

Abstract
The thiazolidinediones (TZDs) are a class of synthetic antidiabetic drugs exerting its action primarily upon acti-vation of the peroxisome proliferator-activated receptor-γ (PPARγ). Given the widespread incidence of diabetes type II and lifelong exposure of these patients to TZDs, there is a possibility that chronic treatment with TZD modifies clinical phenotypes of other common human diseases, for example breast carcinoma. There is evidence that TZDs act as breast carcinoma suppression agents, at least in the in vitro and animal models. Stimulation of the PPARγ by TZDs interferes with oestrogen receptor signalling, STAT5B and NF-κB signalling cascades. On the other hand, TZDs repress TGFβ signalling, a well-known suppressor of the initial stages of breast carcinoma development. Another layer of complexity arises at the later stages of tumour development, when TGFβ acts as a tumour promoter: its overexpression is associated with poor prognosis, higher degree of tumour vascularization and metastasis. Longitudinal studies of breast carcinoma development in chronic TZD users are needed. In this review, we dissect possible interplays between chronic exposure of breast tis-sue to TZDs and TGFβ signalling and predict influence of TZD exposure on cancer-related clinical outcome.
Keywords: thiazolidinediones (TZDs), breast carcinoma, PPARγ, TGFβ, metastasis, proliferation, diabetes, pioglitazone, rosiglitazone
Introduction
Breast cancer is a complex disease that results from the most frequent non-skin cancer to affect women a multi-stage process involving the deregulation of worldwide, and remains one of the top public health a number of different signalling cascades. It is also burdens. There are about 212,930 new cases of breast cancer diagnosed every year, and 40,840 related deaths in the United States alone [1]. Despite a number of recent attempts to stratify breast carcinoma into the transcriptome-based sub-groups [2–4], extreme heterogeneity of these tumours still poses a real challenge. Current therapeutic modalities for breast cancer predominantly employ cytotoxic drugs prescribed with or without adjuvant therapies. Despite the fact that many risk factors have been identified for breast carcinomas, their prognostic and predictive values remain con-troversial. Routine investigations often identify indo-lent precursors for breast cancer, including lobular and ductal carcinomas in situ (LCIS and DCIS) that are often treated by biopsy-like surgery alone [5–7]. In addition, the findings of benign breast lesions, for example ductal hyperplasia and fibroadenoma, are common [8].
Both benign and malignant changes in the breast are commonly found in patients with another wide-spread pathology, diabetes type II. Diabetes patients are routinely treated with thiazolidine-diones (TZDs), class drugs pioglitazone (Actos, Takeda/Lilly) and rosiglitazone (Avandia, Glaxo-SmithKline) exerting glucose-lowering effects. These effects are mediated primarily by decreasing insulin resistance in the muscles and thereby increasing glucose uptake. In addition, TZDs sup-press glucose production in the liver. Recently, the TZD class of medications became a mainstream diabetes therapy [9]. As TZDs are also known to suppress the proliferation and induce apoptosis of breast carcinoma cells in vitro[10], it is likely that the breast epithelium of diabetics exposed to TZDs will also experience those modifying effects. It might translate into the changes of breast lesion inci-dence or in the rates of their malignization in diabet-ics treated with TZDs in comparison with the gener-al population. TZD treatment may also influence the progression of existing invasive lesions that remains yet undiagnosed.
In this review, we attempt to summarize the possi-ble influence of TZD exposure to the molecular circuit-ry involved in the initiation and progression of breast carcinoma. Special attention will be paid to the epige-netic interplay of TZDs with TGFβ signalling, clearly implicated in breast carcinoma development in multi-ple studies.
PPARγ-dependent and -independent action of TZDs
Peroxisome proliferator-activated receptors (PPARs) belong to the superfamily of nuclear hormone recep-tors activated by lipophilic agonists. There are three known isoforms: PPARα, PPARβ (also known PPARδ) and PPARγ[11, 12]. There are two PPARγ isoforms derived from the alternative promoters, PPARγ1 and PPARγ2. PPARγ2 isoform is longer than PPARγ1 by additional 30 N-terminal amino acids [13]. PPARγ2 mostly expresses in adipocytes while PPARγ1 is ubiquitous. Both isoforms have the intrinsic ability to stimulate adipogenesis by induction of the similar changes in the pre-adipocyte expres-sion profile. However, PPARα2 could be activated by lower concentrations of the ligands and is more affine to the components of the DRIP/TRAP coacti-vator complex [13]. The relative expression of the PPARγ isoforms in the human tumours is still under debate [14]. The quantitative and qualitative patterns of their expression in the primary and metastatic breast carcinomas need further investigation.
The general mechanisms of gene transcription modulation by PPARs are quite similar and well understood. After PPAR binds to its specific ligand, it heterodimerizes with retinoid X receptor (RXR) and binds to specific bi-hexametric DNA sequences called PPRE elements. This binding, activation and heterodimerization processes recruit various co-acti-vators and co-repressors that modulate expression of many human genes [15, 16]. Some of the best-known co-activator proteins for PPAR are histone acetyltransferase p300 (CBP), SRC-1, TEF2, Drip205 (Med220) and PGC-1 [17–19]. The latter binds to PPARγ in a ligand-independent manner overlapping the ‘CoR box’ region required for binding of co-repressors [20], recruited by PPARγ in the absence of a ligand. Both silencing mediators of retinoid and thyroid hormone receptors (SMRT) and nuclear receptor co-repressor (NCoR) are capable of down-regulating PPARγ-dependent transcriptional activity. When a suitable ligand binds PPAR, it is com-plex with the co-repressor dissociates [20], thus allow-ing the recruitment of co-activators.
More than 70 PPAR target genes with functional PPREs have been confirmed experimentally (see [21] for the comprehensive list). There is no doubt that this list is skewed towards the targets involved in fatty acid metabolism and adipocyte differentiation, as these two functional pathways serve as the prime focus for recent studies. A recent genome-wide search has been conducted for the high-score PPREs in conserved elements of the 5000 base pairs upstream of all human reference genes [21]. When gene ontology (GO) annotations were retrieved for each human gene returned in the search, fatty acid metabolism appeared at ninth place in the significance table according to associat-ed z-scores. Genes involved in cell cycle arrest, apoptosis and DNA damage response were more likely to be targeted by PPARs than those involved in fatty acids metabolism. Furthermore, five out of eight positions above fatty acid metabolism were occupied by functional categories embracing the regulation of transcription and chromatin remodelling and includ-ed genes HDAC1 and HDAC3. It should be noted that the results of the computational prediction of the transcription factor binding should be used with cau-tion, as even the best algorithms of this kind tend to give many false-positive results. The PPRE data col-lected in [21] await experimental investigation.
It is of interest that most of the PPAR targets revealed by various indirect means, including microarrays, are not found in the list of the genes that contain PPREs [22]. This can be explained by the activation of the PPAR targets through its direct bind-ing to other proteins (protein–protein interactions) that, in turn, indirectly modulate target gene's tran-scription. For example, Hong and colleagues showed that PPAR gamma interacts with both Sp1 and Sp4 proteins in order to activate the p21 promoter [23]. In another cellular system, PPARγ has been shown to bind Sp1 directly and to suppress transcription of the thromboxane receptor gene (TXR) through this inter-action [24]. Another possible explanation of the differ-ences between PPRE-based and microarray-based PPARγ target lists is the ‘two waves’ model of the PPAR-dependent transcription. The first PPAR-dependent wave involves up-regulation of the pri-mary PPARγ targets, including the chromatin-remod-elling proteins necessary for implementation of the broader changes in the cellular transcriptome; these changes are characteristic for the second PPAR-dependent wave, which covers a much larger num-ber of secondary targets.
PPARγ is predominantly expressed in adipocytes, and is also highly expressed in the prostate, breast, colon, liver, skeletal muscles and macrophages [25]. Potential endogenous ligands for PPARγ include polyunsaturated fatty acids (PUFAs) and eicosanoids, which bind to and activate this receptor in various affinities and specificities [26–28]. An anti-inflammatory prostaglandin 15-deoxy-D12,14-PGJ2 (15d-PGJ2) that is formed from PGD2 in vivo is prob-ably the most potent endogenous PPAR ligand [26–28]. Another powerful physiological PPAR stim-ulator is oxidized phosphatidylcholine [29].
A number of chemically synthesized ligands for PPARγ (thiazolidinediones, or TZDs) has been dis-covered and introduced in clinical practice in the late 1990s as insulin sensitizers [9]. One of them, troglitazone, was withdrawn by the FDA in 2000 due to severe hepatotoxic effects it produced in some patients [30]. Two other agents, pioglitazone and rosiglitazone, are commonly used worldwide for the treatment of the insulin resistance. In May 2000, pioglitazone passed the one million prescription mark after only 8 months in the US market. As con-cerns for TZDs side effects remain, particularly in patients predisposed to congestive heart disease, searches for improved synthetic PPARγ activators of non-TZD type are still ongoing [31].
In addition to PPARγ-dependent action, TZDs demonstrate a number of important PPARγ-inde-pendent effects. To name a few examples, the stimu-lation of the proteosomal degradation of cyclins D1 and D3 [32–34], the suppression of the NHE1 chan-nel activity resulting in cellular acidosis [35], the block of G(1)-S transition by inhibiting translation initiation [36] and scavenging effects of some TZDs on reac-tive oxygen species [37] have been demonstrated.
Effects of TZDs in the breast epithelium
Many research experiments have shown that PPARγ ligands suppress the proliferation rates of many types of cancer cells, particularly those derived from liposarcoma, colon cancer, breast cancer, prostate cancer, myeloid leukemia, glioblastoma and many others [38, 39]. Moreover, various in vitro studies have shown that treatment of many types of cancer cells with TZD resulted in the induction of cell differ-entiation or apoptosis, as well as improvement in levelsof various markers for invasion and metastasis [10, 38, 39]. A body of evidence indicates that human breast carcinomas might also be responsive to TZDs [10].
PPARγ is expressed both in the normal breast tis-sue and in many primary breast carcinoma speci-mens [40, 41]. Most likely, PPARγ responsiveness is preserved in clinical breast tumours, as the mutation of the PPARγ gene is a very rare event in human malignancies [42]. Therefore, if PPARγ signalling ever undergoes alterations in the tumour cells, these alterations should be pursued at the epigenetic level. Comparative studies of PPARγ expression in breast carcinoma patients so far produced contradictory results [43–45]. Since complete loss of PPARγ sig-nalling seems to be a rare event, it is likely that TZDs may be able to modify the phenotype of breast carci-noma cells across its histological subtypes. This makes TZDs highly promising adjuvants that could be, in theory, even more practical than hormonal ablation therapy, which largely depends on the pres-ence of the oestrogen receptor (OR) and proges-terone receptors (PR) at the tumour cell surface.
TZDs and 15d-PGJ2 inhibit the growth of both nor-mal human mammary epithelial cells [40] and breast cancer cells [46–48]. Growth suppression by TZDs is mediated by repression of cyclin D1 by PPARγ-dependent transcriptional [49] and proteosome-dependent post-translational [32, 33] mechanisms. Levels of cyclin D3 are also suppressed by similar means in breast carcinoma calls treated with TZDs [34]. Treatment of breast carcinoma MCF7 cells with therapeutic TZD rosiglitazone has been shown to increase levels of mRNA and levels of the tumour suppressor p53 and its effector p21 (WAF1/Cip1) [50]. Interestingly, in this model the binding of PPARγ to the TP53 promoter requires its interaction with nuclear factor κB (NF-κB) binding sequence, an indi-cation of the crosstalk between PPARγ and NF-κB pathways [50]. Another proliferation-related pathway suppressed by rosiglitazone is Akt/PTEN: TZDs increase PTEN expression in MCF7 cells and decrease Akt phosphorylation [51, 52]. In addition to the direct influence on cell proliferation, TZDs inhibit Na+/H+ exchanger (NHE) isoform 1 and therefore induce marked cellular acidosis in breast carcinoma cell lines; this leads to a decreased number of viable cells and suppressed cell proliferation [35]. The latter effect of TZDs is independent of PPARγ transcrip-tional activities [35].
The growth suppressive capabilities of TZDs are complemented by their ability to induce apoptosis. Many breast tumours are naturally resistant to the apoptotic action of the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL). TZDs sensitize these cells to TRAIL at least in part by reducing levels of the anti-apoptotic protein survivin [34]. It is tempt-ing to speculate that TZDs might also sensitize malig-nant cells in newly developed microscopic tumours to endogenous TRAIL, thus preventing further spread. TZDs also synergize with All-trans-retinoic acid (ATRA) in order to induce apoptosis in MCF-7 and pri-mary breast carcinoma cells, but not in the normal breast epithelium. Interestingly, ATRA alone is unable to initiate programmed death in these cells. TZDs/ATRA combination-dependent apoptosis is associated with a dramatic decrease of BCL2 protein levels [48].Troglitazone, but not pioglitazone or rosigli-tazone, up-regulates growth arrest and DNA damage-inducible gene 45 (GADD45) in a time- and dose-dependent manner [53]. It has been shown earlier that GADD45 could be also stimulated by a sudden rise in BRCA1 levels and takes part in BRCA1-induced apoptosis [54]. In turn, expression of BRCA1 itself is enhanced in response to PPARγ activation both by natural (15dPG-J2) and synthetic (rosiglitazone) ligands [55]. Finally, troglitazone directly stimu-lates a promoter of gene POX that encodes proline oxidase, a redox enzyme localized in the mitochondr-ial inner membrane that mediates apoptosis by gen-erating a reactive oxygen species [56].
It seems that PPARγ ligands can also influence the earliest stages of breast carcinoma development, in particular, immortalization. These data were obtained in the experiments with Li-Fraumeni Syndrome (LFS)-derived (p53 +/−, telomerase silent) breast epithelial cells that have been shown to spon-taneously immortalize at a relatively high frequency of approximately 5 × 10−7[57]. Treatment of LFS-derived breast epithelial cells just before crisis with low nontoxic doses of rosiglitazone (10 nM) reduced the frequency of spontaneous immortalization to 1.33 × 10−7. It is of interest that treatments of the same model cells with known chemopreventive agents sulindac sulfide and celecoxib resulted in less pronounced decreases in immortalization [57].
PPARγ agonists stimulate terminal differentiation of the MCF-7 breast cancer cells in vitro[58, 59]. Particularly, treatment of breast carcinoma cells with TZDs causes lipid accumulation and a profound change in breast epithelial gene expression associat-ed with a more differentiated, less malignant state [46]. Nevertheless, expression of aP2 and adipsin, well-established markers of adipogenesis, stays unchanged in these breast carcinoma cells, thus indicating that lipid accumulation in these cells did not result from ‘transdifferentiation’ of carcinoma cells to adipocytes. In addition, the differentiating action of PPARγ ligands on non-malignant stromal cells may also be beneficial for the patients. It is well known that peri- and intra-tumoural fibroblasts provide struc-tural and secretory growth promoting support to tumour tissue [60]. Moreover, malignant breast epithelial cells actively participate in the process of accumulation of stromal fibroblasts around the tumour tissue, known as desmoplastic reaction. Breast carcinoma cells secrete compounds prevent-ing the differentiation of fibroblasts to adipocytes through down-regulation of PPARγ activity in them [61]. Chronic treatment with TZDs may counteract or delay the formation of the scirrhous component of the breast tumours and the subsequent spread of tumour cells.
PPARγ activation by its ligands also possesses anti-invasive activities in tumour cells, as it inhibits gelatinase B (MMP-9) and blocks migration of macrophages and muscle cells. Low concentration treatment of the highly aggressive human breast cancer cell line MDA-MB-231 with pioglitazone, rosiglitazone or 15d-PGJ2 inhibits the invasive capacities of this cell line in a Matrigel™ basement membrane. A mechanism of invasion inhibition in this case is probably linked to the up-regulation of tissue inhibitor of MMP-1/TIMP-1 and subsequent decrease in the gelatinolytic activities [62]. In addition to invasion suppression, PPARγ ligands demonstrate strong anti-angiogenic effects (reviewed in [39]), including direct suppression of the vascular endothe-lial growth factor (VEGF) expression through PPRE [63], repression of the angiopoietin-1 (Ang-1) gene transcription [64], and blocking of the production of the angiogenic ELR+CXC chemokines IL-8 (CXCL8), ENA-78 (CXCL5) and Gro-α (CXCL1) [65]. Rosiglitazone also inhibits VEGF165-induced angio-genesis through increase in NO production, followed by Maxi-K channel opening and vascular cell apopto-sis [59]. On the other hand, there is accumulating evi-dence that in the non-cancerous settings, e.g. in the ischemic brain and gastric ulcers, PPARγ ligands stimulate angiogenesis [66–68]. The contradictory nature of these observations might indicate that PPARγ ligands are capable of counteracting the metastatic process by remodelling the tumour ves-sels, a process known as vascular normalization [69].
Crosstalks of the PPARγ signalling with OR, STAT5B and NF-κB pathways
PPARγ interferes with numerous cellular pathways, particularly OR, STAT5B, TFGβ and NF-κB. Most likely, the interference is mutual, as it is exerted through shared molecular components taking part in the propagation of the signal or the transcriptional regulation (Fig. 1).
1.
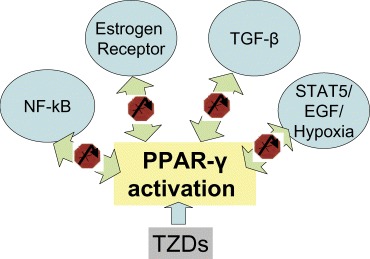
TZDs activate PPAR signalling that interferes with OR, STAT5B, TFGβ and NF-κB pathways. Most likely, this interference is mutual.
A study of the immunolocalization of PPARγ in 238 human breast carcinoma samples showed that PPARγ status is significantly associated with the OR status [49]. That indicates that patients with OR-pos-itive tumours might obtain more pronounced benefits from treatment with TZDs than the patients with OR-negative tumours. This hypothesis is further support-ed by findings made in the breast carcinoma cell line MCF-7 [49]. A treatment of this cell line with PPARγ ligand 15d-PGJ2 significantly inhibited OR element-dependent transcriptional activation by estradiol, which was blocked by the addition of a PPARγ antag-onist GW9662 [47]. Sixteen out of 49 estradiol-dependent genes ceased to respond to that com-pound after treatment with the 15d-PGJ2 ligand [49]. Some important regulators of cell proliferation were found among these genes, including intestinal trefoil factor (TFF1), cyclin D1 (CCND1) and CDK1A1 [47]. The PPARγ signalling also inhibits expression of aromatase (CYP19) that converts androgen to oestro-gen, thus further reducing oestrogenic pressure on the breast epithelium. These effects of PPARγ lig-ands were demonstrated both in breast carcinoma cell lines [70] and in the cultured breast adipose stro-mal cells [71]. Most probably, an interplay between PPARγ and OR signalling involve the recently isolat-ed scaffold attachment factor B1 (SAFB1/HET/HAP); this factor is capable of interacting with both of the above mentioned nuclear receptors and serves as a N-CoR-dependent co-repressor for ERα-dependent transcription [72, 73]. In addition to that, ciglitazone, a prototype TZD, and natural PPARγ ligand 15d-PGJ2 induce proteasome-dependent degradation of the ERα protein [33]. It is likely that binding of therapeu-tic TZDs to PPARγ mediate changes in the OR-medi-ated transcription similar to that of natural PPARγ lig-ands, thus mimicking the effect of oestrogen ablation.
The binding of TZDs to PPARγ suppresses the propagation of signals through the molecular path-ways converging on the transcriptional factor STAT5B [74]. Phosphorylated STAT5B is a critical survival fac-tor for normal, preneoplastic and malignant mammary epithelial cells [75]. In one study, STAT5B was found to be activated and relocated to the nucleus in 76% of breast carcinomas [76]. STAT5B becomes activated in response to epidermal growth factor (EGF) and augments expression of genes taking part in the EGF-induced DNA-synthesis [77]. Hypoxia also stimulates STAT5B-dependent transcription in mammary epithelial cells, particularly, the transcription of cyclin D1 encoding gene [78].As PPARγ agonists sup-press STATB5 activity, they might alleviate effects of EGF and hypoxia on exposed breast epitheliums.
A number of studies demonstrated the ability of PPARγ to suppress NF-αB-dependent transcription. Nuclear staining to NF-αB is a predominant finding in the OR-negative, but not in OR-positive, breast tumours [79]. Activation of NF-αB is linked to resist-ance to neoadjuvant chemotherapy [80]. TZDs induce a transient phosphorylation of PPARγ through the MAP kinase pathway, increasing the physical interaction of PPARγ with p65 and therefore decreasing NF-αB transcriptional activity [81]. In addition, rosiglitazone prompts SUMOylation of the ligand binding domain of the PPARγ[82]. This modification targets PPARγ to NCoR/histone deacetylase-3 (HDAC3) corepressor complexes located on the pro-moters of the quiescent genes that might be stimulat-ed by NF-κB in the absence of PPARγ. Successful initiation of the transcription from these promoters requires the dismissal of the NCoR/HDAC3 complex-es through their Ubc5-dependent ubiquitylation and proteosomal degradation. The recruitment of the Ubc5/19S proteosome machinery to the NCoR/HDAC3 bound promoters is prevented in the presence of the SUMOylated PPARγ. As a result, NCoR/HDAC3 complexes are not cleared from the promoter and NF-κB target genes are maintained in a repressed state [82].
PPARγ activation may interfere with TGFβ signalling
In addition to PPARγ interplays with oestrogen, EGF, NF-κB and other signalling pathways mentioned above, we would like to focus our attention on its crosstalks with transforming growth factor beta (TGFβ) signalling, an indispensable part of the molecular portrait of breast carcinoma. In the normal mammary gland, TGFβ regulates many steps of its development including branching morphogenesis, functional differentiation, cell-lineage decisions and involution [83]. It is generally accepted that TGFβ serves both as a tumour suppressor and as a tumour promoter in different tumour developmental stages and cellular contexts [84, 85]. During the initial phase of breast tumourigenesis, the TGFβ signal inhibits primary tumour development and growth by con-straining cell division and possibly inducing apopto-sis [85, 86]. Eventually, breast carcinoma cells cease to respond to TGFβ due to epigenetic silencing of its type I (RI) and type II (RII) receptors [87] or to aber-rations in downstream SMAD signalling [88]. Both events usually cause the switch of TGFβ's role from tumour suppressor to tumour promotor; the TGFβ overproduced by tumour cells retains its ability to act on tumour stroma [89, 90] as well as on the various cellular components of the immune system [91, 92]. These effects of TGFβ promote the metastatic process by inhibiting host immune surveillance and simultaneously stimulating invasion and angiogenesis.
Accumulating evidence suggests that the activation of the PPARγ interferes with the propagation of TGFβ signalling and decreases the expression of several genes controlled by this cytokine (Fig. 2). For example, pioglitazone attenuates the TGFβ driven induction of alternatively spliced mRNA and the Extra Domain A (EDA)+ protein isoform of fibronectin that are usually seen in fetal cells, tumour cells, and during wound healing, but not in normal adult cells [93]. Fibronectin molecules with EDA domains are significantly more potent in promoting local cell spread than ‘classical’ fibronectin [94]. EDA-contain-ing fibronectin isoforms are present in up to 47% of breast adenocarcinoma cells and in up to 69% of adjacent stromal cells, but never found in fibroadeno-mas or other benign breast conditions [95]. It is pos-sible that pioglitazone counteracts EDA+ fibronectindependent invasion of breast carcinoma through the suppression of TGFβ-mediated signalling.
2.
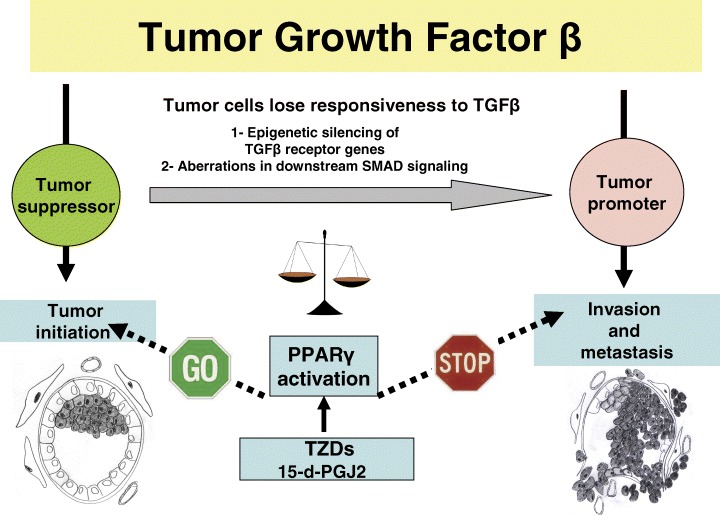
During the initial phase of breast tumourigenesis, the TGFβ signal inhibits primary tumour development. Eventually, breast carcinoma cells cease to respond to TGFβ due to epigenetic silencing of its receptors or to aberrations in downstream SMAD sig-nalling, causing the switch of TGFβ's role from tumour suppressor to tumour promoter. A crosstalk of PPAR signalling with TGFβ pathway most likely interferes with both functions of this molecule. Most likely, the outcome of the PPAR/TGF crosstalk is defined by the net effects of the TZD-related shifts in the balance of the pro-tumourigenic and tumour suppressor molecules that belong to a number of pathways affected by PPAR.
Both natural and synthetic PPARγ agonists sup-press the conversion of fibroblasts into myofibroblasts followed by myofibroblast proliferation, the basis for desmoplastic response to invading breast carcino-mas [96]. The myofibroblastic reaction is mediated by TGFβ serving as a master switch for the general fibrotic program, at least in the context of the tissue fibrosis and in some model tumours [97–99]. Myofibroblastic population is the major stromal source of extracellular matrix proteins, especially col-lagen, as well as of profibrogenic cytokines and chemokines. The production of extracellular mole-cules and the accumulation of fibroblasts and myofi-broblasts result in significant damage to the tissue architecture. PPARγ ligands inhibit both TGFβ-driven myofibroblast differentiation and type I collagen protein production by those cells without affecting their viability [96].
PPARγ activators can counteract angiogenic sig-nals generated by connective tissue growth factor (CTGF) stimulated by TGFβ at both the transcription-al and post-transcriptional levels. Model studies per-formed on hepatic stellate cells revealed that TGFβ-dependent production of CTGF could be suppressed by PPARγ natural ligand 15-d-PGJ2 and by its syn-thetic agonist GW7845 [100]. In one study, overex-pression of CTGF was registered in 55% of the pri-mary breast tumours, where it has been associated with advanced stage of the disease, tumour size, and lymph node status [101]. Whereas the data on CTGF distribution in the breast carcinomas remain contradictory [101, 102], there is no doubt that CTGF pro-motes the proliferation and differentiation of the vas-cular endothelial cells in culture [103, 104]. Furthermore, CTGF also increases the expression of a number of metalloproteinases that play a role in vascular invasive processes and decrease the expression of tissue inhibitors of metalloproteinases (TIMPs) by vascular endothelial cells [105]. A recent study identified CTGF as a central player in the process of breast carcinoma osteolytic metastasis [106]. The treatment of mice injected with MDA231 breast carcinoma cells with CTGF-neutralizing anti-body resulted in a greatly decreased number of oste-olytic bone metastasis as well as in the suppression of microvasculature and osteoclastogenesis. The neutralization of CTGF also inhibits the growth of subcutaneous tumours in vivo and the proliferation and migration of human umbilical vein endothelial cells (HUVECs) in vitro[106]. It is worthy to note that expression level of CYR61, also a member of the CTGF family of growth regulators that promotes aggressive behaviour of breast carcinomas (reviewed in [107]), is also controlled by TGFβ[108]. The possibility that CYR61 production by breast car-cinoma cells may be controlled by PPARγ ligands should be further studied.
PPARγ ligands are also capable of direct suppres-sion of TGFβ production by inhibiting the expression of the TGFβ1 gene. It is interesting that both troglita-zone and rosiglitazone treatments reduce TGFβ1 expression in response to high concentration of glu-cose, but do not change phorbol ester- and hydrogen peroxide-dependent TGFβ1 expression. According to the hypothesis proposed in two recent works [109, 110], one conceivable mechanism of TZD action is the TZD-dependent induction of diacylglycerol (DAG) kinase, which converts DAG to phosphatidic acid (thereby decreasing DAG level), and interferes with protein kinase C (PKC) activation [110]. Other studies indicate that PPARγ ligation may also block phorbol ester stimulated events, for example c-Jun-dependent AP-1 activation of the COX2 promoter in human epithelial cells [111], or ERK1/2 phosphorylation in vascular smooth muscle cells [112]. A recent study performed in mouse fibroblasts demonstrated that activation of PPARγ represses the TGFβ1 gene through dephosphorylation of transcription factor Zf9 (KLF6) [113], which also regulates expression of TGF receptors and collagen α (I). It is probable that active PPARγ exerts its action on Zf9 via induction of tumour suppressor gene PTEN that inhibits p70 ribo-somal S6 Kinase-1 (S6K1) [113]. It is tempting to speculate that in addition to the suppression of the TGFβ1 promoter, inhibition of S6K1 may decrease the signal propagated by mammalian target of rapamycin (mTOR) kinase aberrantly activated in many types of human cancer, including breast carci-noma [114].
TGFβ suppressing action of activated PPARγ could be counteracted by the antagonistic action of TGFβ exerted on PPARγ signalling. Particularly, TGF signals decrease the expression of both C/EBPα and C/EBPβ, which are important regula-tors of PPARγ[115].TGF also increases the level of PPARγ phosphorylation [115] and, therefore, inhibits its function as a transcription regulator. In addition to that, TGFβ suppresses activity of the PPARγ promot-er through its SMAD-binding elements [116]. It is like-ly that the prevalence of the TGFβ or PPARγ signal in a given cell is a result of the tightly balanced molecular control. One possible candidate for the role of the molecular decision controller is Cited2 (CBP/p300-interacting transactivators with glutamic acid (E)/aspartic acid (D)-rich C-terminal domain 2), which is a CBP(p300-binding transcription co-activa-tor without a typical DNA-binding domain. It has been demonstrated recently that Cited2 functions as a transcriptional co-activator for both TGFα[117] and for PPARα/PPARγ[118]. It might be possible that both TGFβ-dependent SMADs and PPARγ compete for the same pool of Cited2 molecules necessary for efficient activation of the target gene transcription by both factors.
The concept of the competitive PPARγ/TGFβ crosstalk became more complex by observations made by VanBuskirk and coauthors [119]. It has been shown that inhibitory effects of TGFβ in acces-sory cells crucial for the re-stimulation of memory cytotoxic T lymphocytes are mediated by the aug-mented expression of PPARγ[119]. Treatment of accessory cells with ciglitazone was found to mimic the above-mentioned effects of TGFβ[119]. In mono-cytes, TGFβ strongly induces PPARγ2 mRNA and protein expression, with a lesser effect on PPARγ1 [120]. That means that TGFβ and PPARγ signalling cascades do not interfere with each other, but rather cooperate, at least in some cellular systems. Other studies indicate that TZDs, particularly pioglitazone, stimulate PPARγ release of TGFβ into the extracellu-lar space and increase the nuclear recruitment of phospho-Smad2 [121]. Outcome of the TGFβ and PPARγ crosstalk may also depend on the nature of the PPARγ activation. For example, 15dPG-J2 inhibits translocation of Smad2 to the nucleus in CHO cells, while rosiglitazone enhances this process [122].
It is important to note that all observations of the crosstalk between TGFβ and PPARγ pathways were made in cultured cells. In our opinion, the outcome of this crosstalk needs to be verified in the in vivo carci-noma models to demonstrate which of the TGFβ effects prevail in the complex signalling milieu of the breast tissue when exposed to increased amounts of PPAR ligands.
Effects of TZDs in rodent models
The first study of the anticarcinogenic effects of TZDs in model animals was performed in 1998 [48], when immunodeficient mice were injected with breast car-cinoma cells MCF-7 and simultaneously treated with troglitazone. TZD treatment alone was shown to sup-press both the tumour volume and weight; observed effects were slightly more pronounced when troglita-zone was used in combination with ATRA [48]. More recent model experiments involved spontaneous development of mammary tumours either in immuno-competent animals treated with various carcinogens or in animals with genetically modified PPARγ sig-nalling. For example, in animals treated with 7,12-dimethylbenz[a]anthracene (DMBA), the experimen-tal decrease in PPARγ signalling (caused by knock-ing out one of the copies of the gene PPARγG) result-ed in a threefold increase in the incidence of mam-mary adenocarcinomas (P < 0.05), an over threefold increase in ovarian granulosa cell carcinomas (P <0.05), an over threefold increase in malignant tumours (P <0.02) and a 4.6-fold increase in metastatic incidence [123]. Yin and co-authors treated FVB/N mice with medroxyprogesterone acetate followed by DMBA administration, then maintained them on either a control diet or a diet containing novel PPARγ agonist GW7845. The latter regimen conferred a chemoprotective effect as demonstrated by the 2-month delay in tumour formation for all ani-mals [124]. These data are in concordance with the results of an earlier study that showed the ability of troglitazone, alone or in combination with RXR lig-ands, to prevent the induction of preneoplastic lesions by DMBA in a mouse mammary gland organ culture model [125]. In the classic rat model of mam-mary tumourigenesis that employs nitrosomethylurea as a carcinogen, GW7845 also significantly reduced both tumour incidence and tumour weight [126].
On the other hand, in the attempt of rosiglitazone chemoprevention of breast carcinogenesis in the MMTV-HER-2/neu transgenic mouse model, no encouraging data were produced [127]. In another study, mice that express a constitutively active form of PPARγ in the mammary gland were bred to trans-genic MMTV-PyV strain prone to mammary cancer development. The development of tumours was greatly accelerated in the PPARγ/MMTV-PyV double transgenic animals, probably due to an increase in Wnt signalling [128]. One explanation for this phe-nomenon is that ligand-activated and Vp16-activated PPARγ may act in dissimilar ways. For example, PPARγ constitutively activated by fusion with herpes simplex virus Vp16 protein may fail to engage coacti-vator protein complexes and/or to displace NCoR/HDAC3 complexes in the same way as ligand-activated PPARγ. Other possible explanations include the lack of the TZD-induced SUMOylation of PPARγ in the Vp16 protein activated PPARγ mutant and the importance of the non-PPARγ mediated effects of TZDs.
Results from experiments with PPARγ chemopre-vention in other tumour models were even more contradictory. For example, in the ApcMin mouse model of colon carcinogenesis, troglitazone mediates an increase in the number and size of colonic tumours [129, 130] as opposed to the results obtained in simi-lar experiments with rats. In the rat model, troglitazone alleviated azoxymethane induction of the aberrant cript foci (ACF) through stimulation of apoptosis in the colonic mucosa [131–133]. One explanation for this paradox is that the effects of PPARγ on pre-cancerous colonic mucosa depend on the state of APC protein [134]. In cells with intact APC, activated PPARγ sup-presses -catenin levels and colon carcinogenesis. In cells with mutated APC, the levels of -catenin are constitutively elevated; so activated PPARγ can no longer serve as a colonic tumour suppressor [134]. In frame of the described model, the enhanced tumouri-genesis in APCmin mice treated with PPARγ ligands observed by some authors [129, 130] could be explained by secondary, non–catenin mediated effects of PPARγ activation or by non-PPARγ effects of troglitazone itself. The hypothesis of APC-depend-ent action of PPARγ activators is supported by recent findings that demonstrated the formation of PPARγ complexes with -catenin and Tcf-4 [135].
Another piece of information that increases the complexity of the relationship between colon carcino-genesis and PPARγ activation came from studies by Niho and co-authors. These authors demonstrated that pioglitazone inhibits formation of intestinal and colonic polyps both in Apc1309 [136] and APCmin [137] mice. Later, the same group of authors showed that polyp suppressive effects of pioglitazone in Apc-deficient mice are not dependent on its PPARγ ago-nistic activities, but rather rely on the non-PPARγ relat-ed boost of lipoprotein lipase activity [138]. The above-mentioned experiments clearly demonstrate that TZD effects on the tumours may be compound specific. Therefore, every potential anticancerous TZD drug needs to be studied individually, both in the chemopre-ventive and therapeutic settings. Precocious general-ization of negative findings should be avoided.
Current and future prospects in the studies of the anticancerous effects of TZDs in human beings
The data describing the influence of the TZDs on the outcome of the pathologies of the human breast are scarce. To date, most encouraging observations were made in the recently completed PROACTIVE Study (PROspective pioglitAzone Clinical Trial In macroVascular Events) [139]. Longitudinal observa-tions of the 5238 diabetic patients treated with piogli-tazone or with placebo revealed, among other find-ings, a non-significant trend towards reduction of breast carcinoma incidence in the pioglitazone-treat-ed group (3 versus 11 cases in equally sized piogli-tazone and placebo arms of the study, respectively).
The only Phase II clinical trial of therapeutical use of the TZDs in patients with breast carcinomas was per-formed on the cohort of patients with advanced breast cancer refractory to at least one chemotherapy regi-men. Daily oral troglitazone treatments (800 mg) of 22 patients were performed for 5 months before troglita-zone withdrawal from the market. No objective responses were observed [140]. Phase I/II trial of pioglitazone in combination with tretinoin is currently underway at the Humboldt University, Germany [10], with no results of the trial made public yet. A number of case-by-case attempts of the off-label use of TZDs for the treatment of other human malignancies have been made. Some beneficial effects of TZDs were revealed in patients with resistant angiosarcomas/hemangioen-dotheliomas [141], Kaposi's sarcoma [142], metastatic melanomas and soft tissue sarcomas [143]. Further research in this direction is undeniably warranted, as mentioned results were collected in non-controlled studies. A number of controlled, randomized trials for TZD therapy of the non-breast human tumours are ongoing (http://www.ClinicalTrials.gov).
In our opinion, the focus of the studies of TZDs in human breast carcinomas should be shifted from the ‘classical’ Phase II trials seeking therapeutic results to longitudinal epidemiological studies that may reveal long-term effects of exposure to TZDs. Such effects may include chemoprevention of the DCIS, LCIS, and various benign lesions of the breast, reduction in the rates of malignization for existing benign lesions, changes in the length of the non-invasive dormancy in breast tumours and modifica-tions of its metastatic behaviour. Also, it is likely that chronic exposure to TZDs may change routes of the tumour progression; such a change might be revealed by comparative studies of the molecular portraits of breast carcinomas developed in patients treated with TZDs or in the general population.
Future longitudinal studies of breast carcinoma development in chronic TZD users need to include the collection of both epidemiological data and tissue samples to allow the profiling of the molecular path-ways. A collaborative INOVA-GMU study of this design is currently underway.
Acknowledgments
This work was supported by Susan G. Komen grant # BCTR0600525, ‘Molecular Network Profiling of DCIS for Patient Stratification and Individualized Therapy’. A.B. was partially covered by NIH R15CA113331-01. The authors are grateful to all INOVA Health System clinicians and staff mem-bers participating in long-term collaboration with GMU. A.B. is grateful to Irene Fanous for excellent help in manuscript proofreading.
References
- 1.Jemal A, Murray T, Ward E, Samuels A, Tiwari R, Ghafoor A, Feuer E, Thun M. Cancer statistics. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Van Laere SJ, Van den Eynden GG, Van der Auwera I, Vandenberghe M, Van Dam P, Van Marck EA, Van Golen KL, Vermeulen PB, Dirix LY. Identification of cell-of-origin breast tumor subtypes in inflammatory breast cancer by gene expression profiling. Breast Cancer Res Treat. 2006;95:243–55. doi: 10.1007/s10549-005-9015-9. [DOI] [PubMed] [Google Scholar]
- 3.Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA. 2003;100:10393–8. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lattimore BS, Crabbe MJ. Expression profiles in the progression of ductal carcinoma in the breast. Comput Biol Chem. 2003;27:115–20. doi: 10.1016/s1476-9271(03)00023-9. [DOI] [PubMed] [Google Scholar]
- 5.Lakhani SR, Audretsch W, Cleton-Jensen AM, Cutuli B, Ellis I, Eusebi V, Greco M, Houslton RS, Kuhl CK, Kurtz J, Palacios J, Peterse H, Rochard F, Rutgers E. The management of lobular carcinoma in situ (LCIS). Is LCIS the same as ductal carcinoma in situ (DCIS)? Eur J Cancer. 2006;42:2205–11. doi: 10.1016/j.ejca.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 6.Erbas B, Provenzano E, Armes J, Gertig D. The nat-ural history of ductal carcinoma in situ of the breast: a review. Breast Cancer Res Treat. 2006;97:135–44. doi: 10.1007/s10549-005-9101-z. [DOI] [PubMed] [Google Scholar]
- 7.Collins LC, Tamimi RM, Baer HJ, Connolly JL, Colditz GA, Schnitt SJ. Outcome of patients with duc-tal carcinoma in situ untreated after diagnostic biopsy: results from the Nurses' Health Study. Cancer. 2005;103:1778–84. doi: 10.1002/cncr.20979. [DOI] [PubMed] [Google Scholar]
- 8.Weaver DL, Rosenberg RD, Barlow WE, Ichikawa L, Carney PA, Kerlikowske K, Buist DS, Geller BM, Key CR, Maygarden SJ, Ballard-Barbash R. Pathologic findings from the Breast Cancer Surveillance Consortium: population-based outcomes in women undergoing biopsy after screening mammography. Cancer. 2006;106:732–42. doi: 10.1002/cncr.21652. [DOI] [PubMed] [Google Scholar]
- 9.Durbin RJ. Thiazolidinedione therapy in the prevention/delay of type 2 diabetes in patients with impaired glucose tolerance and insulin resistance. Diabetes Obes Metab. 2004;6:280–5. doi: 10.1111/j.1462-8902.2004.0348.x. [DOI] [PubMed] [Google Scholar]
- 10.Fenner MH, Elstner E. Peroxisome proliferator-acti-vated receptor-gamma ligands for the treatment of breast cancer. Expert Opin Investig Drugs. 2005;14:557–68. doi: 10.1517/13543784.14.6.557. [DOI] [PubMed] [Google Scholar]
- 11.Lemberger T. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor sig-naling pathway in lipid physiology. Ann Rev Cell Dev Biol. 1996;12:335–63. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- 12.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 13.Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, Hauser S, Rosen ED, Ge K, Roeder RG, Spiegelman BM. Genetic analysis of adipogenesis through peroxi-some proliferator-activated receptor-gamma isoforms. J Biol Chem. 2002;277(44):41925–930. doi: 10.1074/jbc.M206950200. [DOI] [PubMed] [Google Scholar]
- 14.Koeffler HP. Peroxisome proliferator-activated recep-tor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- 15.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–50. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee R, Davies PJA, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamann LG, Boehm CE, Nadzan AM, Paterniti JR, Heyman RA. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature. 1997;386:407–10. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 17.Oberkofler H, Esterbauer H, Linnemayr V, Strosberg AD, Krempler F, Patsch W. Peroxisome proliferator-activated receptor (PPAR) gamma coacti-vator-1 recruitment regulates PPAR subtype specifici-ty. J Biol Chem. 2002;277:16750–7. doi: 10.1074/jbc.M200475200. [DOI] [PubMed] [Google Scholar]
- 18.Leader JE, Wang C, Fu M, Pestell RG. Epigenetic regulation of nuclear steroid receptors. Biochem Pharmacol. 2006;72:1589–1596. doi: 10.1016/j.bcp.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 19.Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor and metabolic dis-ease. Annu Rev Biochem. 2001;70:341–67. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- 20.DiRenzo J, Soderstrom M, Kurokawa R, Ogliastro MH, Ricote M, Ingrey S, Horlein A, Rosenfeld MG, Glass CK. Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol Cell Biol. 1997;17:2166–76. doi: 10.1128/mcb.17.4.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem. 2005;280:13600–5. doi: 10.1074/jbc.M409468200. [DOI] [PubMed] [Google Scholar]
- 22.Lemay DG, Hwang DH. Genome-wide identification of peroxisome proliferator response elements using inte-grated computational genomics. J Lipid Res. 2006;47:1583–7. doi: 10.1194/jlr.M500504-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Hong J, Samudio I, Liu S, Abdelrahim M, Safe S. Peroxisome proliferator-activated receptor gamma-dependent activation of p21 in Panc-28 pancreatic cancer cells involves Sp1 and Sp4 proteins. Endocrinology. 2004;145:5774–85. doi: 10.1210/en.2004-0686. [DOI] [PubMed] [Google Scholar]
- 24.Sugawara A, Uruno A, Kudo M, Ikeda Y, Sato K, Taniyama Y, Ito S, Takeuchi K. Transcription suppres-sion of thromboxane receptor gene by peroxisome proliferator-activated receptor-gamma via an interac-tion with Sp1 in vascular smooth muscle cells. J Biol Chem. 2002;277:9676–83. doi: 10.1074/jbc.M104560200. [DOI] [PubMed] [Google Scholar]
- 25.Fajas L, Auboeuf D. Raspe E, Schoonjans K, Lefebvre AM, Saladin R, Najib J, Laville M, Fruehart JC, Deeb S, Vidal-Puig A, Flier J, Briggs MR, Staels B, Vidal H, Auwerx J. The organization, promoter analysis, and expression of the human PPAR gene. J Biol Chem. 1997;272:18779–89. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- 26.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–9. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 27.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determi-nation factor PPAR gamma. Cell. 1995;83:803–12. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 28.Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, Lehmann JM. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci USA. 1997;94:4318–23. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies SS, Pontsler AV, Marathe GK, Harrison KA, Murphy RC, Hinshaw JC, Prestwich GD, Hilaire AS, Prescott SM, Zimmerman GA, McIntyre TM. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor gamma lig-ands and agonists. J Biol Chem. 2001;276:16015–23. doi: 10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- 30.Shibuya A, Watanabe M, Fujita Y, Saigenji K, Kuwao S, Takahashi H, Takeuchi H. An autopsy case of troglitazone-induced fulminant hepatitis. Diabetes Care. 1998;21:2140–3. doi: 10.2337/diacare.21.12.2140. [DOI] [PubMed] [Google Scholar]
- 31.Usui S, Suzuki T, Hattori Y, Etoh K, Fujieda H, Nishizuka M, Imagawa M, Nakagawa H, Kohda K, Miyata N. Design, synthesis, and biological activity of novel PPARgamma ligands based on rosiglitazone and 15d-PGJ2. Bioorg Med Chem Lett. 2005;15:1547–51. doi: 10.1016/j.bmcl.2005.01.074. [DOI] [PubMed] [Google Scholar]
- 32.Huang JW, Shiau CW, Yang YT, Kulp SK, Chen KF, Brueggemeier RW, Shapiro CL, Chen CS. Peroxisome proliferator-activated receptor gamma-independent ablation of cyclin D1 by thiazolidine-diones and their derivatives in breast cancer cells. Mol Pharmacol. 2005;67:1342–8. doi: 10.1124/mol.104.007732. [DOI] [PubMed] [Google Scholar]
- 33.Qin C, Burghardt R, Smith R, Wormke M, Stewart J, Safe S. Peroxisome proliferator-activated receptor gamma agonists induce proteasome-dependent degradation of cyclin D1 and estrogen receptor alpha in MCF-7 breast cancer cells. Cancer Res. 2003;63:958–64. [PubMed] [Google Scholar]
- 34.Lu M, Kwan T, Yu C, Chen F, Freedman B, Schafer JM, Lee EJ, Jameson JL, Jordan VC, Cryns VL. Peroxisome proliferator-activated receptor gamma agonists promote TRAIL-induced apoptosis by reduc-ing survivin levels via cyclin D3 repression and cell cycle arrest. J Biol Chem. 2005;280:6742–51. doi: 10.1074/jbc.M411519200. [DOI] [PubMed] [Google Scholar]
- 35.Turturro F, Friday E, Fowler R, Surie D, Welbourne T. Troglitazone acts on cellular pH and DNA synthesis through a peroxisome proliferator-activated receptor gamma-independent mechanism in breast cancer-derived cell lines. Clin Cancer Res. 2004;10:7022–30. doi: 10.1158/1078-0432.CCR-04-0879. [DOI] [PubMed] [Google Scholar]
- 36.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidine-diones are independent of peroxisome proliferator-acti-vated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61:6213–18. [PubMed] [Google Scholar]
- 37.Inoue I, Katayama S, Takahashi K, Negishi K, Miyazaki T, Sonoda M, Komoda T. Troglitazone has a scavenging effect on reactive oxygen species. Biochem Biophys Res Commun. 1997;235:113–6. doi: 10.1006/bbrc.1997.6512. [DOI] [PubMed] [Google Scholar]
- 38.Weng JR, Chen CY, Pinzone JJ, Ringel MD, Chen CS. Beyond peroxisome proliferator-activated receptor gamma signaling: the multi-facets of the antitumor effect of thiazolidinediones. Endocr Relat Cancer. 2006;13:401–13. doi: 10.1677/erc.1.01182. [DOI] [PubMed] [Google Scholar]
- 39.Panigrahy D, Huang S, Kieran MW, Kaipainen A. PPARgamma as a therapeutic target for tumor angio-genesis and metastasis. Cancer Biol Ther. 2005;4:687–93. doi: 10.4161/cbt.4.7.2014. [DOI] [PubMed] [Google Scholar]
- 40.Yee LD, Guo Y, Bradbury J, Suster S, Clinton SK, Seewaldt VL. The antiproliferative effects of PPARgamma ligands in normal human mammary epithelial cells. Breast Cancer Res Treat. 2003;78:179–92. doi: 10.1023/a:1022978608125. [DOI] [PubMed] [Google Scholar]
- 41.Papadaki I, Mylona E, Giannopoulou I, Markaki S, Keramopoulos A, Nakopoulou L. PPARgamma expression in breast cancer: clinical value and correla-tion with ERbeta. Histopathology. 2005;46:37–42. doi: 10.1111/j.1365-2559.2005.02056.x. [DOI] [PubMed] [Google Scholar]
- 42.Ikezoe T, Miller CW, Kawano S, Heaney A, Williamson EA, Hisatake J, Green E, Hofmann W, Taguchi H, Koeffler HP. Mutational analysis of the peroxisome proliferator-activated receptor gamma gene in human malignancies. Cancer Res. 2001;61:5307–10. [PubMed] [Google Scholar]
- 43.Badawi AF, Badr MZ. Expression of cyclooxygenase-2 and peroxisome proliferator-activated receptor-gamma and levels of prostaglandin E2 and 15-deoxy-delta12,14-prostaglandin J2 in human breast cancer and metastasis. Int J Cancer. 2003;103:84–90. doi: 10.1002/ijc.10770. [DOI] [PubMed] [Google Scholar]
- 44.Watkins G, Douglas-Jones A, Mansel RE, Jiang WG. The localisation and reduction of nuclear staining of PPARgamma and PGC-1 in human breast cancer. Oncol Rep. 2004;12:483–8. [PubMed] [Google Scholar]
- 45.Jiang WG, Douglas-Jones A, Mansel RE. Expression of peroxisome-proliferator activated recep-tor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int J Cancer. 2003;106:752–7. doi: 10.1002/ijc.11302. [DOI] [PubMed] [Google Scholar]
- 46.Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPAR. Mol Cell. 1998;1:465–70. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- 47.Yin F, Wakino S, Liu Z, Kim S, Hsueh WA, Collins AR, Van Herle AJ, Law RE. Troglitazone inhibits growth of MCF-7 breast carcinoma cells by targeting G1 cell cycle regulators. Biochem Biophys Res Commun. 2001;286:916–22. doi: 10.1006/bbrc.2001.5491. [DOI] [PubMed] [Google Scholar]
- 48.Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, Shintaku P, Said JW, Heber D, Koeftler HP. Ligands for peroxisome proliferator-acti-vated receptor and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cell in vitro and in BNX mice. Proc Natl Acad Sci USA. 1998;95:8806–11. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki T, Hayashi S, Miki Y, Nakamura Y, Moriya T, Sugawara A, Ishida T, Ohuchi N, Sasano H. Peroxisome proliferator-activated receptor gamma in human breast carcinoma: a modulator of estrogenic actions. Endocr Relat Cancer. 2006;13:233–50. doi: 10.1677/erc.1.01075. [DOI] [PubMed] [Google Scholar]
- 50.Bonofiglio D, Aquila S, Catalano S, Gabriele S, Belmonte M, Middea E, Qi H, Morelli C, Gentile M, Maggiolini M, Ando S. Peroxisome proliferator-acti-vated receptor (PPAR) gamma activates p53 gene pro-moter binding to the NF-κB sequence in human MCF7 breast cancer cells. Mol Endocrinol. 2006;20:3083–92. doi: 10.1210/me.2006-0192. [DOI] [PubMed] [Google Scholar]
- 51.Kim KY, Kim SS, Cheon HG. Differential anti-prolifer-ative actions of peroxisome proliferator-activated receptor-gamma agonists in MCF-7 breast cancer cells. Biochem Pharmacol. 2006;72:530–40. doi: 10.1016/j.bcp.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammato-ry actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001;11:764–8. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 53.Yin F, Bruemmer D, Blaschke F, Hsueh WA, Law RE, Herle AJ. Signaling pathways involved in induction of GADD45 gene expression and apoptosis by troglita-zone in human MCF-7 breast carcinoma cells. Oncogene. 2004;23:4614–23. doi: 10.1038/sj.onc.1207598. [DOI] [PubMed] [Google Scholar]
- 54.Harkin DP, Bean JM, Miklos D, Song YH, Truong VB, Englert C, Christians FC, Ellisen LW, Maheswaran S, Oliner JD, Haber DA. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell. 1999;97:575–86. doi: 10.1016/s0092-8674(00)80769-2. [DOI] [PubMed] [Google Scholar]
- 55.Pignatelli M, Cocca C, Santos A, Perez-Castillo A. Enhancement of BRCA1 gene expression by the perox-isome proliferator-activated receptor gamma in the MCF-7 breast cancer cell line. Oncogene. 2003;22:5446–50. doi: 10.1038/sj.onc.1206824. [DOI] [PubMed] [Google Scholar]
- 56.Pandhare J, Cooper SK, Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evi-dence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mecha-nisms. J Biol Chem. 2006;281:2044–52. doi: 10.1074/jbc.M507867200. [DOI] [PubMed] [Google Scholar]
- 57.Herbert BS, Pearce VP, Hynan LS, LaRue DM, Wright WE, Kopelovich L, Shay JW. A peroxisome proliferator-activated receptor-gamma agonist and the p53 rescue drug CP-31398 inhibit the spontaneous immortalization of breast epithelial cells. Cancer Res. 2003;63:1914–9. [PubMed] [Google Scholar]
- 58.Lea MA, Sura M, Desbordes C. Inhibition of cell pro-liferation by potential peroxisome proliferator-activated receptor (PPAR) gamma agonists and antagonists. Anticancer Res. 2004;24:2765–71. [PubMed] [Google Scholar]
- 59.Kim KY, Cheon HG. Antiangiogenic effect of rosiglita-zone is mediated via peroxisome proliferator-activated receptor gamma-activated maxi-K channel opening in human umbilical vein endothelial cells. J Biol Chem. 2006;281:13503–12. doi: 10.1074/jbc.M510357200. [DOI] [PubMed] [Google Scholar]
- 60.Micke P, Ostman A. Tumour-stroma interaction: can-cer-associated fibroblasts as novel targets in anti-can-cer therapy? Lung Cancer. 2004;45:S163–75. doi: 10.1016/j.lungcan.2004.07.977. [DOI] [PubMed] [Google Scholar]
- 61.Meng L, Zhou J, Sasano H, Suzuki T, Zeitoun KM, Bulun SE. Tumor necrosis factor alpha and interleukin 11 secreted by malignant breast epithelial cells inhibit adipocyte differentiation by selectively down-regulating CCAAT/enhancer binding protein alpha and peroxi-some proliferator-activated receptor gamma: mecha-nism of desmoplastic reaction. Cancer Res. 2001;61:2250–5. [PubMed] [Google Scholar]
- 62.Liu H, Zang C, Fenner MH, Possinger K, Elstner E. PPARgamma ligands and ATRA inhibit the invasion of human breast cancer cells in vitro. Breast Cancer Res Treat. 2003;79:63–74. doi: 10.1023/a:1023366117157. [DOI] [PubMed] [Google Scholar]
- 63.Peeters LL, Vigne JL, Tee MK, Zhao D, Waite LL, Taylor RN. PPARgamma represses VEGF expression in human endometrial cells: implications for uterine angiogenesis. Angiogenesis. 2005;8:373–9. doi: 10.1007/s10456-005-9027-4. [DOI] [PubMed] [Google Scholar]
- 64.Fu YG, Sung JJ, Wu KC, Bai AH, Chan MC, Yu J, Fan DM, Leung WK. Inhibition of gastric cancer cells associated angiogenesis by 15d-prostaglandin J(2) through the downregulation of angiopoietin-1. Cancer Lett. 2006;243:246–54. doi: 10.1016/j.canlet.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 65.Keshamouni VG, Arenberg DA, Reddy RC, Newstead MJ, Anthwal S, Standiford TJ. PPAR-gamma activation inhibits angiogenesis by blocking ELR+CXC chemokine production in non-small cell lung cancer. Neoplasia. 2005;7:294–301. doi: 10.1593/neo.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chu K, Lee ST, Koo JS, Jung KH, Kim EH, Sinn DI, Kim JM, Ko SY, Kim SJ, Song EC, Kim M, Roh JK. Peroxisome proliferator-activated receptor-gamma-agonist, rosiglitazone, promotes angiogenesis after focal cerebral ischemia. Brain Res. 2006;1093:208–18. doi: 10.1016/j.brainres.2006.03.114. [DOI] [PubMed] [Google Scholar]
- 67.Brzozowski T, Konturek PC, Pajdo R, Kwiecien SN, Konturek S, Targosz A, Burnat G, Cieszkowski J, Pawlik WW, Hahn EG. Agonist of peroxisome prolifera-tor-activated receptor gamma (PPAR-gamma): a new compound with potent gastroprotective and ulcer healing properties. Inflammopharmacology. 2005;13:317–30. doi: 10.1163/156856005774423908. [DOI] [PubMed] [Google Scholar]
- 68.Pistrosch F, Herbrig K, Oelschlaegel U, Richter S, Passauer J, Fischer S, Gross P. PPARgamma-ago-nist rosiglitazone increases number and migratory activity of cultured endothelial progenitor cells. Atherosclerosis. 2005;183:163–7. doi: 10.1016/j.atherosclerosis.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 69.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 70.Andrews WJ, Winnett G, Rehman F, Shanmugasundaram P, Hagen D, Schrey MP. Aromatase inhibition by 15-deoxy-prostaglandin J(2) (15-dPGJ(2)) and N-(4-hydroxyphenyl)-retinamide (4HPR) is associated with enhanced ceramide produc-tion. J Steroid Biochem Mol Biol. 2005;94:159–65. doi: 10.1016/j.jsbmb.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 71.Rubin GL, Zhao Y, Kalus AM, Simpson ER. Peroxisome proliferator-activated receptor gamma lig-ands inhibit estrogen biosynthesis in human breast adipose tissue: possible implications for breast cancer therapy. Cancer Res. 2000;60:1604–8. [PubMed] [Google Scholar]
- 72.Debril MB, Dubuquoy L, Feige JN, Wahli W, Desvergne B, Auwerx J, Gelman L. Scaffold attach-ment factor B1 directly interacts with nuclear receptors in living cells and represses transcriptional activity. J Mol Endocrinol. 2005;35:503–17. doi: 10.1677/jme.1.01856. [DOI] [PubMed] [Google Scholar]
- 73.Jiang S, Meyer R, Kang K, Osborne CK, Wong J, Oesterreich S. Scaffold attachment factor SAFB1 suppresses estrogen receptor alpha-mediated tran-scription in part via interaction with nuclear receptor corepressor. Mol Endocrinol. 2006;20:311–20. doi: 10.1210/me.2005-0100. [DOI] [PubMed] [Google Scholar]
- 74.Shipley JM, Waxman DJ. Simultaneous, bidirectional inhibitory crosstalk between PPAR and STAT5b. Toxicol Appl Pharmacol. 2004;199:275–84. doi: 10.1016/j.taap.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 75.Nevalainen MT, Xie J, Bubendorf L, Wagner KU, Rui H. Basal activation of transcription factor signal trans-ducer and activator of transcription (Stat5) in nonpreg-nant mouse and human breast epithelium. Mol Endocrinol. 2002;16:1108–24. doi: 10.1210/mend.16.5.0839. [DOI] [PubMed] [Google Scholar]
- 76.Cotarla I, Ren S, Zhang Y, Gehan E, Singh B, Furth PA. Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast can-cers. Int J Cancer. 2004;108:665–71. doi: 10.1002/ijc.11619. [DOI] [PubMed] [Google Scholar]
- 77.Kloth MT, Laughlin KK, Biscardi JS, Boerner JL, Parsons SJ, Silva CM. STAT5b, a mediator of syner-gism between c-Src and the epidermal growth factor receptor. J Biol Chem. 2003;278:1671–9. doi: 10.1074/jbc.M207289200. [DOI] [PubMed] [Google Scholar]
- 78.Joung YH, Lim EJ, Lee MY, Park JH, Ye SK, Park EU, Kim SY, Zhang Z, Lee KJ, Park DK, Park T, Moon WK, Yang YM. Hypoxia activates the cyclin D1 promot-er via the Jak2/STAT5b pathway in breast cancer cells. Exp Mol Med. 2005;37:353–64. doi: 10.1038/emm.2005.45. [DOI] [PubMed] [Google Scholar]
- 79.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci USA. 2004;101:10137–42. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Montagut C, Tusquets I, Ferrer B, Corominas JM, Bellosillo B, Campas C, Suarez M, Fabregat X, Campo E, Gascon P, Serrano S, Fernandez PL, Rovira A, Albanell J. Activation of nuclear factor-kappa B is linked to resistance to neoadjuvant chemotherapy in breast cancer patients. Endocr Relat Cancer. 2006;13:607–16. doi: 10.1677/erc.1.01171. [DOI] [PubMed] [Google Scholar]
- 81.Chen F, Wang M, O'Connor JP, He M, Tripathi T, Harrison LE. Phosphorylation of PPARgamma via active ERK1/2 leads to its physical association with p65 and inhibition of NF-kappabeta. J Cell Biochem. 2003;90:732–44. doi: 10.1002/jcb.10668. [DOI] [PubMed] [Google Scholar]
- 82.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway medi-ates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–63. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Serra R, Crowley MR. Mouse models of transforming growth factor beta impact in breast development and cancer. Endocr Relat Cancer. 2005;12:749–60. doi: 10.1677/erc.1.00936. [DOI] [PubMed] [Google Scholar]
- 84.Muraoka-Cook RS, Dumont N, Arteaga CL. Dual role of transforming growth factor beta in mammary tumorigenesis and metastatic progression. Clin Cancer Res. 2005;11:937s–43s. [PubMed] [Google Scholar]
- 85.Sun L. Tumor-suppressive and promoting function of transforming growth factor beta. Front Biosci. 2004;9:1925–35. doi: 10.2741/1382. [DOI] [PubMed] [Google Scholar]
- 86.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, Wakefield LM. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–24. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ammanamanchi S, Brattain MG. Restoration of transforming growth factor-beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J Biol Chem. 2004;279:32620–5. doi: 10.1074/jbc.M402691200. [DOI] [PubMed] [Google Scholar]
- 88.Xie W, Mertens JC, Reiss DJ, Rimm DL, Camp RL, Haffty BG, Reiss M. Alterations of Smad signaling in human breast carcinoma are associated with poor out-come: a tissue microarray study. Cancer Res. 2002;62:497–505. [PubMed] [Google Scholar]
- 89.Stuelten CH, DaCosta Byfield S, Arany PR, Karpova TS, Stetler-Stevenson WG, Roberts AB. Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-alpha and TGF-beta. J Cell Sci. 2005;118:2143–53. doi: 10.1242/jcs.02334. [DOI] [PubMed] [Google Scholar]
- 90.Vrana JA, Stang MT, Grande JP, Getz MJ. Expression of tissue factor in tumor stroma correlates with progression to invasive human breast cancer: paracrine regulation by carcinoma cell-derived mem-bers of the transforming growth factor beta family. Cancer Res. 1996;56:5063–70. [PubMed] [Google Scholar]
- 91.Ito M, Minamiya Y, Kawai H, Saito S, Saito H, Nakagawa T, Imai K, Hirokawa M, Ogawa J. Tumor-derived TGFbeta-1 induces dendritic cell apop-tosis in the sentinel lymph node. J Immunol. 2006;176:5637–43. doi: 10.4049/jimmunol.176.9.5637. [DOI] [PubMed] [Google Scholar]
- 92.Arteaga CL, Hurd SD, Winnier AR, Johnson MD, Fendly BM, Forbes JT. Anti-transforming growth fac-tor (TGF)-beta antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell(host TGF-beta interactions in human breast cancer progression. J Clin Invest. 1993;92:2569–76. doi: 10.1172/JCI116871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maeda A, Horikoshi S, Gohda T, Tsuge T, Maeda K, Tomino Y. Pioglitazone attenuates TGF-beta(1)-induc-tion of fibronectin synthesis and its splicing variant in human mesangial cells via activation of peroxisome proliferator-activated receptor (PPAR)gamma. Cell Biol Int. 2005;29:422–8. doi: 10.1016/j.cellbi.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 94.Manabe R, Ohe N, Maeda T, Fukuda T, Sekiguchi K. Modulation of cell-adhesive activity of fibronectin by the alternatively spliced EDA segment. J Cell Biol. 1997;139:295–307. doi: 10.1083/jcb.139.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matsumoto E, Yoshida T, Kawarada Y, Sakakura T. Expression of fibronectin isoforms in human breast tis-sue: production of extra domain A+/extra domain B+ by cancer cells and extra domain A+ by stromal cells. Jpn J Cancer Res. 1999;90:320–5. doi: 10.1111/j.1349-7006.1999.tb00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, Phipps RP, Sime PJ. PPARgamma agonists inhibit TGF-beta-induced pul-monary myofibroblast differentiation and collagen pro-duction: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1146–53. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- 97.Reisdorf P, Lawrence DA, Sivan V, Klising E, Martin MT. Alteration of transforming growth factor-beta1 response involves down-regulation of Smad3 signaling in myofibroblasts from skin fibrosis. Am J Pathol. 2001;159:263–72. doi: 10.1016/s0002-9440(10)61692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sam R, Wanna L, Gudehithlu KP, Garber SL, Dunea G, Arruda JA, Singh AK. Glomerular epithelial cells transform to myofibroblasts: early but not late removal of TGF-beta1 reverses transformation. Transl Res. 2006;148:142–8. doi: 10.1016/j.trsl.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 99.Romeo S, Eyden B, Prins FA, Briaire-de Bruijn IH, Taminiau AH, Hogendoorn PC. TGF-beta1 drives partial myofibroblastic differentiation in chondromyxoid fibroma of bone. J Pathol. 2006;208:26–34. doi: 10.1002/path.1887. [DOI] [PubMed] [Google Scholar]
- 100.Sun K, Wang Q, Huang XH. PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective tissue growth factor expression. Acta Pharmacol Sin. 2006;27:715–23. doi: 10.1111/j.1745-7254.2006.00299.x. [DOI] [PubMed] [Google Scholar]
- 101.Xie D, Nakachi K, Wang H, Elashoff R, Koeffler HP. Elevated levels of connective tissue growth factor, WISP-1, and CYR61 in primary breast cancers asso-ciated with more advanced features. Cancer Res. 2001;61:8917–23. [PubMed] [Google Scholar]
- 102.Jiang WG, Watkins G, Fodstad O, Douglas-Jones A, Mokbel K, Mansel RE. Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr Relat Cancer. 2004;11:781–91. doi: 10.1677/erc.1.00825. [DOI] [PubMed] [Google Scholar]
- 103.Shimo T, Nakanishi T, Kimura Y, Nishida T, Ishizeki K, Matsumura T, Takigawa M. Inhibition of endoge-nous expression of connective tissue growth factor by its antisense oligonucleotide and antisense RNA sup-presses proliferation and migration of vascular endothelial cells. Biochemistry. 1998;124:130–40. doi: 10.1093/oxfordjournals.jbchem.a022071. [DOI] [PubMed] [Google Scholar]
- 104.Shimo T, Kubota, Kondo S, Nakanishi T, Sasaki A, Mese H, Matsumura T, Takigawa M. Connective tis-sue growth factor as a major angiogenic agent that is induced by hypoxia in a human breast cancer cell line. Cancer Lett. 2001;174:57–64. doi: 10.1016/s0304-3835(01)00683-8. [DOI] [PubMed] [Google Scholar]
- 105.Kondo S, Kubota S, Shimo T, Nishida T, Yosimichi G, Eguchi T, Sugahara T, Takigawa M. Connective tis-sue growth factor increased by hypoxia may initiate angiogenesis in collaboration with matrix metallopro-teinases. Carcinogenesis. 2002;23:769–76. doi: 10.1093/carcin/23.5.769. [DOI] [PubMed] [Google Scholar]
- 106.Shimo T, Kubota S, Yoshioka N, Ibaragi S, Isowa S, Eguchi T, Sasaki A, Takigawa M. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in oste-olytic metastasis of breast cancer. J Bone Miner Res. 2006;21:1045–59. doi: 10.1359/jbmr.060416. [DOI] [PubMed] [Google Scholar]
- 107.Menendez JA, Mehmi I, Griggs DW, Lupu R. The angiogenic factor CYR61 in breast cancer: molecular pathology and therapeutic perspectives. Endocr Relat Cancer. 2003;10:141–52. doi: 10.1677/erc.0.0100141. [DOI] [PubMed] [Google Scholar]
- 108.Bartholin L, Wessner LL, Chirgwin JM, Guise TA. The human Cyr61 gene is a transcriptional target of transforming growth factor beta in cancer cells. Cancer Lett. 2006;246:230–6. doi: 10.1016/j.canlet.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 109.Isshiki K, Haneda D, Koya S, Maeda T, Sugimoto T, Kikkawa M. Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats. Diabetes. 2000;49:1022–32. doi: 10.2337/diabetes.49.6.1022. [DOI] [PubMed] [Google Scholar]
- 110.Weigert C, Brodbeck K, Bierhaus A, Haring HU, Schleicher ED. c-Fos-driven transcriptional activation of transforming growth factor beta-1: inhibition of high glucose-induced promoter activity by thiazolidinediones. Biochem Biophys Res Commun. 2003;304:301–7. doi: 10.1016/s0006-291x(03)00599-0. [DOI] [PubMed] [Google Scholar]
- 111.Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ. Peroxisome proliferator-activated receptor c ligands suppress the transcriptional activation of cyclooxyge-nase-2. Evidence for involvement of activator protein-1 and CREB-binding protein(p300. J Biol Chem. 2001;276:12440–8. doi: 10.1074/jbc.M007237200. [DOI] [PubMed] [Google Scholar]
- 112.Hattori Y, Akimoto K, Kasai K. The effects of thiazo-lidinediones on vascular smooth muscle cell activation by angiotensin II. Biochem Biophys Res Commun. 2000;273:1144–9. doi: 10.1006/bbrc.2000.3084. [DOI] [PubMed] [Google Scholar]
- 113.Lee SJ, Yang EK, Kim SG. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediat-ed p70 ribosomal S6 kinase-1 inhibition: role for Zf9 dephosphorylation. Mol Pharmacol. 2006;70:415–25. doi: 10.1124/mol.106.022954. [DOI] [PubMed] [Google Scholar]
- 114.Hynes NE, Boulay A. The mTOR pathway in breast cancer. J Mammary Gland Biol Neoplasia. 2006;11:53–61. doi: 10.1007/s10911-006-9012-6. [DOI] [PubMed] [Google Scholar]
- 115.Ahdjoudj S, Kaabeche K, Holy X, Fromigue O, Modrowski D, Zerath E, Marie PJ. Transforming growth factor-beta inhibits CCAAT/enhancer-binding protein expression and PPARgamma activity in unloaded bone marrow stromal cells. Exp Cell Res. 2005;303:138–47. doi: 10.1016/j.yexcr.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 116.Zheng S, Chen A. Disruption of transforming growth factor-beta signaling by curcumin induces gene expression of peroxisome proliferator-activated recep-tor-gamma in rat hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2006;292:G113–23. doi: 10.1152/ajpgi.00200.2006. [DOI] [PubMed] [Google Scholar]
- 117.Chou YT, Wang H, Chen Y, Danielpour D, Yang YC. Cited2 modulates TGF-beta-mediated upregulation of MMP9. Oncogene. 2006;25:5547–60. doi: 10.1038/sj.onc.1209552. [DOI] [PubMed] [Google Scholar]
- 118.Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated recep-tor alpha coregulator. Biol Chem. 2004;279:24053–63. doi: 10.1074/jbc.M401489200. [DOI] [PubMed] [Google Scholar]
- 119.VanBuskirk AM, Lesinski GB, Nye KJ, Carson WE, Yee LD. TGF-beta inhibition of CTL re-stimulation requires accessory cells and induces peroxisome-pro-liferator-activated receptor-gamma (PPAR-gamma) Am J Transplant. 2006;6:1809–19. doi: 10.1111/j.1600-6143.2006.01387.x. [DOI] [PubMed] [Google Scholar]
- 120.Kintscher U, Wakino S, Bruemmer D, Goetze S, Graf K, Hsueh WA, Law RE. TGF-beta(1) induces peroxisome proliferator-activated receptor gamma1 and gamma2 expression in human THP-1 monocytes. Biochem Biophys Res Commun. 2002;297:794–9. doi: 10.1016/s0006-291x(02)02264-7. [DOI] [PubMed] [Google Scholar]
- 121.Redondo S, Ruiz E, Santos-Gallego CG, Padilla E, Tejerina T. Pioglitazone induces vascular smooth muscle cell apoptosis through a peroxisome prolifera-tor-activated receptor-gamma, transforming growth factor-beta1, and a Smad2-dependent mechanism. Diabetes. 2005;54:811–7. doi: 10.2337/diabetes.54.3.811. [DOI] [PubMed] [Google Scholar]
- 122.Ye F, Sun T, Luo H, Ding H, Chen K, Shen X, Jiang H. Quantitative characterization of 15-deoxy-delta(12,14)-prostaglandin J2 in regulating EGFPSmad2 translocation in CHO cells through PPARgamma(TGFbeta(Smad2 pathway. Cell Physiol Biochem. 2006;18:143–50. doi: 10.1159/000095183. [DOI] [PubMed] [Google Scholar]
- 123.Nicol CJ, Yoon M, Ward JM, Yamashita M, Fukamachi K, Peters JM, Gonzalez FJ. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–55. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- 124.Yin Y, Russell RG, Dettin LE, Bai R, Wei ZL, Kozikowski AP, Kopelovich L, Glazer RI. Peroxisome proliferator-activated receptor delta and gamma agonists differentially alter tumor differentia-tion and progression during mammary carcinogenesis. Cancer Res. 2005;65:3950–7. doi: 10.1158/0008-5472.CAN-04-3990. [DOI] [PubMed] [Google Scholar]
- 125.Mehta RG, Williamson E, Patel MK, Koeffler HP. A ligand of peroxisome proliferator-activated receptor gamma, retinoids, and prevention of preneoplastic mammary lesions. J Natl Cancer Inst. 2000;92:418–23. doi: 10.1093/jnci/92.5.418. [DOI] [PubMed] [Google Scholar]
- 126.Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, Sporn MB. A new ligand for the perox-isome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogene-sis. Cancer Res. 1999;59:5671–3. [PubMed] [Google Scholar]
- 127.Yee LD, Young DC, Rosol TJ, Vanbuskirk AM, Clinton SK. Dietary (n-3) polyunsaturated fatty acids inhibit HER-2/neu-induced breast cancer in mice inde-pendently of the PPARgamma ligand rosiglitazone. J Nutr. 2005;135:983–8. doi: 10.1093/jn/135.5.983. [DOI] [PubMed] [Google Scholar]
- 128.Saez E, Rosenfeld J, Livolsi A, Olson P, Lombardo E, Nelson M, Banayo E, Cardiff RD, Izpisua-Belmonte JC, Evans RM. PPAR gamma signaling exacerbates mammary gland tumor development. Genes Dev. 2004;18:528–40. doi: 10.1101/gad.1167804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–7. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 130.Pino MV, Kelley MF, Jayyosi Z. Promotion of colon tumors in C57BL/6J-APC(min)/+ mice by thiazolidinedione PPARgamma agonists and a structurally unrelated PPARgamma agonist. Toxicol Pathol. 2004;32:58–63. doi: 10.1080/01926230490261320. [DOI] [PubMed] [Google Scholar]
- 131.Osawa E, Nakajima A, Wada K, Ishimine S, Fujisawa N, Kawamori T, Matsuhashi N, Kadowaki T, Ochiai M, Sekihara H, Nakagama H. Peroxisome proliferator-activated receptor gamma ligands sup-press colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003;124:361–7. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- 132.Kohno H, Yoshitani S, Takashima S, Okumura A, Hosokawa M, Yamaguchi N, Tanaka T. Troglitazone, a ligand for peroxisome proliferator-activated receptor gamma, inhibits chemically induced aberrant crypt foci in rats. Jpn J Cancer Res. 2001;92:396–403. doi: 10.1111/j.1349-7006.2001.tb01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tanaka T, Kohno H, Yoshitani S, Takashima S, Okumura A, Murakami A, Hosokawa M. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001;61:2424–8. [PubMed] [Google Scholar]
- 134.Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, Nambiar P, Rosenberg DW, Bronson RT, Edelmann W, Kucherlapati R, Gonzalez FJ, Spiegelman BM. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci USA. 2002;99:13771–6. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, Pettersson S. The Wnt(beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc Natl Acad Sci USA. 2005;102:1460–5. doi: 10.1073/pnas.0405928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Niho N, Takahashi M, Kitamura T, Shoji Y, Itoh M, Noda T, Sugimura T, Wakabayashi K. Concomitant suppression of hyperlipidemia and intestinal polyp for-mation in Apc-deficient mice by peroxisome prolifera-tor-activated receptor ligands. Cancer Res. 2003;63:6090–5. [PubMed] [Google Scholar]
- 137.Niho N, Takahashi M, Shoji Y, Takeuchi Y, Matsubara S, Sugimura T, Wakabayashi K. Dose-dependent suppression of hyperlipidemia and intestinal polyp for-mation in Min mice by pioglitazone, a PPAR gamma ligand. Cancer Sci. 2003;94:960–4. doi: 10.1111/j.1349-7006.2003.tb01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mutoh M, Niho N, Wakabayashi K. Concomitant sup-pression of hyperlipidemia and intestinal polyp forma-tion by increasing lipoprotein lipase activity in Apc-defi-cient mice. Biol Chem. 2006;387:381–5. doi: 10.1515/BC.2006.051. [DOI] [PubMed] [Google Scholar]
- 139.Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L, Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. PROactive investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–89. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 140.Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat. 2003;79:391–7. doi: 10.1023/a:1024038127156. [DOI] [PubMed] [Google Scholar]
- 141.Vogt T, Hafner C, Bross K, Bataille F, Jauch KW, Berand A, Landthaler M, Andreesen R, Reichle A. Antiangiogenetic therapy with pioglitazone, rofecoxib, and metronomic trofosfamide in patients with advanced malignant vascular tumors. Cancer. 2003;98:2251–6. doi: 10.1002/cncr.11775. [DOI] [PubMed] [Google Scholar]
- 142.Coras B, Hafner C, Reichle A, Hohenleutner U, Szeimies RM, Landthaler M, Vogt T. Antiangiogenic therapy with pioglitazone, rofecoxib, and trofosfamide in a patient with endemic kaposi sarcoma. Arch Dermatol. 2004;140:1504–7. doi: 10.1001/archderm.140.12.1504. [DOI] [PubMed] [Google Scholar]
- 143.Reichle A, Bross K, Vogt T, Bataille F, Wild P, Berand A, Krause SW, Andreesen R. Pioglitazone and rofe-coxib combined with angiostatically scheduled trofos-famide in the treatment of far-advanced melanoma and soft tissue sarcoma. Cancer. 2004;101:2247–56. doi: 10.1002/cncr.20574. [DOI] [PubMed] [Google Scholar]