PIKE isoforms could be accounted by their structural variation in the PH domain. When compared with that of PIKE-L, the PH domain of PIKE-S is truncated which composed of the first 75 amino acid of PIKE-L PH domain. On the other hand, PH domain from PIKE-A omits 20 amino acids in the C-terminal which is the result of an exon skipping during transcription (Fig. 1).
The presence of all PIKE-L structural domains is not necessary for its GTPase activity as the GTP-binding domain of PIKE-L alone possesses intrinsic enzymatic activity [29]. However, occurrence of other structural domains together with the GTP-binding motif would greatly enhance its GTPase activity. It was found that the addition of Arf-GAP domain markedly increased its hydrolytic activity. This activity could also be increased when PI-3,4,5-P3 binds to the PH domain. Taken together, these observations suggest that phosphatidylinositol lipids may regulate PIKE-L conformation through the PH domain, leading to the C-terminal Arf-GAP domain accessible to its GTPase domain and accelerating its GTPase acivity. In fact, similar finding has been reported by Xia et al. [26] that the Arf-GAP domain has a regulatory effect to the GTPase activity of PIKE-A.
Not only the GTPase activity of PIKE-L but also its cellular localization is regulated by the phosphatidylinositol. Surprisingly, the PH domain of PIKE-L was exclusively distributed in the nucleus but the full-length PIKE-L was predominantly located in the nucleus with small amount found in the cytoplasm [29]. Mutation of the consensus amino acid of the PH domain (K679,687N) which abrogated its phosphatidylinositol-binding activity redirected the PIKE-L into the cytoplasm. It is therefore suggested that nuclear phosphoinositol lipids might tether PIKE-L in the nucleus thorough binding to the PH domain. However, such mutation in the PH domain of PIKE-A did not alter its cytoplasmic localization. In fact, sequence analysis revealed that PH domain in rat PIKE-L contained two NLS motifs. The first one locates between amino acid 679 to 692 which is conserved in both PIKE-A and PIKE-L and the second lies between amino acid 847 to 866 which is absent from PIKE-S and PIKE-A. Because the PH domain from PIKE-A resided exclusively in the cytoplasm, it was suggested that integrity of both NLS motifs is necessary for PIKE nuclear localization. Results from the mutation studies of the PH domain in PIKE-L further supported this idea as the K679,687N mutant disrupted the first NLS [29]. Nevertheless, it is noteworthy that PIKE-S, the PIKE isoforms which contains truncated PH domain, is nuclear specific [23]. Apparently, the PH domain is not the sole factor that controls the cellular localization of PIKEs and other structural domain in the N-terminal of PIKE-S might play roles in confining its cellular localization.
Anti-apoptotic activity of PIKE-A in cancers
PIKE-A cDNA has been identified a decade ago but its function remained unknown until its isoform PIKE-S was isolated [24, 25]. Previous studies only concentrated on its tissue distribution as well as its GTPase activity [26]. However, it has been noted that amplification of gene locus that including the CENTG1 and CDK4 (cyclin-dependent-kinase 4) sequences was frequently observed in sarcomas and brain tumours [25]. Ahn et al. [22, 27] further demonstrated that PIKE-A expression was substantially amplified in neuroblastoma cell line NGP-127, glioblastoma cell line LN-Z308, CRL-2061, SF188, SF767, TP366 and primary brain tumours. Using TP366 cells as example, it was found that this amplification was possibly due to chromosomal rearrangement as amplified CENTG1 could be found in non-chromosome-12 region whereas the endogenous CENTG1 was located in 12q13.3 of human chromosome. Cancer cells with this amplification are more resistance to apoptotic stimuli when compared with the cancer cells with normal CENTG1 copy number [27]. The anti-apoptotic role of PIKE-A was further confirmed from the studies using the cells whose endogenous PIKE-A is not detectable (In SF763 and LN229). In these cells, over-expressing PIKE-A remarkably inhibited staurosporine-induced apoptosis [27]. Moreover, knocking down of PIKE-A caused more cells with apoptotic features in cells with normal PIKE-A copy number than those with PIKE-A amplification [27]. The findings that expression of PIKE-A and p110α catalytic subunit of PI3K were significantly enhanced in primary glioblastomas further support the role of PIKE-A and PI3K in the molecular pathogenesis of primary glioblastomas [70].
Unlike PIKE-L and PIKE-S, PIKE-A is not able to bind and activate PI3K because of the absence of the N-terminal domains which are necessary for PI3K interaction [23]. Therefore, the anti-apoptotic function of PIKE-A must act through other signal transduction components. It is thus suggested that PIKE-A might interact directly with Akt, a downstream effector of PI3K which is activated by the phosphoinositol products produced by PI3K, to perform its anti-apoptotic function. This assumption is proved from two pieces of evidence. First, immuno-precipitation using anti-Akt antibody leads to the co-precipitation of PIKE-A in cell-line with over-expressed endogenous PIKE-A but not in those with normal PIKE-A expression [22]. In non-cancerous cells, the interaction of PIKE-A and Akt was growth factor dependent which suggested that PIKE-A only interacted with activated-Akt. Moreover, this interaction was GTP dependent as GTP pre-treatment of GST-PIKE-A strongly enhanced its interaction with Akt [22]. Further studies using in vitro binding assay suggested that PIKE-A interacted with activated Akt through its extreme N-terminus (amino acid 1–72) and a portion of the GTPase domain [61]. Interestingly, the GTPase domain alone was sufficient to bind Akt and was influenced by neither the presence of GTP nor the Akt activation [22]. These results suggest that the interaction of PIKE-A and Akt is somehow regulated by the overall structure of PIKE-A as the presence of other structural domains modified the binding pattern between PIKE-A and Akt. Presumably, the binding domains in full-length PIKE-A protein might be sheltered from Akt in the absence of guanine nucleotides. When bound to GTP or GDP, the conformation of PIKE-A might be changed which makes the binding site accessible to active Akt.
Similar to other PIKE isoforms, PIKE-A serves as an activator to its partner. In HEK293 cells, the epi-dermal growth factor (EGF) elicited Akt activity was further enhanced after over-expressing PIKE-A but not its GTP-binding mutated PIKE-A (K84A,S85N). Moreover, over-expressing the PIKE-A(K84A,S85N) mutant reduced the basal Akt activity [22]. Further evidence for the essentialness of PIKE-A in Akt activation came from the fact that EGF-stimulated Akt phosphorylation was substantially diminished in PIKE-A-knockdown cells. Indeed, the augmentation of Akt activity by PIKE-A depended on whether PIKE-A was GTP-bound. Binding of GTP to PIKE-A greatly enhanced the Akt activity indicating that activating the PIKE-A is a pre-requisite for the Akt activity augmentation. Nevertheless, the molecular mechanism of how PIKE-A regulates Akt remains elusive. A scenario is proposed that PIKE-A preferentially binds to phosphorylated (active) Akt which mask the phosphorylated region from phsophatase, so that the phosphorylated Akt is preferentially accumulated [22].
PIKE-A amplification not only protects the cancer cells from apoptosis but also increased their invasion. Over-expressing the PIKE-A in U87MG cells which normally do not have PIKE-A expression elicited a doubling of invasive cell number [22]. On the other hand, infecting the cells with PIKE-A(K84A,S85N) mutant reduced the cell invasion significantly. This reduces in cellular invasion was a result of reduced Akt activity as infecting the cells with the domain-negative Akt (K179A,T308A,S473A) diminished cell invasion in both cells with no PIKE-A expression (U87MG cells) or amplified PIKE-A expression (LN-Z308) [22].
PIKE-A as the physiological substrate of Fyn
Phosphorylation of a protein is a common mechanism to modify its activity. After growth factor stimulation, numerous proteins are phosphorylated in an orchestrated fashion to achieve the growth promotion. It has been reported that PIKE-A could also be phosphorylated after growth factor stimulation [71]. EGF triggered tyrosine phosphorylation of PIKE-A at its C-terminal which exclude the GTPase domain. Mutation analysis of the potential phosphorylation site revealed that Y682 and Y774 are the phosphorylation sites in PIKE-A after EGF stimulation. This phosphorylation, however, was not induced by the EGF-R receptor directly as neither an interaction between EGF-R and PIKE-A nor a direct phosphorylation could be seen [71]. These results indicated the phosphorylation of PIKE-A was mediated by an unknown kinase that could be activated by EGF-R signaling. In search for such kinase, Tang et al. [72] tested a variety of protein including focal adhesion kinase, Src and Fyn for their ability to phosphorylate PIKE-A. It was found that both active Fyn A and Src A were the kinases responsible for the EGF induced phosphorylation. However, it was Fyn that was responsible for the Y682 and Y774 phosphorylation as mutation of both sites abolished the phosphorylation of PIKE-A by Fyn but not Src. Both kinases belong to the src family of non-receptor protein tyrosine kinases which play an important role in cell cycle control, cell adhesion, cell proliferation and differentiation [73]. Further studies revealed that Fyn A and PIKE-A form a complex through the interaction between the Arf-GAP domain on PIKE-A and the SH1 (kinase domain) of Fyn [72]. In fact, the interaction of PIKE-A and Fyn depended on both the phosphorylation of PIKE-A and the kinase activity of Fyn. Interestingly, The phosphorylation of PIKE-A by Fyn A has no effect to its ability to Akt binding which suggested that phosphorylation of PIKE-A was unnecessary for its interaction with Akt.
Functional analysis indicated phosphorylation of PIKE-A prevented the caspases induced cleavage under apoptotic condition. It has been reported that the C-terminal of PIKE-A which contains the PH, Arf-GAP and the ANK domains could be cleaved by active apoptosomes isolated from HEK293 cells [71]. Point mutation of potential aspartate into alanine indicated that D474 and D592 were the major apop-totic cleavage sites [72]. While the Y682F and Y774F mutants enhanced the apoptotic degradation, the cleavage could be alleviated in vivo after the cells were stimulated with EGF suggesting that phospho-rylation of PIKE-A might prevent the cleavage possibly through Fyn A phosphorylation [73]. The role of Fyn in protecting PIKE-A from apoptotic cleavage was further confirmed from the studies using Fyn knockout (Fyn –/–) mouse embryonic fibroblast (MEF). When compared with control MEF, degradation of PIKE-A was significantly enhanced even in the absence of apoptosis inducing agent [73]. Moreover, EGF treatment could not remedy the basal PIKE-A cleavage in (Fyn–/–) MEF. The phosphorylation of PIKE-A by Fyn is also important for its anti-apoptotic function. When co-transfected with Fyn A and wild-type PIKE-A, the transfected Hela cells showed less DNA fragmentation than those transfected with Fyn A-phosphorylation crippled PIKE-A. Moreover, in Hela cells over-expressing PIKE-A mutant (D747,592A) which is caspase-resistant, decreased DNA fragmentation and capsase-3 activity were observed [73]. Collectively, Fyn phosphorylation of PIKE-A prevents its apoptotic degradation which leads to the promotion of cell survival.
Perspective remarks
Although PIKE proteins are crucial in anti-apoptotic action, our understanding on these proteins is still limited. The exploration of more physiological functions of PIKE proteins would definitely be one of the future directions in this research area. Since PI3K is a converge point of several signal transduction cascades triggered by stimulations other than cell survival signals, its upstream effector PIKE might also be involved in these physiological functions. For example, PI3K has been reported to be critical in memory formation and retrieval [74, 75], it is thus reasonable to speculated that PIKE might play a role in this process. This hypothesis is further supported by the fact that mGluR-I, the binding partner of PIKE-L, contribute to at least one form of activity dependent synaptic plasticity [76].
Another fundamental question regarding the PIKE signaling is whether PIKE activation is growth factor specific, i.e. only NGF/EGF could trigger PIKE activation. This question is partially answered by the studies of Rong et al. [21] which showed that PIKE-L could be activated by glutamate receptor agonist. Nevertheless, no studies have been performed to investigate the role of PIKE in those factors which relies heavily in PI3K/Akt activity. One of these examples is insulin. Although insulin is best known for controlling the glucose homeostasis in peripheral tissues, its role in neuronal physiology has now been recognized. Similar to that found in peripheral tissues, the signaling pathway triggered by insulin in neuron relies on the PI3K/Akt cascade [77]. Indeed, the function of insulin in brain is multivalent which includes the control of neurotransmitter receptor activity, neuronal-outgrown and cellular survival [78–80]. Our understanding of neuronal physiological functions would thus be enhanced if an establishment was made between PIKE and the factors using PI3K/Akt as their major signaling pathway.
It is also interesting to note that amplification of PIKE-A but not other PIKE isoforms are found in some of the glioblastomas like LN-Z308 and CRL-2061 [22, 70]. Provided that the three PIKE isoforms are coming from the same CENTG1 gene, it is thus logical to infer that a specific transcriptional regulatory mechanism to control the expression of each PIKE proteins must exist in these cells. The identification of responsible transcription factors in this unique mechanism would enhance our understanding in the PIKE signaling and be beneficial to control the growth of brain tumours.
In conclusion, the identification of the anti-apoptotic function of PIKE proteins reveals only a start of this novel research area. Further studies on PIKE signaling are necessary which not only facilitate our understanding in the neurobiology but also the cancer biology.
References
- 1.Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuropprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- 2.Arévalo JC, Wu SH. Neurotrophin signaling: many exciting surprises. Cell Mol Life Sci. 2006;63:1523–37. doi: 10.1007/s00018-006-6010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katso R, Okkenhaug K, Ahmadi K, White S, Timms S, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implication for development, immunity, homeostatsis and cancer. Annu Rev Cell Dev Biol. 2001;17:615–75. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 4.Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- 5.Holgado-Madruga M, Moscatello DK, Emlet DR, Diererich R, Wong AJ. Grb2-associated binder-1 mediates phosphatidylinositol-3-OH kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–24. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 7.Datta SR, Dedek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 8.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 9.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 10.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-βB by the Akt/PKB kinase. Curr Biol. 1999;9:601–4. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 11.Neri LM, Milani D, Bertolaso L, Stroscio M, Bertagnolo V, Capitani S. Nuclear translocation of phosphatidylinositol 3-kinase in rat pheochromocytoma PC 12 cells after treatment with nerve growth factor. Cell Mol Biol. 1994;40:619–26. [PubMed] [Google Scholar]
- 12.Lu PJ, Hsu AL, Wang DS, Yan HY, Yin HL, Chen CS. Phosphoinositide 3-kinase in rat liver nuclei. Biochemistry. 1998;37:5738–45. doi: 10.1021/bi972551g. [DOI] [PubMed] [Google Scholar]
- 13.Kim SJ. Insulin rapidly induces nuclear translocation of PI3-kinase in HeG2 cells. Biochem Mol Biol Int. 1998;46:187–96. doi: 10.1080/15216549800203692. [DOI] [PubMed] [Google Scholar]
- 14.Martelli AM, Borgatti P, Bortul R, Manfredini M, Massari L, Capitani S, Neri LM. Phosphatidylinositol 3-kinase translocates to the nucleus of osteoblast-like MC3T3-E1 cells in response to insulin-like growth factor I and platelet-derived growth factor but not to the proapoptotic cytokine tumor necrosis factor alpha. J Bone Miner Res. 2000;15:1716–30. doi: 10.1359/jbmr.2000.15.9.1716. [DOI] [PubMed] [Google Scholar]
- 15.Didichenko SA, Thelen M. Phosphatidylinositol 3-kinase C2 contains a nuclear localization sequence and associates with nuclear speckles. J Biol Chem. 2001;276:48135–42. doi: 10.1074/jbc.M104610200. [DOI] [PubMed] [Google Scholar]
- 16.Metjian A, Roll RL, Ma AD, Abrams CS. Agonists cause nuclear translocation of phosphatidylinositol 3-kinaseγ: a Gβγ-dependent pathway that requires the p110γ amino terminus. J Biol Chem. 1999;274:27943–7. doi: 10.1074/jbc.274.39.27943. [DOI] [PubMed] [Google Scholar]
- 17.Ahn JY, Rong R, Liu X, Ye K. PIKE/nuclear PI 3-kinase signaling mediates the anti-apoptotic actions of NGF in the nucleus. EMBO J. 2004;23:3995–4006. doi: 10.1038/sj.emboj.7600392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn JY, Liu X, Cheng D, Peng J, Chan PK, Wade PA, Ye K. Nucleophosmin/B23, a nuclear PI(3,4,5)P3 receptor, mediates the antiapoptotic actions of NGF by inhibiting CAD. Mol Cell. 2005;18:435–45. doi: 10.1016/j.molcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 19.York JD, Majerus PW. Nuclear phosphatidylinositols decrease during S-phase of the cell cycle in HeLa cells. J Biol Chem. 1994;269:7847–50. [PubMed] [Google Scholar]
- 20.Neri LM, Bortul R, Tabellini G, Borgatti P, Baldini G, Celeghini C, Capitani S, Martelli AM. Erythropoietin-induced erythroid differentiation of K562 cells is accompanied by the nuclear translocation of phosphatidylinositol 3-kinase and intranuclear generation of phosphatidylinositol (3,4,5) trisphosphate. Cell Signal. 2002;14:21–9. doi: 10.1016/s0898-6568(01)00224-8. [DOI] [PubMed] [Google Scholar]
- 21.Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Synder SH, Ye K. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–61. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- 22.Ahn JY, Rong R, Kroll TG, Van Meir EG, Snyder SH, Ye K. PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J Biol Chem. 2004;279:16441–51. doi: 10.1074/jbc.M312175200. [DOI] [PubMed] [Google Scholar]
- 23.Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ, Blackshaw S, Ferris CD, Snyder SH. Pike: a nuclear GTPase that enhances PI3 kinase activity and is regulated by protein 4.1N. Cell. 2000;103:919–30. doi: 10.1016/s0092-8674(00)00195-1. [DOI] [PubMed] [Google Scholar]
- 24.Nagase T, Seki N, Ishikawa K, Tanaka A, Nomura N. Prediction of the coding sequences of 40 new genes (KIAA0161-KIAA0200) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1996;3:17–24. doi: 10.1093/dnares/3.1.17. [DOI] [PubMed] [Google Scholar]
- 25.Elkahloun AG, Krizman DB, Wang Z, Hofmann TA, Roe B, Meltzer PS. Transcript mapping in a 46-kb sequenced region at the core of 12q13.3 amplification in human cancers. Genomics. 1997;42:295–301. doi: 10.1006/geno.1997.4727. [DOI] [PubMed] [Google Scholar]
- 26.Xia C, Ma W, Stafford LJ, Liu C, Gong L, Martin JF, Liu M. GGAPs, a new family of bifunctional GTP-binding and GTPase-activating proteins. Mol Cell Biol. 2003;23:2476–88. doi: 10.1128/MCB.23.7.2476-2488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahn JY, Hu Y, Kroll TG, Allard P, Ye K. PIKE-A is amplified in human cancers and prevents apoptosis by up-regulating Akt. Proc Natl Acad Sci USA. 2004;101:6993–8. doi: 10.1073/pnas.0400921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Hu Y, Hao C, Rempel S, Ye K. PIKE-A is a proto-oncogene, promoting cell growth, transformation and invasion. Oncogene. 2007 doi: 10.1038/sj.onc.1210290. in press. [DOI] [PubMed] [Google Scholar]
- 29.Hu Y, Liu Z, Ye K. Phosphoinositol lipids bind to phosphatidylinositol 3 (PI3)-kinase enhancer GTPase and mediates its stimulatory effect on PI3-kinase and Akt signalings. Proc Natl Acad Sci USA. 2005;102:16853–8. doi: 10.1073/pnas.0507365102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic. 2005;6:187–98. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- 31.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 32.Mattagajasingh SN, Huang SC, Hartenstein JS, Snyder M, Marchesi VT, Benz EJ. A nonerythroid iso-form of protein 4.1R interacts with the nuclear mitotic apparatus (NuMA) protein. J Cell Biol. 199(145):29–43. doi: 10.1083/jcb.145.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lallena MJ, Martinez C, Valcarcel J, Correas I. Functional association of nuclear protein 4.1 with pre-mRNA splicing factors. J Cell Sci. 1998;111:1963–71. doi: 10.1242/jcs.111.14.1963. [DOI] [PubMed] [Google Scholar]
- 34.Takakuwa Y. Protein 4.1, a multifunctional protein of the erythrocyte membrane skeleton: structure and functions in erythrocytes and nonerythroid cells. Int J Hematol. 2000;72:298–309. [PubMed] [Google Scholar]
- 35.Ye K, Compton DA, Lai MM, Walensky LD, Snyder SH. Protein 4.1N binding to nuclear mitotic apparatus protein in PC12 cells mediates the antiproliferative actions of nerve growth factor. J Neurosci. 1999;19:10747–56. doi: 10.1523/JNEUROSCI.19-24-10747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Cell. 1994;76:933–45. [Google Scholar]
- 37.Li SS. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem J. 2005;390:641–53. doi: 10.1042/BJ20050411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye K, Aghdasi B, Luo HR, Moriarity JL, Wu FY, Hong JJ, Hurt KJ, Base SS, Suh P, Synder SH. Phospholipase Cγ1 is a physiological guanine nucleotide exhange factor for the nuclear GTPase PIKE. Nature. 2002;415:541–44. doi: 10.1038/415541a. [DOI] [PubMed] [Google Scholar]
- 39.Smith MR, Liu YL, Matthews NT, Rhee SG, Sung WK, Kung HF. Phospholipase C-gamma 1 can induce DNA synthesis by a mechanism independent of its lipase activity. Proc Natl Acad Sci USA. 1994;91:6554–8. doi: 10.1073/pnas.91.14.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang PS, Davis L, Huber H, Goodhart PJ, Wegrzyn RE, Oliff A, Heimbrook DC. An SH3 domain is required for the mitogenic activity of microinjected phospholipase C-gamma 1. FEBS Lett. 1995;358:287–92. doi: 10.1016/0014-5793(94)01453-8. [DOI] [PubMed] [Google Scholar]
- 41.Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 42.Manzoli L, Martelli AM, Billi AM, Faenza I, Fiume R, Cocco L. Nuclear phospholipase C: Involvement in signal transduction. Prog Lip Res. 2005;44:185–206. doi: 10.1016/j.plipres.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 43.Klein C, Gensburger C, Freyermuth S, Nair BC, Labourdette G, Malviya AN. A 120 kDa nuclear phospholipase C(1 protein fragment is stimulated in vivo by EGF signal phosphorylating nuclear membrane EGFR. Biochemistry. 2004;43:15873–83. doi: 10.1021/bi048604t. [DOI] [PubMed] [Google Scholar]
- 44.Malviya AN, Klein C. Mechanism regulating nuclear calcium signaling. Can J Physiol Pharmacol. 2006;84:403–22. doi: 10.1139/y05-130. [DOI] [PubMed] [Google Scholar]
- 45.Gomes DA, Leite MF, Bennett AM, Nathanson MH. Calcium signaling in the nucleus. Can J Physiol Pharmacol. 2006;84:325–32. doi: 10.1139/y05-117. [DOI] [PubMed] [Google Scholar]
- 46.Vaudry D, Stork PJS, Lazarovici, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648–9. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- 47.Beneken J, Tu JC, Xiao B, Nuriya M, Yuan JP, Worley PF, Leahy DJ. Structure of the Homer EVH1 domain-peptide complex reveals a new twist in polyproline recognition. Neuron. 2000;26:143–54. doi: 10.1016/s0896-6273(00)81145-9. [DOI] [PubMed] [Google Scholar]
- 48.Ango F, Pin JP, Tu JC, Xiao B, Worley PF, Bockaert J, Fagni L. Dendritic and axonal targeting of type 5 metabotropic glutamate receptor is regulated by homer1 proteins and neuronal excitation. J Neurosci. 2000;20:8710–6. doi: 10.1523/JNEUROSCI.20-23-08710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–26. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 50.Feng W, Tu J, Yang T, Vernon PS, Allen PD, Worley PF, Pessah IN. Homer regulates gain of ryanodine receptor type 1 channel complex. J Biol Chem. 2002;277:44722–30. doi: 10.1074/jbc.M207675200. [DOI] [PubMed] [Google Scholar]
- 51.Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, Dehoff MH, Schwarz MK, Seeburg PH, Muallem S, Worley PF. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell. 2003;114:777–89. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
- 52.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–92. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 53.Xiao B, Tu JC, Worley PF. Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol. 2000;10:370–4. doi: 10.1016/s0959-4388(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 54.Copani A, Bruno VM, Barresi V, Battaglia G, Condorelli DF, Nicoletti F. Activation of metabotropic glutamate receptors prevents neuronal apoptosis in culture. J Neurochem. 1995;64:101–8. doi: 10.1046/j.1471-4159.1995.64010101.x. [DOI] [PubMed] [Google Scholar]
- 55.Vincent AM, Maiese K. The metabotropic glutamate system promotes neuronal survival through distinct pathways of programmed cell death. Exp Neurol. 2000;166:65–82. doi: 10.1006/exnr.2000.7487. [DOI] [PubMed] [Google Scholar]
- 56.Rong R, Tang X, Gutmann DH, Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc Natl Acad Sci USA. 2004;101:18200–5. doi: 10.1073/pnas.0405971102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, Pulst SM, Lenoir G, Bijlsma E, Fashold R, Dumanski J, De Jong P, Parry D, Eldrige R, Aurias A, Delattre O, Thomas G. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–21. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 58.Twist EC, Ruttledge MH, Rousseau M, Sanson M, Papi L, Merel P, Delattre O, Thomas G, Rouleau GA. The neurofibromatosis type 2 gene is inactivated in schwannomas. Hum Mol Genet. 1994;3:147–51. doi: 10.1093/hmg/3.1.147. [DOI] [PubMed] [Google Scholar]
- 59.Bianchi AB, Hara T, Ramesh V, Gao J, Klein-Szanto AJP, Morin F, Menon AG, Trofatter JA, Gusella JF, Seizinger BR, Kley KN. Mutations in transcript iso-forms of the neurofibromatosis 2 gene in multiple human tumour types. Nat Genet. 1994;6:185–92. doi: 10.1038/ng0294-185. [DOI] [PubMed] [Google Scholar]
- 60.Lasota J, Fetsch JF, Wozniak A, Wasag B, Sciot R, Miettinen M. The neurofibromatosis type 2 gene is mutated in perineurial cell turmors: A molecular genetic study of eight cases. Am J Pathol. 2001;158:1223–9. doi: 10.1016/S0002-9440(10)64072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheikh HA, Tometsko M, Niehouse L, Aldeeb D, Swalsky P, Finkelstein S, Barnes EL, Hunt JL. Molecular genotyping of medullary thyroid carcinoma can predict tumor recurrence. Am J Surg Pathol. 2004;28:101–6. doi: 10.1097/00000478-200401000-00012. [DOI] [PubMed] [Google Scholar]
- 62.Pineau P, Marchio A, Nagamori S, Seki S, Tiollais P, Dejean A. Homozygous deleltion scanning in hepato-biliary tumor cell lines reveals alternative pathways for liver carcinogenesis. Hepatology. 2003;37:852–61. doi: 10.1053/jhep.2003.50138. [DOI] [PubMed] [Google Scholar]
- 63.Schulze KM, Hanemann CO, Muller HW, Hanenberg H. Transduction of wild-type merlin into human schwannoma cells decreases schwannoma cell growth and induces apoptosis. Hum Mol Genet. 2002;11:69–76. doi: 10.1093/hmg/11.1.69. [DOI] [PubMed] [Google Scholar]
- 64.Xiao GH, Gallagher R, Shetler J, Skele K, Altomare DA, Pestell RG, Jhanwar S, Testa JR. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Cell Biol. 2005;25:2384–94. doi: 10.1128/MCB.25.6.2384-2394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–9. doi: 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 66.Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, O'Bryan JP, Gupta V, Ratner N, Der CJ, Jacks T, McClatchey AI. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev Cell. 2001;1:63–72. doi: 10.1016/s1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 67.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–76. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 68.Touhara K, Inglese J, Pitcher JA, Shaw G, Lefkowitz RJ. Binding of G protein beta gamma-subunits to pleckstrin homology domains. J Biol Chem. 1994;269:10217–20. [PubMed] [Google Scholar]
- 69.Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–44. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Knobbe CB, Trampe-Kieslich A, Reifenberger G. Genetic alteration and expression of the phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropath Appl Neurobiol. 2005;31:486–90. doi: 10.1111/j.1365-2990.2005.00660.x. [DOI] [PubMed] [Google Scholar]
- 71.Tang X, Ye K. PIKE tyrosine phosphorylation regulates its apoptotic cleavage during programmed cell death. Advan Enzyme Regul. 2006;46:289–300. doi: 10.1016/j.advenzreg.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 72.Tang X, Feng Y, Ye K. Src-family tyrosine kinase Fyn phosphrylates phosphatidylinositol 3-kinase enhancer-activating Akt, preventing its apoptotic cleavage and promoting cell survival. Cell Death Differ. 2007;14:368–77. doi: 10.1038/sj.cdd.4402011. [DOI] [PubMed] [Google Scholar]
- 73.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–58. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 74.Horwood JM, Dufour F, Laroche S, Davis S. Signalling mechanisms mediated by the phospho-inositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur J Neurosci. 2006;23:3375–84. doi: 10.1111/j.1460-9568.2006.04859.x. [DOI] [PubMed] [Google Scholar]
- 75.Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–31. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- 76.Eckert MJ, Racine RJ. Metabotropic glutamate receptors contribute to neocortical synaptic plasticity in vivo. Neuroreport. 2004;15:2685–9. doi: 10.1097/00001756-200412030-00027. [DOI] [PubMed] [Google Scholar]
- 77.Van Der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol. 2006;79:205–21. doi: 10.1016/j.pneurobio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 78.Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. 1997. Recruitment of functional GABA (A) receptors to postsynaptic domains by insulin. Nature. 1997;388:686–90. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- 79.Mill JF, Chao MV, Ishii DN. Insulin, insulin-like growth factor II, and nerve growth factor effects on tubulin mRNA levels and neurite formation. Proc Natl Acad Sci USA. 1985;82:7126–30. doi: 10.1073/pnas.82.20.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tanaka M, Sawada M, Yoshida S, Hanaoka F, Marunouchi T. Insulin prevents apoptosis of external granular neurons in rat cerebellar slice cultures. Neurosci Lett. 1995;199:37–40. doi: 10.1016/0304-3940(95)12009-s. [DOI] [PubMed] [Google Scholar]