

Abstract
Background: To investigate the cardiotoxic role of reactive oxygen species (ROS) and of products derived from catecholamines auto-oxidation, we studied: (1) the response of antioxidant cardiac cellular defence systems to oxidative stress induced by norepinephrine (NE) administration, (2) the effect of NE administration on cardiac β1-adrenergic receptors by means of receptor binding assay, (3) the cellular morphological alterations related to the biologically cross-talk between the NE administration and cytokines [tumor necrosis factor-alpha (TNF-α), monocyte chemotactic protein-1 (MCP-1), interleukins IL6, IL8, IL10]Methods and Results: A total of 195 male rats was used in the experiment. All animals underwent electrocardiogram (EKG) before being sacrificed. The results obtained show that NE administration influences the antioxidant cellular defence system significantly increasing glutathione peroxidase (GPx) activity, glutathione reductase (GR) and superoxide dismutase (SOD). The oxidized glutathione (GSH/GSSG) ratio significantly decreases and malondialdehyde (MDA) levels increase showing a state of lipoperoxidation of cardiac tissue. We describe a significant apoptotic process randomly sparse in the damaged myocardium and the effect of ROS on the NE-mediated TNF-α, MCP-1, and IL6, IL8, IL10 production. Conclusions: Our results support the hypothesis that catecholamines may induce oxidative damage through reactive intermediates resulting from their auto-oxidation, irrespective of their interaction with adrenergic receptors, thus representing an important factor in the pathogenesis of catecholamines-induced cardiotoxicity. The rise of the cardioinhibitory cytokines may be interpreted as the adaptive response of jeopardized myocardium with respect to the cardiac dysfunction resulting from NE injection.
Keywords: norepinephrine, cardiac antioxidant cellular defence system, β1-adrenergic receptors, cytokines
Introduction
It has been noticed that the excessive endogenous release or the excessive exogenous administration of catecholamines causes cardiotoxicity [1]. The pathogenesis of cardiac damage has been ascribed to intracellular Ca2+ overload through catecholamines-induced oxidative mechanisms [2, 3]. The molecular mechanisms by which oxidative stress is produced and affects cells and tissues, the cellular sources of oxygen free radicals and the role of specific antioxidants are currently under intensive investigation. Catecholamines can easily undergo oxidation with production of unstable catecholamines-O-quinones that in turn give rise to the respective adrenochromes and subsequent production of oxygen free radicals like superoxide radicals. The superoxide radicals can be reduced by superoxide dismutase to hydrogen peroxide, which damages the membrane integrity. The superoxide radical was proposed as responsible for the damage observed in the catecholamine-induced cardiomyopathy [4, 5]. Catecholamines toxicity through auto-oxidation enzymatically or metal catalyzed, with the concomitant production of reactive intermediates and free radicals, seems to involve particularly cardiac and nervous tissues [6, 7].
To investigate the possible cardiotoxic role of reactive oxygen species (ROS) and of products derived from catecholamines auto-oxidation, we studied: (1) the response of antioxidant cardiac cellular defence systems to oxidative stress induced by norepinephrine (NE) administration; (2) the effect of NE administration on cardiac β1-adrenergic receptors by means of receptor binding assay; (3) the cellular morphological alterations related to the biological cross-talk between the NE administration and cytokines [tumor necrosis factor-alpha (TNF-α), monocyte chemotactic protein-1 (MCP-1), IL6, IL8, IL10].
Materials and methods
The experimental procedures followed the ‘Principles of laboratory animal care’ (NIH publication 85-23, revised 1985) and were approved by the University of Siena Committee for animal experiments.
Animal model and experimental protocol
Male albino Wistar rats (180–200 g) were obtained from Harlan (Milan, Italy). They were allowed to acclimatize to their surrounding conditions (MIL, Morini diet; water ad libitum, room temperature of 22–24°C, alternate 12-hrs light/dark cycles) for 7 days before experiments. One hundred and ninety-five male rats (Wistar, Charles River) were used in the experiment to analyse the effect of NE administration, alone and in presence of adrenergic receptors antagonists–propranolol and prazosinon rats' heart. After the treatment all the animals, controls included, were maintained individually in their respective cages for avoiding possible sources of stress. In the group treated with NE, the animals were sacrificed by decapitation at the following times: 1, 4, 8, 24, 48 hrs, 10 and 30 days after treatment while in the group treated with adrenergic receptors antagonists the rats were sacrificed by decapitation 4 hrs after administration. All samples were used to determine biochemical parameters, for β1-binding study, and for morphological examination.
Biochemical analysis
Oxidative stress evaluation
NE group: Oxidative stress was evaluated in 72 rats (eight groups) weighing 200–250 g. NE 3 mg/kg i.p. was administered to seven groups of animals consisting of nine rats each. The eighth group was used as control; for every time examined, we have sacrificed at least one animal of control (total nine animals).
Norepinephrine + Propranolol + Prazosin (NEPP) group: Oxidative stress was evaluated in 36 rats weighing 200–250 g divided in four groups of nine animals each.
Group 1: Treated with propranolol 2mg/kg i.v., with prazosin 0.3 mg/kg i.v. and after 10 min with NE 3 mg/kg i.p., sacrificed 4 hrs after treatment (NEPP).
Group 2: Treated with propranolol 2mg/kg i.v. and with prazosin 0.3 mg/kg i.v. and sacrificed 4 hrs after treatment (PP).
Group 3: Treated with NE 3 mg/kg i.p. and sacrificed 4 hrs after treatment (NE).
Group 4: Control group, nine rats were treated with saline, 0.5 ml i.p. and all sacrificed to the same time (4 hrs) after administration.
All animals were sacrificed at 3–5 p.m. of the day to avoid possible variations ascribed to circadian rhythms (e.g. glutathione). The hearts of the treated and control animals were immediately resected and frozen at –80°C up to the time of the determination of reduced and oxidized glutathione (GSH, GSSG), malondialdehyde (MDA) and ascorbic acid (AA) levels and of superoxide dismutase (SOD), glutathione peroxidase (GPx) and glutathione reductase (GR) enzymatic activities.
GSH/GSSG ratio and protein determination
Heart tissue was homogenized in ethylenediaminetetraacetic acid (EDTA) K+–phosphate buffer, pH 7.4 (1:3, w/v) at 0°C. Total GSH was analysed as described by Tietze [8] and GSSG was determined according to Griffith's method [9]. Proteins were assayed according to the Lowry's method [10].
SOD, GPx and GR assessment
In order to measure cytosolic enzyme activity, the hearts' samples were homogenized according to Whanger and Butler [11]. GPx activity was measured according to Paglia and Valentine [12]. GR activity was analysed as described by Goldberg and Spooner [13]. Total superoxide dismutase (Cu/Zn superoxide dismutase and Mn-superoxide dismutase) was assayed by spectrophotometric method based on the inhibition of a superoxide-induced reduced nicoti-namide adenin dinucleotide (NADH) oxidation according to Paoletti et al.[14].
The cytosolic protein concentration was determined using the Lowry's method with bovine serum albumin (BSA) as standard [10].
MDA assessment
The extent of lipid peroxidation in the rats' heart was estimated by calculation of MDA levels as described by Shara et al.[15].
AA assay
Hearts' tissues were homogenized in EDTA K+- phosphate buffer pH 7.4 (1:4, w/v) at 0°C and analysed as described by Ross [16].
Control groups
For every parameter, the values obtained from every control groups used were not statistically different among them, therefore they have been united in a pool and the mean value was used as control value.
β1-binding study
NE group: Cardiac β1-adrenoreceptor density (Bmax) was determined on 28 rats (eight groups) weighing 200–250 g. NE 3 mg/kg i.p. was administered to seven groups of animals of three rats each. The eighth group (seven rats) was used as control.
The hearts of the treated and control animals were immediately resected and frozen at 80°C until the radioreceptor assay (RRA). After thawing at room temperature, myocardium was homogenized, in 10 volumes (w/v) of ice-cold 50 mM Tris buffer, pH 7.4, containing 150 mM NaCl and 1 mM MgCl2 Aliquots (0.5 ml) of membrane suspensions were kept at 80°C until use for binding assays as described by Schwinger [17]. The proteins' content was measured according to Lowry et al. with bovine serum albumin as standard [10].
Myocardial β1-adrenoreceptor assay
Myocardial β1-adrenoreceptor density was investigated using [125I] iodocyanopindolol (specific activity 2000 Ci/mmol) as the radiolabelled ligand [18]. The maximum number of receptor-binding sites (Bmax) and the dissociation constant (Kd) were calculated using Kell software (version 6, Biosoft, Cambridge, UK).
Electrocardiographic (EKG) measurements
All animals treated with NE, underwent EKG (Cardiette, Medical & Device SPA, Milano, Italy) 1 hr before being sacrificed; all animals treated with NE, propranolol and prazosin underwent EKG 5 min before being sacrificed.
Morphological examination
NE group: The histopathological examination of the heart was carried out on 40 rats, weighing 200–250 g, divided in eight groups of five animals each. Seven groups were treated with NE 3 mg/kg i.p., while one group was used as control.
The hearts of treated and control animals were immediately resected and weighed and, after removal of the apex, they were fixed in 10% buffered formalin for 45 hrs and fixed in paraffin. Paraffin-embedded tissue specimens of hearts were sectioned at 4 μm and stained with haematoxylin and eosin. In addition, immunohistochemical investigation of hearts' samples was performed with an antibody anti-TNF-α, anti-MCP-1 (Santa Cruz, CA, USA), anti-IL-6, IL-8, IL-10, and a TUNEL assay (Chemicon, Temecula, CA, USA). We used 4-μm-thick paraffin sections mounted on slides covered with 3, amminopropyl-triethoxysilane (Fluka, Buchs, Switzerland). A pre-treatment was necessary to facilitate antigen retrieval and to increase membrane permeability to antibodies anti-TNF-α boiling in 0.1 M citric acid buffer, for MCP-1 boiling 0.25 M EDTA buffer, for IL-6-8-10 (5 min proteolytic enzyme Dako, Copenhagen, Denmark, 20°C), and for TdT enzyme the sections were immerged in proteinase K (20 μg/ml of TRIS) for 15 min at 20°C. The primary antibody was applied in 1:600 ratio TNF-α, in a 1:2000 ratio IL-6 (Santa Cruz), in a 1:500 ratio IL-8 (Abcam, Cambridge, UK), and MCP-1 (Santa Cruz) in a 1:2000 ratio IL-10 (Peprotec, London, UK), and incubated for 120 min at 20°C. The sections were covered with the TdT enzyme, diluted in a ratio of 30% in reaction buffer (Apotag Plus Peroxidase In Situ Apoptosis Detection Kit, Chemicon) and incubated for 60 min at 38°C. The positive reaction was visualized by 3,3 diaminobenzidine (DAB) peroxidation, according to standard methods. The sections were counterstained with Mayer's haematoxylin, dehydrated, coverslipped and observed in a Leica DM4000B optical microscope (Leica, Cambridge, UK).
Using Leica system the total area in mm2 was measured in one superior and one inferior heart sections. By total screening of the histological area, number of necrotic foci and myocardial cells were counted and their frequency referred to 100 mm2. The hearts of animals dead spontaneously were fixed in 10% buffered formalin and examined as the hearts of sacrificed rats.
The samples were also examined under a confocal microscope and a three-dimensional reconstruction was performed (True Confocal Scanner, Leica TCS SP2).
Catecholamines determination
Catecholamines plasma levels were determined by high-performance liquid chromatography (HPLC) method according to Ueyama et al.[19].
Statistical analysis
Values are presented as means ± S.D. The unpaired 2-way Student's t-test was used to compare the results obtained for treated rats with the control group. A value of p < 0.05 was accepted as indicative of significant difference among groups.
Results
Enzymatic and non-enzymatic antioxidant cellular defence system evaluation
NE treatment
The results obtained (Fig. 1) showed that NE administration influences the antioxidant cellular defence system significantly increasing, compared to controls, GPx activity starting from 4 hrs up to 30 days after administration (Fig. 1A).
1.
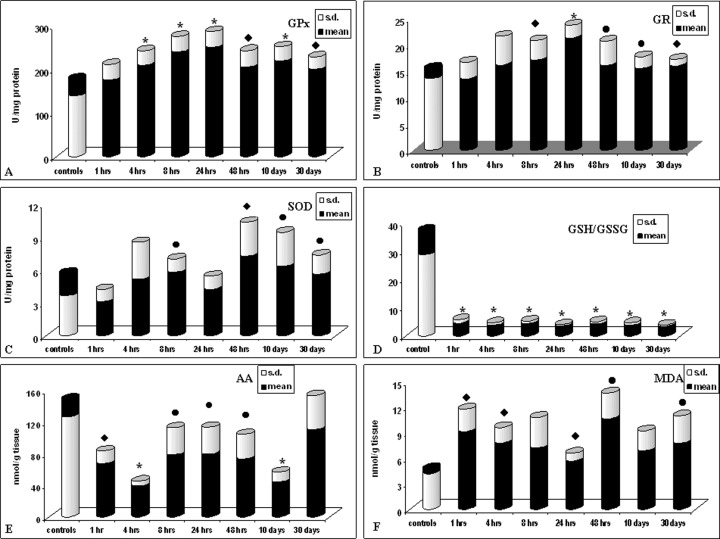
Effect of norepinephrine exposure (3 mg/kg i.p.) on cardiac antioxidant cellular systems. Values are presented as the mean (S.D.) of nine rats • p < 0.05, ♦ p < 0.01 and ★ p < 0.001 compared with control group.
Also GR (Fig. 1B) and SOD (Fig. 1C) showed after 4 hrs a tendency towards increase, which becomes statistically significant after 8 hrs, with a maximum (100%) for SOD after 48 hrs. This increase remains still significantly high, compared to controls, after 30 days for both the enzymes.
The GSH/GSSG ratio (Fig. 1D) significantly decreases starting from 1 hr up to 30 days, and AA (Fig. 1E) is significantly depleted at every time of examination until 10 days after NE administration. MDA levels (Fig. 1F) increase starting from 1 hr up to 30 days showing a state of lipoperoxidation of cardiac tissue.
NE and adrenergic antagonists treatment
The effect of NE administration on cellular redox state was further evaluated in presence of an α1 antagonist (prazosin) and of a β antagonist (propranolol) to check if oxidative stress was produced irrespective of adrenergic activation.
The results obtained 4 hrs after propranolol, prazosin and subsequent NE administration (NEPP group) (Fig. 2) show that also in this case the antioxidant cardiac cellular enzymatic defence system was influenced: in fact GPx (Fig. 2A) and GR (Fig. 2B) activities were increased compared to controls (p < 0.001 and p < 0.05, respectively), whereas SOD activity shows no statistically significant increase (Fig. 2C).
2.
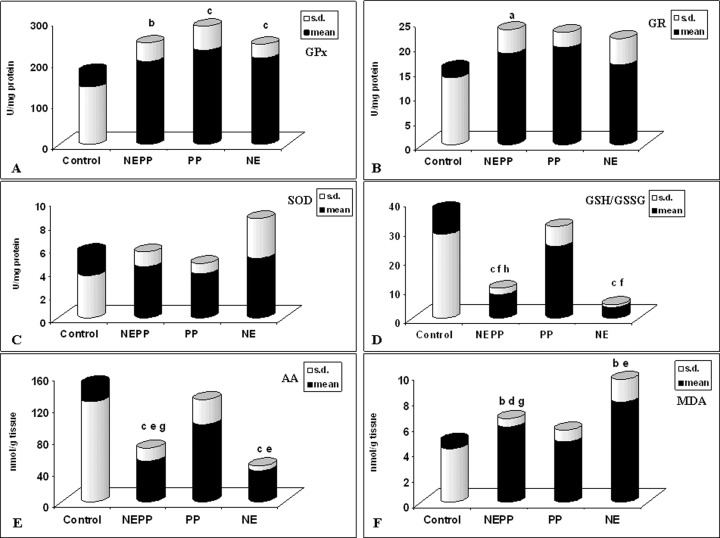
Effect of propranolol (2 mg/kg i.v.), prazosin (0.3 mg/kg i.v.) and norep-inephrine (3 mg/kg i.p.) exposure on cardiac antioxidant cellular systems. Values are presented as the mean (S.D.) of nine rats. PP = treated with propranolol and prazosin, NE = treated with norepinephrine, NEPP = treated with norepinephrine, propranolol and prazosin.
A= p < 0.05 compared with control group B= p < 0.01 compared with control group C= p < 0.001 compared with control group D= p < 0.05 compared with PP group E= p < 0.01 compared with PP group F= p < 0.001 compared with PP group G= p < 0.05 compared with NE group H= p < 0.01 compared with NE group
The data obtained in the NEPP group 4 hrs after treatment (Fig. 2D) show a significant decrease of GSH/GSSG ratio (p < 0.001) compared to controls and a statistically significant (p < 0.001) increase compared to the group treated only with NE (NE group). Determination of AA concentrations 4 hrs after treatment (Fig. 2E) shows a significant decrease compared to controls in the NEPP group (p < 0.0001) and a significant increase compared to the NE group (p < 0.05); adrenergic blockers administration alone does not affect AA levels.
Evaluation of MDA concentrations in the different treated groups (Fig. 2F) shows an increase in the NEPP group, which is significantly different compared to controls (p < 0.01) and to PP group (p < 0.05) and a statistically significant decrease compared to NE group (p < 0.05). The analysis of the results thus obtained shows anyway the presence of oxidative stress in cardiac cells also after adrenergic blockers administration.
β1-adrenoceptor binding sites
The iodocyanopindolol-binding assay (Table 1) demonstrated a saturable and highly specific binding pattern. Total myocardial β1-adrenoceptor density (Bmax) was increased after 1 hr (1194 ± 111.9 pmol/mg) and 4 hrs (1279 ± 178.4 pmol/mg) compared to control rats (960 ± 31.7 pmol/mg; p < 0.05). The receptor affinity decreased, i.e. dissociation constant (Kd) increased after 1 and 4 hrs, Kd in the control was 0.97 ± 0.454 pM compared with 1 hr (5.28 ± 0.116 pM; p < 0.001) and 4 hrs (2.60 ± 0.840 pM; p < 0.05).
1.
Mean characteristics of β1-adrenoceptor binding sites in myocardial membranes homogenates from NE treated and control rats
| Bmax | Controls | 1 hr | 4 hrs | 8 hrs | 24 hrs | 48 hrs |
|---|---|---|---|---|---|---|
| Mean | 959.67 | 1194.00 | 1278.67 | 1215.00 | 1089.00 | 976.00 |
| S.D. | 31.660 | 111.906 | 178.374 | 184.724 | 111.879 | 30.116 |
| p< | 0.025 | 0.038 | 0.078 | 0.126 | 0.553 | |
| Kd | Controls | 1 hr | 4 hrs | 8 hrs | 24 hrs | 48 hrs |
| Mean | 0.97 | 5.28 | 2.60 | 1.55 | 1.16 | 1.47 |
| S.D. | 0.454 | 0.116 | 0.840 | 1.076 | 0.799 | 0.935 |
| p< | 0.0001 | 0.042 | 0.438 | 0.743 | 0.452 |
Bmax (pmoli/mg membrane protein); Kd (pM); values are the means ± S.D. of three animals. t-test: p < 0.05 was considered significant.
Cardiac function (EKG)
Basal conditions
In the treated group, baseline EKG showed a base-line sinus rhythm at a mean rate of 542 ± 14 beats/min (Fig. 3A). The control group presented a baseline sinus rhythm at an average rate of 528 ± 16 beats/min. No significant difference in heart rate was present between the two groups.
3.
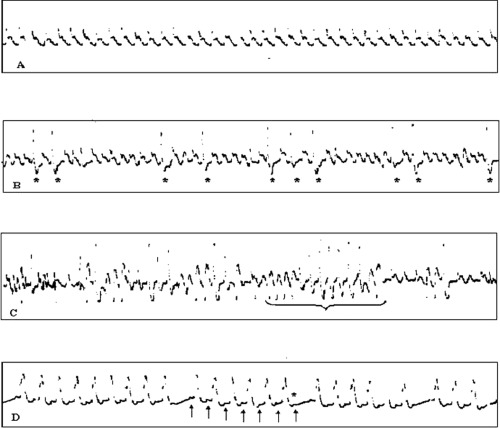
A: sinusal rhythm. B: frequent ventricular ectopic beats (VEB) (*), often in bigeminism. C: episodes of non-sustained ventricular tachy-cardia (VT). The longest one consists of 13 polymorphic beats. D: brady-cardia due to atrio-ventricular block of second degree with the Wenckebach's phenomenon: progressive prolongation of PR interval and then a P wave (*) is not conducted to the ventricule.
Time course of EKG changes after NE infusion
After NE administration, we observed a prompt increase in heart rate reaching progressively maximal value at the 15th minute (576 ± 53 beats/min). Later on, during the first hour, we found a plateau phase in which the heart rate remained steadily higher than that at baseline (i.e. 570 ± 60 at the 30th minute and 564 ± 68 at the 60th minute). Various ventricular arrhythmias were registered throughout this period. More in detail, all rats showed frequent ventricular ectopic beats (VEB) either alone (often in bigeminism) (Fig. 3B), or coupled, and in three rats we documented repeated episodes of unsustained ventricular tachycardia (VT) (Fig. 3C). Blood samples collected during the first hour after NE infusion demonstrated very high levels of circulating cate-cholamines, especially NE (412.1 ± 212.98 ng/ml, with respect to 2.9 ± 1.29 ng/ml in the control group), presumably responsible for an intense cardiac β-receptor stimulation, notwithstanding the observed fivefold reduction in the receptor affinity (Kd = 5.28 ± 0.116 versus 0.97 ± 0.454 pM in the controls).
Four hours after NE infusion, EKG tracings demonstrated an evident reduction in heart rate with bradycardia (480 ± 30 beats/min). It must be stressed that one rat showed a more severe bradycardia because of the onset of an intermittent sinusal block, and another one died after a progressive degree of sinus bradycardia resulting in a cardiac arrest. In these rats, NE plasma levels decreased dramatically with respect to the values detected during the first hour after infusion, ranging in an order similar to that observed in the controls (9.1 ± 2.17 ng/ml). On the other side, the restoration of a normal β-adrenoceptor affinity proceeded more slowly, as demonstrated by the high values of Kd also detectable in this phase (Kd = 2.6 ± 0.84 pM). On this basis, it can be hypothesized that the observed bradycardia may be related to a transient, functional hypostimulation of cardiac β-adrenoceptor, as a consequence of the long lasting reduction of receptor affinity associated with quite low NE plasma levels, thus actually mimicking the effect of a pharmacologic β-blocking.
From the eighth hour, EKG evaluation showed the restoration of a sinus rhythm at a mean rate similar to that of baseline (543 ± 30 beats/min). Similar values of mean heart rate were observed also in the following records after 24 hrs, 48 hrs, 10 days and 1 month, respectively. At all of these times, we observed a concomitant normalization of both NE plasma levels and β-adrenoceptor affinity.
Effect of adrenergic antagonists
The EKG tracings performed 4 hrs after the co-administration of prazosin and propranolol showed sinus bradycardia (470 ± 69 beats/min), similar to that observed with NE infusion after the fourth hour.
Finally, when we evaluated the electrocardiography findings of the rats treated concomitantly with NE, prazosin and propranolol, a more marked brady-cardia was documented (365 ± 33 beats/min). In this group, a rat had a severe bradycardia due to the onset of an atrio-ventricular block of second degree with the Wenckebach's phenomenon (Fig. 3D). A possible interpretation of these effects may involve the concomitant occurrence of the relative hyporesponsiveness of β-adrenoceptors observed 4 hrs after the NE infusion and the actual pharmacological block of the receptors due to adrenergic antagonists.
Morphology
The heart weight and its percentage of the body weight were greater in the NE group (0.80 ± 0.103 g and 239 ± 23 g) than in the control group (0.78 ± 0.107 and 257 ± 35, respectively).
Histology
The only lesion found were scattered foci of contraction band necrosis (CBN), typical of norepinephrine necrosis (NN) [20].This lesion was already visible in rats sacrificed at 1 hr and consisted of foci of few hypercontracted myocardial cells, intensively stained by eosin. This first change was associated with foci of myocardial cells with ruptured myofibrils and characterized by hypereosinophilic bands of hypercontracted sarcomeres alternated by clear empty spaces (Fig. 4A–D). Subsequently, after 8 hrs, a scanty infiltrate of macrophagic monocytes around the necrotic myocardial cells was observed. Within 24 hrs this infiltrate became more widespread with an increased number of elements and an early removal of the necrotic material. When the latter was completely removed, the myocardial cell appeared as an empty sarcolemmal tubes that proved the presence of macrophages, resulting in an “alveolar pattern”. At 30 days we were unable to see a clearcut fibrous repair of the necrotic foci that maintained the previously described changes. The number of necrotic foci and myocardial cells × 100 mm2 of the histological area are reported in Table 2.
4.
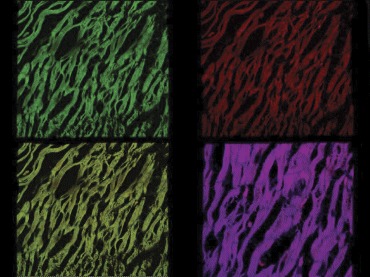
Confocal laser scanning microscope: foci of myocardial cells with ruptured myofibrils and characterized by hypereosinophilic bands of hypercontracted sarcomeres demonstrated by granular cytoplasm.
2.
Number of foci and necrotic cardiac myocytes × 100 mm2±S.E. in rats following intraperitoneal injection of 3 mg/kg of norepinephrine
| Groups | Total rats | Number | Percentage | ||||
|---|---|---|---|---|---|---|---|
| Sacrificed | Foci | Myocytes | Apoptosis % | ||||
| 1 hr | 5 | 205 ± 73 | 1424 ± 1121 | 3 ± 1 | |||
| 4 hrs | 5 | 163 ± 35 | 2616 ± 710 | 3 ± 2 | |||
| 8 hrs | 5 | 150 ± 77 | 2388 ± 1047 | 12 ± 8 | |||
| 1 day | 5 | 110 ± 30 | 944 ± 281 | 38 ± 18 | |||
| 2 days | 5 | 145 ± 86 | 924 ± 653 | 45 ± 17 | |||
| 10 days | 5 | 204 ± 20 | 3645 ± 714 | 60 ± 18 | |||
| 30 days | 5 | 194 ± 31 | 2161 ± 228 | 70 ± 13 | |||
| Grand total | 35 | 167 ± 20 | 2015 ± 272 | 33 ± 11 | p < 0.0001 | ||
| Dead spontaneously | |||||||
| <1 hr | 11 | 33 ± 19 | 96 ± 68 | 3 ± 1 | |||
| 1–5 hrs | 13 | 17 ± 34 | 35 ± 12 | 3 ± 2 | |||
| Grand total | 24 | 25 ± 10 | 66 ± 35 | 3 ± 1.5 | |||
| Controls | 5 | 75 ± 34 | 382 ± 105 | 3 ± 1 | |||
In rats dead spontaneously as well as in controls, monocytic infiltrates were absent and the number of foci and myocytes with catecholamine necrosis were significantly less (p < 0.001) than those found in treated and sacrificed animals. No interstitial haemor-rhages within necrotic foci were observed. Occasionally, a small subendocardial haemorrhage without relationship with myocardial necrotic foci was present.
In order to obtain the determination of the fraction of myocyte nuclei labelled by TUNEL, the number of myocyte nuclei per unit area of tissue was determined by counting an average of 10 fields, 1.4 mm2 each, at a magnification of 10 in each area of myocardium sampled. The percentage of apoptotic myocyte nuclei was determined.
The immunohistochemical study revealed an intensive positive result to TUNEL assay (Fig. 5A–D): approximately 3 ± 1, 3 ± 2, 12 ± 8, 38 ± 18, 45 ± 17, 60 ± 18 and 70 ± 13% apoptotic cells were observed after 1, 4, 8, 24, 48 hrs, 10 and 30 days (Table 2). TNF-α was demonstrated to significantly increase in heart specimens 4 hrs after NE infusion with the strongest evidence from the eighth hour to the 48th hour. On the 10th day, TNF-α significantly decreased in treated rats (Fig. 6A–D). MCP-1 demonstrated a gradually strong positive reaction from the first hour until 24th hour, to decrease until normalization after 48 hrs (Fig. 7A, B, C and D). IL8 was rapidly positive with a peak of evidence on eighth hour to decrease on 24th hour. On the 48th hour, both IL6 and IL10 demonstrated the strongest evidence but they differed among one another with respect to a gradual decrease for IL6 on 10th day when IL10 was strongly positive until 30 days (Table 3) (Fig. 8A–D).
5.
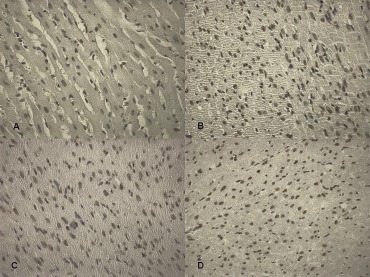
Determination of apoptotic cardiomyocytes nuclei by TUNEL assay: (A) control, (B) 8 hrs after treatment, (C) 24 hrs after treatment, (D) 10 days after treatment.
6.
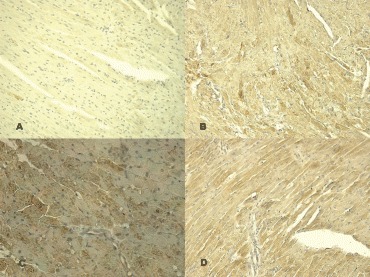
Immunohistochemical detection of TNFα with positive reaction after treatment: control (A), 4 hrs after treatment (B), 8 hrs after treatment (C), 24 hrs after treatment (D).
7.
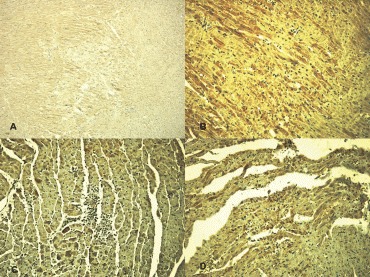
MCP-1 expression: control (A), 8 hrs after treatment (B), 24 hrs after treatment (C), and 10 days after treatment (D).
3.
Time course of cytokines response in heart specimens
| TNF-α | MCP-1 | IL-6 | IL-8 | IL-10 | |
|---|---|---|---|---|---|
| 1 hr | ++ | +++ | + | ++ | _ |
| 4 hrs | +++ | ++ | + | +++ | _ |
| 8 hrs | ++++ | ++ | ++ | +++ | _ |
| 24 hrs | ++ | +++ | ++ | ++ | + |
| 48 hrs | + | ++ | +++ | + | ++ |
| 10 days | _ | ++ | + | + | +++ |
8.
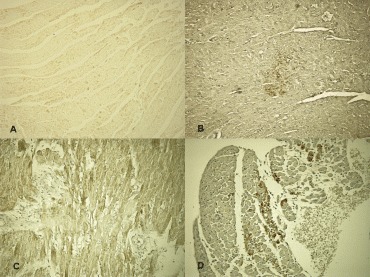
Immunohistochemical detection of the time course of the cardioinhibitory cytokines: control (A), IL6 at 1 hrs after treatment (B), IL8 at 24 hrs after treatment (C), IL10 at 24 hrs after treatment (D).
Confocal laser scanning microscope
The confocal laser scanning microscope aspect of the early lesion confirmed the pattern seen histologically and is already described in dog after intravenous infusion of isoproterenol and NE [21]. Furthermore, in order to establish morphologically the existence of apoptosis, in each group of sacrificed rats, nuclei of normal myocardial cells surrounding foci of necrotic myocytes were examined. In apoptotic cardiomyocytes, nuclei appeared to be of abnormal shape with nonlinear contours. Condensation of chromatin was evident in every case, with preferential peripheral condensation in a circular pattern (in a ‘doughnut-like’ pattern) and intranuclear clumping. Impending nuclear rupture with detachment of the nuclear membrane was occasionally seen [22].
Catecholamines dosage
The NE plasma levels decreased dramatically with respect to the values detected during the first hour after infusion, ranging in an order similar to that observed in the controls after the fourth hour (Table 4).
4.
Norepinephrine and epinephrine plasma levels
| Groups | Total rats | Number | Nore. ng/ml | Epin. ng/ml | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 hr | 5 | 1 | 714,3 | 41.4 | |||||||
| 2 | 364.4 | 51.4 | |||||||||
| 3 | 537.4 | 52.5 | |||||||||
| 4 | 215.5 | 16.4 | |||||||||
| 5 | 229.1 | 23.3 | |||||||||
| 4 hrs | 5 | 6 | 5.25 | 7.1 | |||||||
| 7 | 10.26 | 12.4 | |||||||||
| 8 | 10.01 | 11.7 | |||||||||
| 9 | 9.97 | 11.0 | |||||||||
| 10 | 10.14 | 12.1 | |||||||||
| 8 hrs | 5 | 11 | 0.48 | 4.9 | |||||||
| 12 | 2.97 | 6.0 | |||||||||
| 13 | 2.41 | 7.4 | |||||||||
| 14 | 4.28 | 4.8 | |||||||||
| 15 | 38.0 | 12.1 | |||||||||
| 24 hrs | 5 | 16 | 6.90 | 17.2 | |||||||
| 17 | 15.1 | 12.9 | |||||||||
| 18 | 14.8 | 4.7 | |||||||||
| 19 | 8.1 | 8.2 | |||||||||
| 20 | 4.4 | 29.3 | |||||||||
| 48 hrs | 5 | 21 | 1.53 | 4.1 | |||||||
| 22 | 5.3 | 2.5 | |||||||||
| 23 | 1.6 | 1.2 | |||||||||
| 24 | 1.8 | 2.5 | |||||||||
| 25 | 2.8 | 12.4 | |||||||||
| 10 days | 5 | 26 | 3.0 | 1.8 | |||||||
| 27 | 3.11 | 8.1 | |||||||||
| 28 | 2.2 | 1.9 | |||||||||
| 29 | 1.1 | 8.2 | |||||||||
| 30 | 1.1 | 8.9 | |||||||||
| 30 days | 5 | 31 | 1.63 | 9.1 | |||||||
| 32 | 1.61 | 15.4 | |||||||||
| 33 | 2.0 | 4.8 | |||||||||
| 34 | 2.1 | 4.2 | |||||||||
| 35 | 6.1 | 33.3 | |||||||||
| Control | 5 | 36 | 4.8 | 36.8 | |||||||
| 37 | 1.2 | 13.4 | |||||||||
| 38 | 2.6 | 31.9 | |||||||||
| 39 | 3.3 | 21.7 | |||||||||
| 40 | 2.8 | 27.0 | |||||||||
Discussion
In this study we showed that acute administration of NE produces alterations of the cellular redox state and lipoperoxidation in cardiomyocytes, thus suggesting an important role played by intermediates derived from catecholamines auto-oxidation and by ROS in the mechanism of cellular damage.
NE and oxidative stress
The results obtained in this study support the hypothesis regarding the role of oxidative mechanism in NE-induced cardiac damage. The analysis of the biochemical parameters examined clearly shows the involvement of the antioxidant cellular defence system in response to an evident oxidative stress. The GSH/GSSG ratio, which is a good estimation of the redox state as well as oxidative stress in the cell, decreases starting from 1 hr up to 30 days after NE administration showing a GSH depletion which might be the result of conjugation with the produced o-quinone and/or of its oxidation to GSSG; the latter does not seem to undergo the ‘sparing’ effect of AA, also strongly depleted at every time of examination. GSH oxidation to GSSG might result, for example, from reduction of ROS like H2O2, occurring in the presence of Cu2+ or from direct reduction of Cu2+ to Cu+[23–25]. The increase of GPx activity 4 hrs after NE administration may contribute, with its action on H2O2, to GSH consumption and to the formation of GSSG. Also SOD and GR increase their activity, but only 8 hrs after treatment, showing the tendency to contrast a still operating oxidative stress. This delayed increase of antioxidant enzymes activity might explain the accumulation, after 4 hrs, of O2−and GSSG due to its insufficient reduction to GSH by GR. In vitro studies with purified enzymes showed that GR activity was inhibited by o-quinones and aminochromes [2].
Finally, MDA significantly increased in the heart of rats treated with NE, compared to controls, after 1 hr and at all times of examination, showing that, despite antioxidant enzymes increased activity, ROS were not sufficiently neutralized, making possible the disproportionate reaction of O2 and H2O2 to OH radical and 1O2 (singlet oxygen;Haber-Weiss reaction).Theoretically, OH may result in vivo also by NE non-enzymatic oxidation [26]. Moreover, singlet oxygen is a highly reactive specie and may cause damage to membrane lipids and inactivation of several enzymes.It has been shown [27] that 1O2 is the ROS responsible of the alteration of Ca2+-ATPase in the cardiac sarcoplasmic reticulum; also high levels of H2O2 cause inhibition of Ca2+-ATPase activity [28, 29] and this is probably related to –SH groups oxidation. The final event of these processes may consist in functional and morphological alterations of myocytes like CBN resulting from intracellular calcium overload caused by ROS activity [5]. This is actually the only morphological alteration detected at the histological and ultrastructural examination in hearts of rats treated with NE.
NE and β1-adrenoceptor density
In our study, NE increased Kd and myocardial β1-adrenoceptor density 1 and 4 hrs after treatment. In two studies, myocardial β1-adrenoceptor density also increased in the rats infused with NE for 1 week [30] as in dog after short-term infusion [31]. In contrast, chronic administration of NE for 8 weeks has been shown to reduce myocardial β1-adrenoceptor density [32, 33].
Previous work demonstrated that the β1-adrenoceptor can exist in two affinity states for agonist.The finding of increase of both β1-adrenoceptor and Kd may be explained by agonists-induced conversion of β1-adrenoceptor from a native form with high affinity for agonists to a form with much lower affinity for agonists [34].
Haenen et al. reported that oxidative stress increased β-adrenoceptor number and reduced cyclic adenosine monophosphate (c-AMP) formation in the ventricle membrane and reduced β-adrenoceptor function [35]. Furthermore, intense exposure of β-adrenoceptor to an agonist leads to a decrease in receptor responsiveness, as is the case for most G protein-coupled receptors. These might represent physiological mechanisms that protect the heart against excessive β1-adrenoceptor stimulation.
Cardiac function (EKG)
About the EKG alterations due to NE administration, we can hypothesize that the observed bradycardia may be related to a transient, functional hypostimulation of cardiac β-adrenoceptor, as a consequence of the long-lasting reduction of receptor affinity associated with quite low NE plasma levels, thus actually mimicking the effect of a pharmacologic β-blocking. When we evaluated the electrocardiography findings of the rats treated concomitantly with NE, prazosin, and propranolol, a more marked bradycardia was documented. A possible interpretation of these effects may involve the concomitant occurrence of the relative hyporesponsiveness of β-adrenoceptors observed 4 hrs after the NE infusion and the actual pharmacological block of the receptors due to adrenergic antagonists.
NE and morphology
The concept that CBN is a result of direct cate-cholamine toxicity is based on experimental cate-cholamine infusion.The lesion is visible within 5–10 min of perfusion in the presence of normal vessels. Previous experimental studies and clinical work demonstrated that the intramyocardial release of cat-echolamines may result from mechanisms other than asynergic myocardium any time the cardiac nerves are involved, e.g. inflammation around nerves of the tunica media of an atherosclerotic plaque [21]. They may trigger a catecholamine myotoxicity linked with ventricular fibrillation and acting through free-radical-mediated lipid peroxidation with intramyocellular Ca2+ influx [36]. Growing evidence demonstrate that ROS may mediate cardiomyocyte apoptosis [37]. Recently, it has been demonstrated that β-adrenergic receptor-stimulated apoptosis is mediated by ROS-dependent pathway in rat cardiomyocytes [38]. Our results confirm that ROS could act as the upstream initiators of NE-induced apoptosis in cardiomyocytes and we demonstrate that ROS are involved in the regulation of TNF-α production and apoptosis upon NE treatment [37, 39]. TNF-α has been shown to evoke decrease in calcium fluxes through a sphingosine signalling intermediate, suppression of mito-chondrial metabolism, activation of apoptosis and enhancement of deposition of collagen in transforming growth factor (TGF)–mediated pathway [40]. The present study demonstrated a strong reaction in heart specimens of cytokines with different time course.The rise of the cardioinhibitory cytokines may be interpreted as the adaptive response of jeopardized myocardium with respect to the cardiac dysfunction resulting from NE injection [40].
In conclusion, our results support the hypothesis that catecholamines may induce oxidative damage through reactive intermediates resulting from their auto-oxidation, irrespective of their interaction with adrenergic receptors, thus representing an important factor in the pathogenesis of catecholamines induced cardiotoxicity [41–44]. We describe a significant apoptotic process randomly sparse in the damaged myocardium and the effect of ROS on the NE-mediated TNF-α, MCP-1, and IL6, IL8, IL10 production [45]. The rise of the cardioin-hibitory cytokines may be interpreted as the adaptive response of jeopardized myocardium with respect to the cardiac dysfunction resulting from NE injection [40, 45]. Our findings further suggest that antioxidant therapy may be beneficial in congestive heart failure in which cardiac NE release is increased [43]. Contrary to the general opinion that excess catecholamines produce cardiotoxicity mainly through binding to adrenoceptors, we demonstrate that catecholamine-induced deleterious actions occur through oxidative mechanisms. Plasma NE correlates with the severity of the left ventricular dysfunction [46–47].
References
- 1.Rona G. Catecholamine cardiotoxicity. J Mol Cell Cardiol. 1985;17:291–306. doi: 10.1016/s0022-2828(85)80130-9. [DOI] [PubMed] [Google Scholar]
- 2.Remiao F, Carmo H, Carvalho FD, Bastos ML. Inhibition of glutathione reductase by isoproterenol oxi-dation products. J Enzyme Inhib. 2000;15:47–61. [PubMed] [Google Scholar]
- 3.Bindoli A, Rigobello MP, Deeble DJ. Biochemical and toxicological properties of the oxidation products of cat-echolamines. Free Radic Biol Med. 1992;13:391–405. doi: 10.1016/0891-5849(92)90182-g. [DOI] [PubMed] [Google Scholar]
- 4.Behonick GS, Novak MJ, Nealley EW, Baskin SI. Toxicology update: the cardiotoxicity of the oxidative stress metabolites of catecholamines (aminochromes) J Appl Toxicol. 2001;21:S15–22. doi: 10.1002/jat.793. [DOI] [PubMed] [Google Scholar]
- 5.Tappia PS, Hata T, Hozaima L, Sandhu MS, Panagia V, Dhalla NS. Role of oxidative stress in catecholamine-induced changes in cardiac sarcolemmal Ca2+ transport. Arch Biochem Biophys. 2001;387:85–92. doi: 10.1006/abbi.2000.2234. [DOI] [PubMed] [Google Scholar]
- 6.Bolli R. Oxygen-derived free radicals and myocardial reperfusion injury: an overview. Cardiovasc Drugs Ther. 1991;5:249–68. doi: 10.1007/BF00054747. [DOI] [PubMed] [Google Scholar]
- 7.Smythies J, Galzigna L. The oxidative metabolism of catecholamines in the brain: a review. Biochim Biophys Acta. 1998;1380:159–62. doi: 10.1016/s0304-4165(97)00131-1. [DOI] [PubMed] [Google Scholar]
- 8.Tietze F. Enzymatic method for quantitative determination of nanogram amounts of total and oxidized gluthatione. Application to mammalian blood and other tissues. Anal Biochem. 1969;27:502–22. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 9.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–12. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 10.Lowry OH, Rosebrough HJ, Farr AC, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 11.Whanger PD, Butler JA. Effects of various dietary levels of selenium as selenite or selenomethionine on tissue selenium levels and glutathione peroxidase activity in rats. J Nutr. 1988;118:846–52. doi: 10.1093/jn/118.7.846. [DOI] [PubMed] [Google Scholar]
- 12.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–69. [PubMed] [Google Scholar]
- 13.Goldberg DM, Spooner RG. Glutathione reductase. In: Bergmeyer J, Grabl M, editors. Methods of enzymatic analysis. Deerfield Beach: Chemie; 1983. pp. 258–65. [Google Scholar]
- 14.Paoletti F, Aldinucci D, Mocali A, Caparrini A. A sensitive spectrophotometric method for the determination of superoxide dismutase activity in tissue extracts. Anal Biochem. 1986;154:536–41. doi: 10.1016/0003-2697(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 15.Shara MA, Dickson PH, Bagchi D, Stohs SJ. Excretion of formaldehyde, malondialdehyde, acetaldehyde and acetone in the urine of rats in response to 2,3,7,8-tetrachlorodibenzo-P-dioxin, paraquat, endrin and carbon tetrachloride. J Chromatogr. 1992;576:221–33. doi: 10.1016/0378-4347(92)80196-w. [DOI] [PubMed] [Google Scholar]
- 16.Ross MA. Determination of ascorbic acid and uric acid in plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1994;657:197–200. doi: 10.1016/0378-4347(94)80087-1. [DOI] [PubMed] [Google Scholar]
- 17.Schwinger RH, Bohm M, Pieske B, Erdmann E. Different beta-adrenoceptor-effector coupling in human ventricular and atrial myocardium. Eur J Clin Invest. 1991;21:443–51. doi: 10.1111/j.1365-2362.1991.tb01393.x. [DOI] [PubMed] [Google Scholar]
- 18.Anthonio RL, Brodde OE, Van Veldhuisen DJ, Scholtens E, Crijns HJ, Van Gilst WH. Beta-adreno-ceptor density in chronic infarcted myocardium: a sub-type specific decrease of beta1-adrenoceptor density. Int J Cardiol. 2000;72:137–41. doi: 10.1016/s0167-5273(99)00181-3. [DOI] [PubMed] [Google Scholar]
- 19.Ueyama J, Kitaichi K, Iwase M, Takagi K, Takagi K, Hasegawa T. Application of ultrafiltration method to measurement of catecholamines in plasma of human and rodents by high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;798:35–41. doi: 10.1016/j.jchromb.2003.08.045. [DOI] [PubMed] [Google Scholar]
- 20.Baroldi G, Mittleman RE, Parolini M, Silver MD, Fineschi V. Myocardial contraction bands. Definition, quantification and significance in forensic pathology. Int J Legal Med. 2001;115:142–51. doi: 10.1007/s004140100229. [DOI] [PubMed] [Google Scholar]
- 21.Fineschi V, Baroldi G, Silver MD. Forensic Pathology and Sudden death in forensic medicine. Boca Raton: Taylor & Francis; 2006. pp. 134–56. [Google Scholar]
- 22.Takemura G, Fujiwara H. Morphological aspects of apoptosis in heart disease. J Cell Mol Med. 2006;10:56–75. doi: 10.1111/j.1582-4934.2006.tb00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rupp H, Dhalla KS, Dhalla NS. Mechanisms of cardiac cell damage due to catecholamines: significance of drugs regulating central sympathetic outflow. J Cardiovasc Pharmacol. 1994;24:S16–24. doi: 10.1097/00005344-199424001-00004. [DOI] [PubMed] [Google Scholar]
- 24.Remiao F, Carvalho M, Carmo H, Carvalho F, Bastos ML. Cu2+-induced isoproterenol oxidation into isoprenochrome in adult rat calcium-tolerant cardiomyocytes. Chem Res Toxicol. 2002;15:861–9. doi: 10.1021/tx025518q. [DOI] [PubMed] [Google Scholar]
- 25.Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S. doi: 10.1016/0002-9343(91)90279-7. [DOI] [PubMed] [Google Scholar]
- 26.Obata T, Yamanaka Y. Cardiac microdialysis of sali-cylic acid. OH generation on nonenzymatic oxidation by norepinephrine in rat heart. Biochem Pharmacol. 1997;53:1375–8. doi: 10.1016/s0006-2952(96)00870-2. [DOI] [PubMed] [Google Scholar]
- 27.Kukreja RC, Hess ML. The oxygen free radical system: from equations through membrane-protein interactions to cardiovascular injury and protection. Cardiovasc Res. 1992;26:641–55. doi: 10.1093/cvr/26.7.641. [DOI] [PubMed] [Google Scholar]
- 28.Kukreja RC, Weaver AB, Hess ML. Sarcolemmal Na(+)-K(+)-ATPase: inactivation by neutrophil-derived free radicals and oxidants. Am J Physiol. 1990;259:H1330–6. doi: 10.1152/ajpheart.1990.259.5.H1330. [DOI] [PubMed] [Google Scholar]
- 29.Khaper N, Rigatto C, Seneviratne C, Li T, Singal PK. Chronic treatment with propranolol induces antioxidant changes and protects against ischemia-reperfusion injury. J Mol Cell Cardiol. 1997;29:3335–44. doi: 10.1006/jmcc.1997.0558. [DOI] [PubMed] [Google Scholar]
- 30.Zhao M, Hagler HK, Muntz KH. Regulation of alpha 1-, beta 1-, and beta 2-adrenergic receptors in rat heart by norepinephrine. Am J Physiol. 1996;271:H1762–8. doi: 10.1152/ajpheart.1996.271.5.H1762. [DOI] [PubMed] [Google Scholar]
- 31.Vatner DE, Vatner SF, Nejima J, Uemura N, Susanni EE, Hintze TH, Homcy CJ. Chronic norepinephrine elicits desensitization by uncoupling the beta-receptor. J Clin Invest. 1989;84:1741–8. doi: 10.1172/JCI114357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong E, Yatani A, Mohan A, Liang CS. Myocardial beta-adrenoceptor down-regulation by norepinephrine is linked to reduced norepinephrine uptake activity. Eur J Pharmacol. 1999;384:17–24. doi: 10.1016/s0014-2999(99)00652-4. [DOI] [PubMed] [Google Scholar]
- 33.Barki-Harrington L, Perrino C, Rockman HA. Network integration of the adrenergic system in cardiac hypertrophy. Cardiovasc Res. 2004;63:391–402. doi: 10.1016/j.cardiores.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Toews ML. Comparison of agonist-induced changes in beta- and alpha 1-adrenergic receptors of DDT1 MF-2 cells. Mol Pharmacol. 1987;31:58–68. [PubMed] [Google Scholar]
- 35.Haenen GR, Veerman M, Bast A. Reduction of beta-adrenoceptor function by oxidative stress in the heart. Free Radic Biol Med. 1990;9:279–88. doi: 10.1016/0891-5849(90)90002-z. [DOI] [PubMed] [Google Scholar]
- 36.Mak IT, Weglicki WB. Protection by beta-blocking agents against free radical-mediated sarcolemmal lipid peroxidation. Circ Res. 1988;63:262–6. doi: 10.1161/01.res.63.1.262. [DOI] [PubMed] [Google Scholar]
- 37.Fu YC, Chi CS, Yin SC, Hwang B, Chiu YT, Hsu SL. Norepinephrine induced apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species— TNFalpha-caspase signaling pathway. Cardiovasc Res. 2004;62:558–67. doi: 10.1016/j.cardiores.2004.01.039. [DOI] [PubMed] [Google Scholar]
- 38.Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, Singh K, Colucci WS. Beta-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res. 2003;92:136–8. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 39.Aikawa R, Nitta-Komatsubara Y, Kudoh S, Takano H, Nagai T, Yazaki Y, Nagai R, Komuro I. Reactive oxygen species induce cardiomyocyte apoptosis partly through TNF-alpha. Cytokine. 2002;18:179–83. doi: 10.1006/cyto.2001.1007. [DOI] [PubMed] [Google Scholar]
- 40.Kosmala W, Przewlocka-Kosmala M, Mazurek W. Proinflammatory cytokines and myocardial viability in patients after acute myocardial infarction. Int J Cardiol. 2005;101:449–56. doi: 10.1016/j.ijcard.2004.03.067. [DOI] [PubMed] [Google Scholar]
- 41.Hori M, Gotoh K, Kitakaze M, Iwai K, Iwakura K, Sato H, Koretsune Y, Inoue M, Kitabatake A, Kamada T. Role of oxygen-derived free radicals in myocardial oedema and ischemia in coronary microvascular embolization. Circulation. 1991;84:828–40. doi: 10.1161/01.cir.84.2.828. [DOI] [PubMed] [Google Scholar]
- 42.Lameris TW, De Zeeuw S, Alberts G, Boomsma F, Duncker DJ, Verdouw PD, Veld AJ, Van Den Meiracker AH. Time course and mechanism of myocardial catecholamine release during transient ischemia in vivo. Circulation. 2000;101:2645–50. doi: 10.1161/01.cir.101.22.2645. [DOI] [PubMed] [Google Scholar]
- 43.Qin F, Rounds NK, Mao W, Kawai K, Liang CS. Antioxidant vitamins prevent cardiomyocyte apoptosis produced by norepinephrine infusion in ferrets. Cardiovasc Res. 2001;51:736–48. doi: 10.1016/s0008-6363(01)00323-6. [DOI] [PubMed] [Google Scholar]
- 44.Fineschi V, Baroldi G, Centini F, Cerretani D, Fiaschi AI, Micheli L, Parolini M, Turillazzi E, Giorgi G. Markers of cardiac oxidative stress and altered morphology after intraperitoneal cocaine injection in a rat model. Int J Legal Med. 2001;114:323–30. doi: 10.1007/s004140000194. [DOI] [PubMed] [Google Scholar]
- 45.Fedak PW, Verma S, Weisel RD, Li RK. Cardiac remodeling and failure: from molecules to man (Part I) Cardiovasc Pathol. 2005;14:1–11. doi: 10.1016/j.carpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Colucci WS, Sawyer DB, Singh K, Communal C. Adrenergic overload and apoptosis in heart failure: implications for therapy. J Card Fail. 2000;6:1–7. [PubMed] [Google Scholar]
- 47.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–34. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]