

Abstract
Purine bias, which is usually referred to as an “ancestral codon”, is known to result in short-range correlations between nucleotides in coding sequences, and it is common in all species. We demonstrate that RWY is a more appropriate pattern than the classical RNY, and purine bias (Rrr) is the product of a network of nucleotide compensations induced by functional constraints on the physicochemical properties of proteins. Through deductions from universal correlation properties, we also demonstrate that amino acids from Miller’s spark discharge experiment are compatible with functional primeval proteins at the dawn of living cell radiation on earth. These amino acids match the hydropathy and secondary structures of modern proteins.
Keywords: genomics, ancestral codon, purine bias, short-range correlations, protein features
Introduction
The genome of a given organism is a plastic structure, but it is nonetheless highly ordered. Shaping factors act on the structure and composition of coding and noncoding regions.1,2 The most important factor that acts on protein coding sequences (CDSs) is undoubtedly the genetic code, which is basically a set of rules relating codons to amino acids. One of the most important properties of the genetic code is its universality, ie, it is the same (or almost the same) in all organisms ranging from prokaryotes to eukaryotes with minor exceptions, and most of these exceptions occur in organelle genomes.
A universal characteristic of the CDSs is their three-base periodicity. The three-base periodicity is induced by the purine bias (Rrr), which was recognized by Shepherd3 and proposed by him has a universal fingerprint of CDSs. According to the purine bias, the relative frequency of purines (adenine and guanine, ie, A and G, respectively) is larger in the first codon position than in the second and third codon positions, which justifies the logo Rrr for codons to indicate the larger than expected purine frequency (R) in first codon position and the lower than expected purine frequency (r) in the two other codon positions. As a consequence of the purine bias, one has that 1) the product of purine probabilities is the largest for the first codon position (PA1PG1); 2) the product of the probabilities PC1PG2PA3 of cytosine (C) at the first codon position (C1), G in the second codon position (G2), and A in the third codon position (A3) has the lowest value in the coding frame compared to the other frames4; 3) the relative frequency of G1 is larger than that of G2; and 4) the relative frequency of thymine (T) in the first codon position (T1) is lower than that of A in the second codon position (A2). The formulation of CDS features, which are observed in the coding frame (≥150 bp) of all living organisms regardless of codon usage, according to the universal feature method (UFM),4,5 allows the coding versus noncoding classifications over six frames for open reading frames (ORFs) of sizes larger than ~300 bp.
The cause of the three-base periodicity is not trivial given 1) the large interval of variation in average guanine plus cytosine (GC) covered by coding DNA in all living forms and 2) the set of codons available in the genetic code allowing for random nucleotide distribution. Based on these two lines of evidence, one could expect any or no periodicity in coding DNA within a species. The existence of a three-base periodicity implies the existence of a codon preference responsible for that periodicity that is common to all living forms. This codon preference has been shown to be induced by selective pressure on the secondary structures and the physicochemical properties of amino acids in proteins.6 The purine bias observed in coding DNA in modern organisms results from a balance in selective pressures acting on protein functionality, the energy cost of amino acid synthesis, and ribosomal evolution given thermodynamic constraints on translation processivity and accuracy.7 In reality, the number of amino acids most frequently found in α-helices, β-sheets, and aperiodic (turns or coils) secondary structures in modern proteins is small (alanine, glycine, valine), and all are G1.
Because the selective pressures acting on protein functionality are not expected to have changed since the prebiotic era, one can ask what can be inferred about the primeval genetic code from the amino acid distribution in modern proteins. First, let us briefly review the main ideas that were animating the research on the origin of the genetic code until now.
The RNA world coined by Gilbert,8 which has largely been supported by success stories9 with the systematic evolution of ligands by exponential enrichment (SELEX) technique,10 is gradually replaced by the notion that nucleotide polymers must have existed together with ancillary peptides.11 Despite hot debates,12 the idea that both nucleotide and amino acid polymers might have been produced by a proto-metabolism that might have evolved naturally from autocatalytic physicochemical reactions on the hypercycle model13–15 emerged as the most robust one. The trend in today’s scientific community is to consider that a few triplets, each of which coding a corresponding amino acid, must have been at the beginning of the genetic codes and that it, then, evolved step by step16 by mutation following a scheme already intuited by Woese.17 The hierarchical step-by-step acquisition of complexity generates a dependency from inherited mechanisms with regard to parental ones, a common property of evolving beings, which prompted Crick18 to declare the modern genetic code as a “frozen accident” since there is no way back without the extinction of present life. Based on considerations on primeval metabolism and conserved base stretches in tRNA, Hartman19,20 proposed that the genetic code might have started with the two letters G and C. Under that hypothesis, arginine or lysine, which are expected to have come late because of the large number of catalytic steps needed for them to be synthesized from a primeval citric acid cycle, might be replaced by ornithine.21 However, ornithine might not even be necessary for short proteins to have catalytic activities.22 Hypothetically, the minimal size of proto-tRNA might be a 17-nucleotide aptamer with 7 nucleotides in the “anticodon” loop and the 10 remaining ones for the double helical arm as well as the amino acid attachment region of 4 nucleotides.21 Such a pro-to-tRNA might have co-evolved into the modern tRNA, following the scheme proposed by Di Giulio,23 together with the nucleic acid24,25 polymers26–28 and amino acids29 available in the prebiotic soup.30 It has been shown that the prebiotic soup did not likely include more than 10 among the amino acid list used by modern cells29 and that it should be those described by Miller.31,32 Later on, it was shown that aminoacyl-tRNA synthetases are divided into two classes that match the early and late amino acids.33 The prevalence of RNY codons3 in coding sequences has been seen as a remnant of a simpler code encoding primeval proteins due to the fact that modern proteins use a large proportion of amino acids whose codons start with purines.34 This observation has motivated a decomposition of the evolutionary steps the primeval code could have gone through to gain its today’s figure.35
In investigating the evolutionary origin of the genetic code, Ikehara et al.7 showed that it may have originated from a system of four amino acids: the GNC code. This GNC code (G for guanine, N for any of the four nucleotides, C for cytosine) is capable of encoding [GADV]-proteins (G for glycine, A for alanine, D for aspartic acid, V for valine) with appropriate three-dimensional structures including the characteristics of water-soluble globular proteins such as hydropathy, α-helices, β-sheets, β-turns, and catalytic activities.22 According to Ikehara et al.7, this primitive code may have first evolved into a code containing 16 codons and 10 amino acids, the so-called SNS (S for “strong” ie, G or C) and then the RNY (R for purines, Y for pyrimidines) ancestral codon suggested by Shepherd3 and revisited by Brooks and Fresco.36 The RNY pattern is responsible for the three-base periodicity in coding sequences by inducing short-range correlations (nucleotide correlations on a distance shorter than average CDS size).37,38
Short-range correlations among nucleotides in CDSs can be analyzed in heterogeneous genomes, ie, genomes whose CDSs cover a wide range of GC variation (>50%) in third codon position (GC3), such as the Homo sapiens and Oryza sativa genomes, or homogeneous genomes (GC3 variation <50%) at different positions of the universal correlation regression line (see Fig. 1 in Ref. 39). The universal correlation, shown for the first time by Sueoka,40 describes the linear relationship that species display when their genomes are plotted for the average GC3 versus GC in the second codon position (GC2) of their CDSs. In eukaryotes, the genomes at both boundaries of this relationship are Plasmodium falciparum (AT-rich) and Chlamydomonas reinhardtii (GC-rich).
Figure 1.
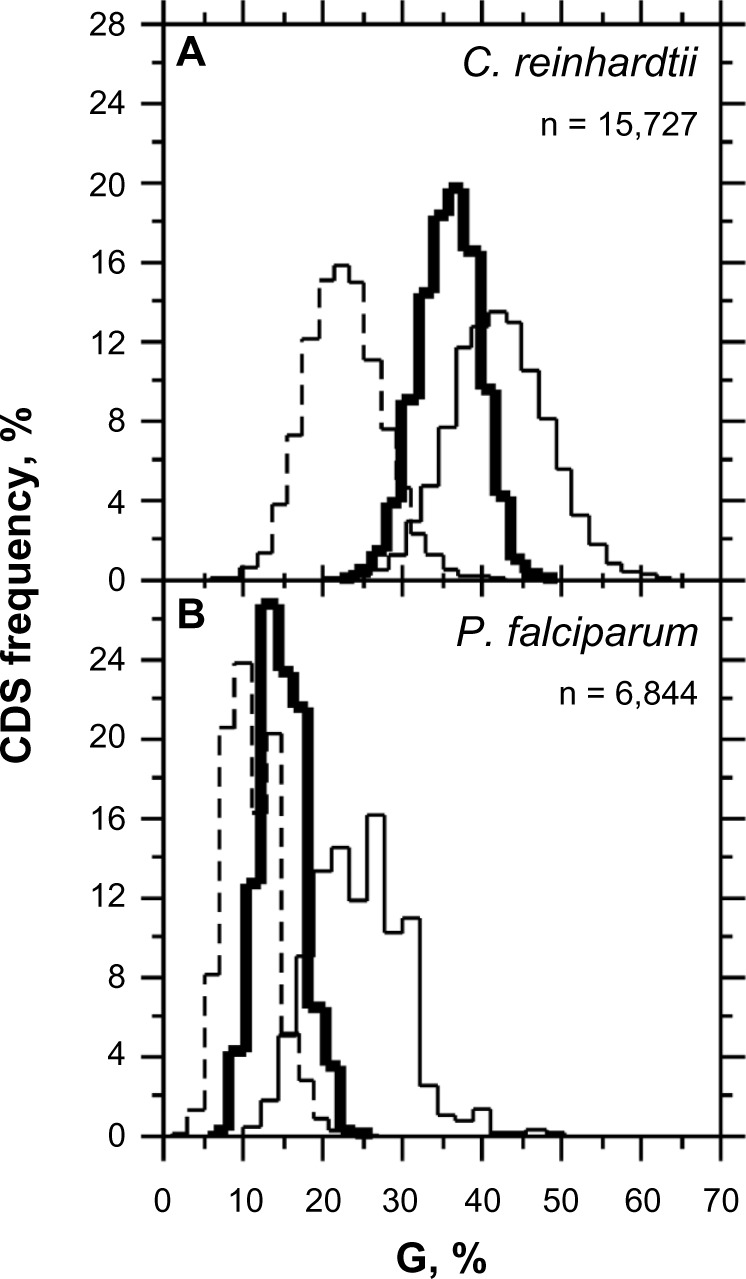
Distribution of CDSs according to G (average G1, G2, G3), G1, G2 in C. reinhardtii (A) and P. falciparum (B). Bold lines are for G, thin lines are for G1, and dash lines are for G2.
In this paper, we show that the ancestral codon was RWY (RWr) and not the commonly accepted pattern RNY (“W” is for “weak”, ie, A or T) because of short-range correlations. Here, we distinguish between the “Y” and “r” in the third codon position because codon asymmetry is induced by purine bias in such a way that R1 > R2, R1 > R3, G1 > G2, and G1 > G3. This finding has the consequence that pyrimidines most likely have a “compensatory” role, which is consistent with DNA structural constraints, and a wobble base at the first anti-codon position. We also show that the pattern of the ancestral codon is compatible with the synthesis of primeval proteins based on Miller’s41 amino acids list. In Miller’s list41 (reanalyzed by Johnson et al.42 and Parker et al.43), the most representative amino acids of the ancestral codon RWY are Asp, Val, and isoleucine (Ile). The amino acid composition in primeval proteins based on Miller’s list complies with the hydropathy and secondary structure of modern proteins, which is in line with ongoing pressures on protein functionality since their appearance on earth.
Materials and Methods
In this study, we focused on eukaryote CDSs; however, it would not make difference to sample prokaryote CDSs because the universal correlation for prokaryotes matches that of eukaryotes.39 This is the reason why this correlation can be called universal. One benefit of eukaryote CDSs is their larger sample size per genome, which allows a reduction in the number of genome to be analyzed to cover most of the universal correlation without loss of statistical significance. Thus, we used CDS datasets from 1) H. sapiens (GC3 = 30–90%)44 and O. sativa (GC3 = 30–100%),45 which have broad internal GC variations between GC-poor and GC-rich CDSs, and 2) P. falciparum (GC3 = 0–30%), which is GC-poor,46 and C. reinhardtii (GC3 = 60–100%), which is GC-rich47, the last two species having homogeneous genomes.
The human CDS sample was obtained from Fedorov’s group (n = 23,366),48,49 which is stored in the file hs37p1.EID.tar.gz (may be downloaded from http://www.utoledo.edu/med/depts/bioinfo/database.html). To link this CDS sample with experimental evidence, we compared the protein sequence homology of the CDSs of this sample with protein sequences in PDB (E ≤ 0.0001). Homologous hits were then filtered so that only the best hit was retained (n = 13,672) for each human accession. We then filtered the list, keeping pairs with identities ≥40%, and we used the accession identifiers to retrieve the corresponding DNA sequences from the original CDS file. Finally, trivial redundancy was eliminated by discarding sequences with identical values for size, GC1, GC2, and GC3 (or a difference of GC3 <2%). The final sample size was n = 10,892.
Complete nuclear CDSs from O. sativa (n = 89,665) and P. falciparum (n = 10,823) were retrieved from GenBank (release 194, February 15, 2013) using ACNUC50 with the filtering options “t = cds”, “no k = partial”, and “no o = mitochondrion” (plus “no o = chloroplast” in the case of O. sativa). The C. reinhardtii CDS samples (n = 19,526) were obtained from ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v9.0/Creinhardtii/annotation/Creinhardtii_236_cds.fa.gz.51 Similar to humans, we also compared the CDS samples of O. sativa, P. falciparum, and C. reinhardtii with proteins in the PDB using the same filtering process. For O. sativa, we obtained n = 8,643 CDSs as a final sample, but for P. falciparum and C. reinhardtii the number of CDSs was too small (nPf = 2,100 and nCr = 2,100) for proper statistical analysis. To obtain larger and unbiased CDS samples, we filtered all the CDS samples from P. falciparum and C. reinhardtii with UFM5 to remove conflicting CDSs with regard to strand allocation, CDSs out of coding frame, or CDSs including in-frame stop codons. The sizes of the CDS samples obtained by this procedure were nPf = 6,844 and nCr = 15,727 for P. falciparum and C. reinhardtii, respectively.
To calculate the relative frequencies of amino acids for each protein secondary structure (α-helix, β-sheet, and aperiodic, which we termed H, E, and A, respectively), we used a dataset from Ponce de Leon et al.6 This dataset was obtained from a set of 10,731 nonredundant proteins for which the three-dimensional structures have been experimentally determined. This set of proteins was selected from the RCSB Protein Data Bank (PDB, release 3.2). The average protein size for this dataset was 282 amino acids (σ = 154) accounting for 3,025,111 amino acids in total, with 948,410 for H (31%), 633,489 for E (21%), and 1,443,212 for A (48%). Based on the hypothesis that the distribution of amino acids for each secondary structure follows a similar proportion in modern and primeval proteins, we considered the relative frequencies of amino acids per secondary structure as an indication for their likelihood in primeval proteins. This hypothesis is based on the fact that the constraints on functional proteins do not change over time because there is no reason why the physicochemical characteristics of amino acids would have changed. Only the number of amino acids have increased over time; however, the amino acids that came later were more expensive to synthesize (in terms of energy), and for this reason the contribution of the amino acids in Miller’s list41 to modern proteins is supposed to be similar to that of the amino acids actually available at the time of primeval proteins.
All relative nucleotide frequencies were calculated as a ratio of a given occurrence to the number of contiguous triplet N = n/3. The correlations were obtained with the classical formula r = cov(X,Y)/σXσY and orthogonal regression lines as reported by Jolicoeur.52 The relative frequencies were obtained by direct counting from sequence files (FASTA format) using Perl scripts, and further calculations were performed in Excel. Histograms and plots were obtained in StatView and edited in Canvas 6.
Results
Under unbiased nucleotide distribution, one would expect a similar guanine frequency in the three codon positions as would match their average value for CDSs. However, in P. falciparum, G2 (G2 = 10.946, σG2 = 3.268) < G (G = 14.881, σG = 2.842) < G1 (G1 = 24.525, σG1 = 5.924) (Fig. 1A) in such a way that null hypotheses of G1 = G or G2 = G must be rejected according to Student's t-test (α = 0.05 and 0.01), which is also true in C. reinhardtii where G2 (G2 = 22.832, σG2 = 5.047) < G (G = 36.026, σG = 3.825) < G1 (G1 = 42.451, σG1 = 6.141) (Fig. 1B). For sake of completeness, let us note here for G calculation that G3 = 9.171 (σG3 = 2.746) in P. falciparum and G3 = 42.795 (σG3 = 5.993) in C. reinhardtii (data not shown). Thus, the purine bias is such that the G1 and G2 levels (%) are, respectively, higher and lower than expected in P. falciparum, which is AT-rich, as well as in C. reinhardtii, which is by contrast GC-rich (Fig. 1A, C, E) with the consequence that G1 > G2. G2 is negatively correlated with T2 (Fig. 2B, D, F); a negative correlation was also found between C2 and A2 (Table 1). The negative correlations between R2 and Y2 agree with the observation that AT2 is generally larger than GC2. In reality, the average GC2 level in C. reinhardtii is 53.70% (σ = 8.31), which indicates that the AT2 level is 46.30% (σ = 8.31); thus, this highly GC-rich eukaryote has GC2 ≈ AT2 ≈ 50% (Fig. 3). By contrast, the average GC2 level in P. falciparum is 25.51% (σ = 6.84), which indicates that the AT2 level is 74.49% (σ = 6.86), and according to the universal correlation, the average GC2 level in H. sapiens is 42.54% (σ = 6.62), which falls between that of P. falciparum and C. reinhardtii. Thus, one can reasonably draw the relationship between GC2 and AT2 for any biological species as W2 ≥ S2. Because A1 is positively correlated with A2 (Fig. 4A, D, G), A2 and T2 tend to increase together on average (Fig. 4B, E, H), and R1 > R2 is generally true in coding frames ≥300 bp.4 This situation occurs at the cost of C1 because A1 and C1 are negatively correlated (Table 1). A3 has a strong negative correlation with C3 (r < −0.9), and the second position is connected to the third via the negative correlation between A2 and C3 (Fig. 4C, F, I). Because A1 is positively correlated with A2 (Fig. 4A, D, G), A1 is also negatively correlated with C3 and A3 (Table 1).
Figure 2.
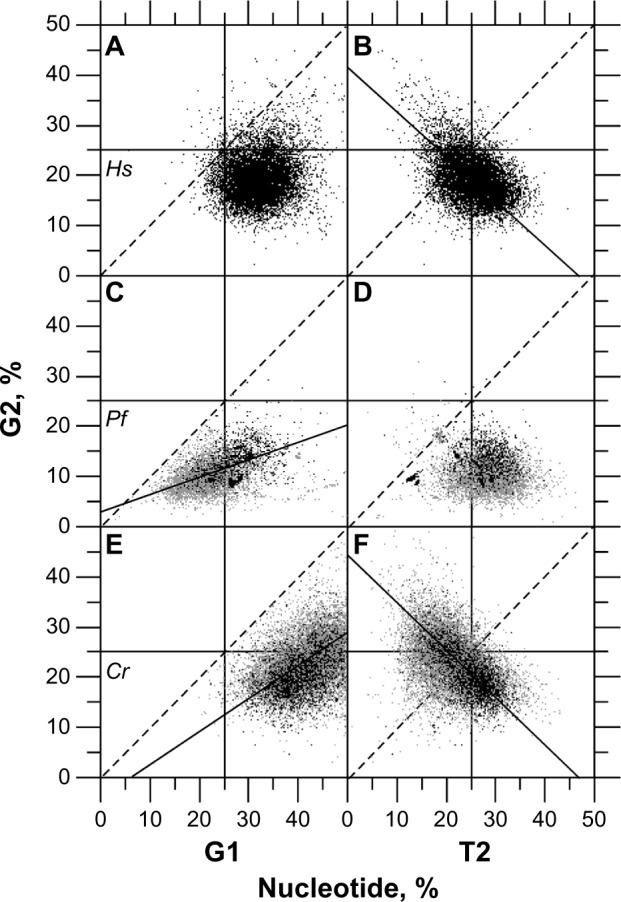
Correlations between G2 and G1 (panels A, C, E), G2 and T2 (panels B, D, F), in H. sapiens (Hs, n = 10,892, panels A, B), P. falciparum (Pf, n = 6,844, panels C, D), and C. reinhardtii (Cr, n = 15,727, panels E, F). r stands for the correlation coefficient and P for the statistical significance. Each r coefficient is associated with a P-value <0.001. Gray dots are for UFM-certified CDSs, and black dots are for CDSs homologous to proteins from PDB. (A) r = 0.12. (B) r = −0.43, y = −0.86x + 41.64. (C) rUFM = 0.43, rpdb = 0.41, y = 0.31x + 3.39. (D) rUFM = 0.09, rpdb = 0.19. (E) rUFM = 0.38, rpdb = −0.40, y = 0.61x −3.05. (F) rUFM = −0.48, rpdb = −0.53, y = −099x + 44.71.
Table 1.
Correlations (r) between nucleotide composition in the three positions of codons in H. sapiens (Hs, n = 10,892), O. sativa (Os, n = 8,643), and P. falciparum (n = 6,844) plus C. reinhardtii (n = 15,727) (Pf + Cr, n = 22,571). The gray boxes are for r ≥ +0.55 or r ≤ −0.55.
| SP | A1 | A2 | A3 | C1 | C2 | C3 | G1 | G2 | G3 | T1 | T2 | T3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hs | A1 | 1 | |||||||||||
| A2 | +0.57** | 1 | |||||||||||
| A3 | +0.58** | +0.44** | 1 | ||||||||||
| C1 | −0.72** | −0.46** | −0.58** | 1 | |||||||||
| C2 | −0.40** | −0.59** | −0.21** | +0.43** | 1 | ||||||||
| C3 | −0.52** | −0.48** | −0.92** | +0.55** | +0.28** | 1 | |||||||
| G1 | −0.43** | −0.12** | −0.20** | −0.05** | +0.12** | +0.12** | 1 | ||||||
| G2 | −0.44** | −0.50** | −0.31** | +0.31** | +0.11** | +0.35** | +0.12** | 1 | |||||
| G3 | −0.53** | −0.23** | −0.83** | +0.57** | +0.12** | +0.68** | +0.34** | +0.17** | 1 | ||||
| T1 | +0.19** | +0.03** | +0.28** | −0.37** | −0.20** | −0.21** | −0.56** | +0.04** | −0.50** | 1 | |||
| T2 | +0.14** | −0.13** | −0.03* | −0.16** | −0.41** | −0.02* | −0.07** | −0.43** | 0.00 | +0.13** | 1 | ||
| T3 | +0.52** | +0.34** | +0.84** | −0.59** | −0.22** | −0.88** | −0.24** | −0.25** | −0.88** | +0.42** | +0.06** | 1 | |
| Os | A1 | 1 | |||||||||||
| A2 | +0.56** | 1 | |||||||||||
| A3 | +0.50** | +0.41** | 1 | ||||||||||
| C1 | −0.53** | −0.26** | −0.30** | 1 | |||||||||
| C2 | −0.49** | −0.61** | −0.35** | +0.28** | 1 | ||||||||
| C3 | −0.48** | −0.44** | −0.91** | +0.30** | +0.36** | 1 | |||||||
| G1 | −0.61** | −0.35** | −0.46** | −0.09** | +0.30** | +0.41** | 1 | ||||||
| G2 | −0.34** | −0.47** | −0.27** | +0.15** | +0.02** | +0.29** | +0.24** | 1 | |||||
| G3 | −0.43** | −0.28** | −0.79** | +0.30** | +0.31** | +0.60** | +0.44** | +0.21** | 1 | ||||
| T1 | +0.12** | +0.04** | +0.35** | −0.27** | −0.06** | −0.28** | −0.53** | −0.04** | −0.41** | 1 | |||
| T2 | +0.23** | −0.02* | +0.18** | −0.14** | −0.43** | −0.16** | −0.15** | −0.44** | −0.24** | +0.08** | 1 | ||
| T3 | +0.50** | +0.42** | +0.88** | −0.34** | −0.38** | −0.91** | −0.44** | −0.28** | −0.82** | +0.36** | +0.22** | 1 | |
| Pf | A1 | 1 | |||||||||||
| + | A2 | +0.89** | 1 | ||||||||||
| Cr | A3 | +0.84** | +0.81** | 1 | |||||||||
| C1 | −0.89** | −0.80** | −0.84** | 1 | |||||||||
| C2 | −0.82** | −0.86** | −0.71** | +0.76** | 1 | ||||||||
| C3 | −0.77** | −0.79** | −0.94** | +0.78** | +0.69** | 1 | |||||||
| G1 | −0.89** | −0.78** | −0.74* * | +0.67** | +0.78** | +0.67** | 1 | ||||||
| G2 | −0.81** | −0.83** | −0.71** | +0.70** | +0.64** | +0.69** | +0.76** | 1 | |||||
| G3 | −0.88** | −0.80** | −0.93** | +0.87** | +0.72** | +0.80** | +0.80** | +0.70** | 1 | ||||
| T1 | +0.74** | +0.66** | +0.75** | −0.76** | −0.73** | −0.68** | −0.84** | −0.65** | −0.80** | 1 | |||
| T2 | +0.46** | +0.32** | +0.33** | −0.41** | −0.62** | −0.30** | −0.57** | −0.53** | −0.35** | +0.62** | 1 | ||
| T3 | +0.88** | +0.83** | +0.93** | −0.87** | −0.74** | −0.92** | −0.79** | −0.73** | −0.94** | +0.79** | +0.35** | 1 |
Notes: () for a value of r that is not statistically significant at α€ = €0.05. (*) for a value of r that is statistically significant at probability level α < 0.05. (**) for a value of r that is statistically significant at probability level α < 0.01.
Figure 3.
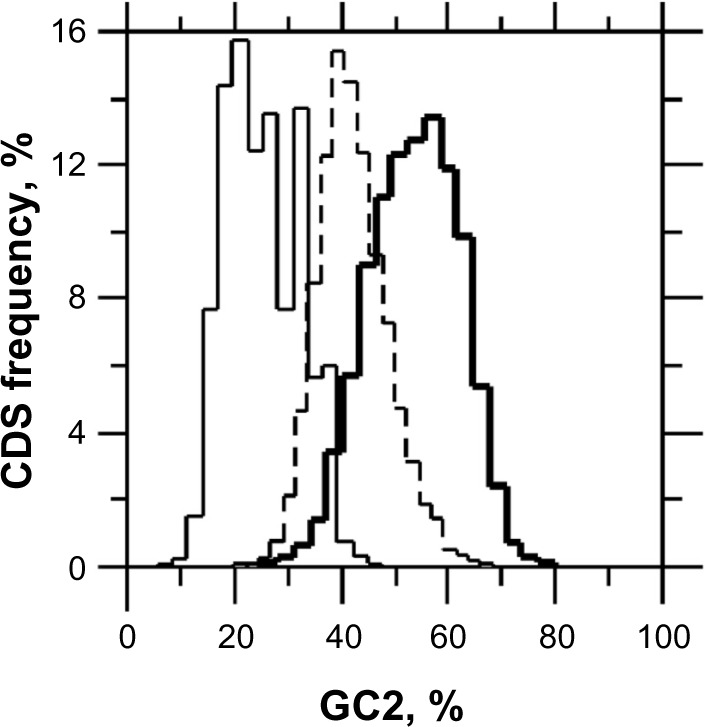
GC2 in P. falciparum (n = 6,844), H. sapiens (n = 10,892), and C. reinhardtii (n = 15,727). The thin line is for P. falciparum with an average GC2 of 25.51% (σ = 6.84), the dot line is for H. sapiens with an average GC2 of 42.54% (σ = 6.61), and the bold line is for C. reinhardtii with an average GC2 of 53.70% (σ = 8.31).
Figure 4.

Correlations between A2 and A1 (panels A, D, G), A2 and T2 (panels B, E, H), A2 and C3 (panels C, F, I) in H. sapiens (Hs, n = 10,892, panels A, B, C), P. falciparum (Pf, n = 6,844, panels D, E, F), and C. reinhardtii (Cr, n = 15,727, panels G, H, I). r stands for the correlation coefficient and P for the statistical significance. Each r coefficient is associated with a P-value <0.001. Gray dots are for UFM-certified CDSs, and black dots are for CDSs homologous to proteins from PDB. (A) r = 0.57, y = 1.16x + 0.70. (B) r = −0.13. (C) r = −0.48, y = −0.35x + 42.39. (D) rUFM = 0.49, rpdb = 0.49, y = 1.2x – 4.6. (E) rUFM = 0.43, rpdb = −0.57, y = −32.48x + 926.73. (F) rUFM = −0.05, rpdb = 0.25. (G) rUFM = 0.48, rpdb = 0.63, y = 1.4x – 2.5. (H) rUFM = 0.18, rpdb = 0.28. (I) rUFM = 0.07, rpdb = 0.19.
In homogeneous genomes (eg, P. falciparum, A. thaliana, D. melanogaster, and C. reinhardtii), nucleotide correlations within CDSs may not be detectable because of the small range of variation in nucleotide composition and the relatively large data noise. In contrast, in heterogeneous genomes (H. sapiens and O. sativa), these correlations may become apparent (Table 1) because heterogeneous genomes have genes with different codon usage. When comparing two genomes with different average GC levels, one observes a similar trend as found in heterogeneous genomes where greater differences in GC levels lead to higher correlation values and greater statistical significance. Table 1 shows that neat correlations are obtained in inter-genomic comparisons between P. falciparum and C. reinhardtii, and these correlations have the same sign as obtained in the intra-genomic context of H. sapiens and O. sativa. When comparing the correlation coefficients of H. sapiens and O. sativa, we found that 63 of 66 (95.5%) had the same sign, and the three sign discrepancies involved correlations (A2 vs T2, T1 vs G2, and T2 vs G3) with coefficients close to zero. When comparing P. falciparum plus C. reinhardtii with H. sapiens and O. sativa, we found 61/66 (92.4%) and 64/66 (97.0%), respectively. The sign discrepancies found were for the same reasons as outlined above for the H. sapiens versus O. sativa comparison (Table 1).
The exercise of comparing inter-genomic (P. falciparum plus C. reinhardtii) with intra-genomic correlations (H. sapiens or O. sativa) is allowed because of the existence of the universal correlation.39 Evidence for this statement appears in Figure 2 where the relative frequencies of nucleotides in CDSs from P. falciparum plus C. reinhardtii and H. sapiens are plotted together. Data from P. falciparum and C. reinhardtii are at the extremes of this relationship, while data from H. sapiens fall in between these two but remaining on the line joining these two extremes. Even in the relationship between A2 and T2, one can see that the H. sapiens data are between the P. falciparum and C. reinhardtii extremes. A similar A2 versus T2 relationship occurs for O. sativa, but the data are not shown in this study to avoid unnecessary redundancy. Interestingly, Table 1 also shows that T2 has lower correlation coefficients with other nucleotides in the different codon positions than the general trend of P. falciparum plus C. reinhardtii. This relationship is expected from the significant correlation of T2 with the physicochemical characteristics of proteins such as secondary structures and hydropathy, which indicates that T2 is primarily constrained by characteristics acting on proteins but not on DNA.6 On average, T2 is higher in P. falciparum (T2 = 41.64%, σT2 = 4.82) than in C. reinhardtii (T2 = 22.19%, σT2 = 5.08; Fig. 4E, H) with the consequence that P. falciparum proteins are more hydrophobic and contain more E’s, on average, compared with those in C. reinhardtii.
From the observations reported above, one can deduce that the ancestral codon RNY can indeed be written as RWY and should ideally match R(T|A)Y, ie, encode Asp, Val, and Ile. However, RWY is a statistical concept, which does not prohibit the existence of other codons in the sequence provided that RWY remains prominent. In actuality, if one considers that the amino acids available in prebiotic conditions are those in Table 3,41 one may note that an adequate proportion of all of the amino acids in this table may satisfy the ancestral codon RWY. The proportion of amino acids in secondary structures in modern proteins (Table 2) allows better inference of the amino acid distribution in primeval proteins. The identification of amino acids in Miller’s list in Table 2 (bold-italic) shows that, in most cases, they remain the most abundant amino acids in modern proteins (columns Ept, Hpt, Apt) and have a specific pattern of preference for specific secondary structures (columns Ess, Hss, Ass).
Table 3.
Codon usage of ancestral codons RWr in relation to amino acid (aa) availability in primeval terrestrial conditions, aa hydropathy, and secondary structure of modern proteins. In adequate proportion, all the aa of this table may satisfy the ancestral codon RWr; more specifically 1) the white background indicates codons that do not match the ancestral codon RWr, 2) the light gray background indicates codons that imperfectly match the ancestral codon RWr, and 3) the dark gray background indicates the aa that exactly match the ancestral codon RWr. Black rectangle are for values larger than 2.
| AA | MILLER1 (μM) | CARB. LAT.2 | HYDROP.3 | CODON | SPLIT | DEGENER. | E4 | H | A |
|---|---|---|---|---|---|---|---|---|---|
| Gly | 440.0 | 0 | −0.4 | GG(A|C|G|T) | Quartet | 1.03 | 1.05 | 5.44 | |
| Ala | 790.0 | 1 | 1.8 | GC(A|C|G|T) | Quartet | 1.35 | 3.77 | 3.24 | |
| Ser | 5.0 | 1 | −0.8 | TC(A|C|G|T) | AG(C|T) | Sextet | 0.99 | 1.39 | 3.43 |
| Thr | 0.8 | 2 | −0.7 | AC(A|C|G|T) | Quartet | 1.32 | 1.31 | 2.73 | |
| Asp | 34 | 2 | −3.5 | GA(C|T) | Duet | 0.68 | 1.48 | 3.69 | |
| Val | 19.5 | 3 | 4.2 | GT(A|C|G|T) | Quartet | 2.97 | 2.04 | 2.22 | |
| Glu | 7.7 | 3 | −3.5 | GA(A|G) | Duet | 0.99 | 2.98 | 3.00 | |
| Ile | 4.8 | 4 | 4.5 | AT(A |C|T) | Triplet | 2.19 | 1.96 | 1.72 | |
| Leu | 11.3 | 4 | 3.8 | CT(A|C|G|T) | TT(A|G) | Sextet | 2.22 | 3.83 | 3.15 |
| Pro | 1.5 | 6 | −1.6 | CA(A|C|G|T) | Quartet | 0.42 | 0.64 | 3.62 |
Table 2.
Distribution of amino acids (aa) in secondary structures of proteins, ie, β-sheet (E), α-helix (H), and aperiodic (A). The dataset of nonredundant proteins is from Ponce de Leon et al.6
| AA | EPT1 | HPT | APT | SUM2 | ESS3 | HSS | ASS | PAV4 |
|---|---|---|---|---|---|---|---|---|
| Ala5 | 1.35 | 3.77 | 3.24 | 8.36 | 6.44 | 12.04 | 6.79 | 8.36 |
| Cys | 0.33 | 0.31 | 0.54 | 1.18 | 1.58 | 1.00 | 1.13 | 1.18 |
| Asp | 0.68 | 1.48 | 3.69 | 5.85 | 3.24 | 4.73 | 7.74 | 5.85 |
| Glu | 0.99 | 2.98 | 3.00 | 6.97 | 4.72 | 9.50 | 6.29 | 6.97 |
| Phe | 1.20 | 1.23 | 1.55 | 3.99 | 5.73 | 3.93 | 3.26 | 3.99 |
| Gly | 1.03 | 1.05 | 5.44 | 7.52 | 4.94 | 3.34 | 11.41 | 7.52 |
| His | 0.47 | 0.62 | 1.19 | 2.28 | 2.26 | 1.97 | 2.50 | 2.28 |
| Ile | 2.19 | 1.96 | 1.72 | 5.86 | 10.45 | 6.24 | 3.60 | 5.86 |
| Lys | 0.94 | 2.10 | 2.82 | 5.86 | 4.48 | 6.71 | 5.92 | 5.86 |
| Leu | 2.22 | 3.83 | 3.15 | 9.20 | 10.60 | 12.21 | 6.61 | 9.20 |
| Met | 0.48 | 0.85 | 0.96 | 2.29 | 2.27 | 2.71 | 2.01 | 2.29 |
| Asn | 0.53 | 0.93 | 2.67 | 4.13 | 2.51 | 2.98 | 5.60 | 4.13 |
| Pro | 0.42 | 0.64 | 3.62 | 4.67 | 1.98 | 2.03 | 7.58 | 4.67 |
| Gln | 0.55 | 1.42 | 1.59 | 3.55 | 2.60 | 4.52 | 3.33 | 3.55 |
| Arg | 0.95 | 1.96 | 2.28 | 5.19 | 4.55 | 6.25 | 4.77 | 5.19 |
| Ser | 0.99 | 1.39 | 3.43 | 5.81 | 4.74 | 4.42 | 7.18 | 5.81 |
| Thr | 1.32 | 1.31 | 2.73 | 5.36 | 6.31 | 4.17 | 5.73 | 5.36 |
| Val | 2.97 | 2.04 | 2.22 | 7.23 | 14.18 | 6.51 | 4.65 | 7.23 |
| Trp | 0.34 | 0.44 | 0.51 | 1.29 | 1.64 | 1.39 | 1.06 | 1.29 |
| Tyr | 1.00 | 1.05 | 1.36 | 3.41 | 4.78 | 3.36 | 2.85 | 3.41 |
| Sum | 20.94 | 31.35 | 47.71 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
Notes:
In the columns with “pt” as subscript, the frequencies in the table are given relative (%) to the total number of aa (n = 3,025,111) in the protein samples (n = 10,731) analyzed. The dataset of nonredundant proteins is from Ponce de Leon et al.6
The sum is over the columns Ept, Hpt, Apt and gives the average amino acid per protein.
In the columns with “ss” as subscript, the frequencies in the table are given relative (%) to the number of aa per secondary structure.
Pav is for the average of columns Ess, Hss, Ass weighted with their average representativeness of these secondary structures in proteins (20.94, 31.35, 47.71, respectively) showing the consistency of the calculation.
Bold-italic amino acids indicate the amino acids from the Miller’s experiment (1992). The numbers on dark gray background are for values larger than 3 for Ept, Hpt, Apt and larger than 10 for Ess, Hss, Ass. The numbers on light gray background are for values in the range 2–3 for Ept, Hpt, Apt and in the range 5–10 for Ess, Hss, Ass. The numbers on white background are for values lower than 2 for Ept, Hpt, Apt and lower than 5 for Ess, Hss, Ass.
Interestingly, rescaling the frequency of the numbers in Table 3 to their sum (66.85) allows inferring a hypothetical contribution of secondary structures to primeval proteins. By summing columns E, H, and A after rescaling, we calculated 21.2%, 30.1%, and 48.2%, respectively, as the proportions of secondary structures, almost identical to those found in modern proteins. Similarly, rescaling Table 3 to the sum (47.16) of RNN codons (eliminating Ser and typical YNN codons such as Leu and Pro) led to a similar proportion of secondary structures as found in modern proteins, ie, 22.3%, 30.9%, and 46.7% for E, H, and A, respectively.
These considerations suggest that the RNN codons (Gly, Ala, Thr, Asp, Val, Glu, Ile) are sufficient to support the necessary catalytic activity and hydropathy of primeval proteins, with Asp, Val, and Ile being the most representative in the GWY ancestral codon. The amino acids encoded by these RNN codons are characterized by low complexity residues that are no larger than four bonds.
Discussion
With regard to the nucleotide correlations in CDSs, there are at least three important observations to consider: 1) mutations can be biased toward the accumulation of GCs (as in H. sapiens, D. melanogaster, O. sativa, and C. reinhardtii) or ATs (as in A. thaliana and P. falciparum); 2) the GC level may vary between 12% (P. falciparum) and 100% (O. sativa) in the third codon position based on mutation bias; and 3) the purine bias (Rrr) is universal to life and is a specific characteristic of CDSs regardless of the taxonomic position of a species.5,36,53 Related questions include the following: 1) How do the characteristics listed above manage to coexist? 2) What is responsible for the nucleotide correlations in CDSs when the genetic code, in principle, allows for random nucleotide distribution and large mutational bias?
The range of significant correlations among CDS nucleotides in homogeneous genomes is smaller than that for heterogeneous genomes. In fact, a homogeneous genome behaves as a compositional “point” on the universal correlation regression line. In comparison, heterogeneous genomes cover more than one point on the universal correlation regression line. For example, rice has two gene classes that correspond to two different codon usages within the same genome.54 The search for compositional correlations implies the need for variations in nucleotide composition large enough for correct testing of nucleotide interdependency. The nucleotide compositional correlations found between codon positions at the intra-genomic level in a heterogeneous genome can be lost when analyzing homogeneous genomes at the same compositional interval as that of heterogeneous genomes. In that case, inter-genomic nucleotide compositional correlations between homogeneous genomes can be substituted for intra-genomic correlations corresponding to the compositional interval of heterogeneous genomes. This exercise can be performed because translational machinery is conserved across all modern cellular life forms55 and because the protein code56 is something that is absolute in essence.57,58 The protein code is partially due to the periodicity of secondary structures, but it is also due to the periodicity of hydrophobic amino acids in proteins.59 P. falciparum (GC-poor) and C. reinhardtii (GC-rich) have genomes that exist at both extremities of GC variation among eukaryotes, while the H. sapiens genome occurs between these extremes. Given the universal correlation,39 these three genomes are sufficient for describing nucleotide correlations in eukaryote CDSs. The correspondence between indications for correlation in H. sapiens (heterogeneous genome) on one hand and P. falciparum plus C. reinhardtii on the other confirms the consistency of this reasoning. In reality, such a correspondence between indications for correlation cannot be obtained by chance because the species under comparison here are separated by at least one billion years of evolution from their common ancestor and show different characteristics.
As derived from purine bias, the nucleotide composition in CDSs is constrained by the codon position. Compensation for nucleotide constraint occurs in a network of correlations whose final product is purine bias regardless of the GC composition in the third codon position. Given that AT2 ≥ GC2, purine bias may be alternatively characterized by the logo RWY or even GWY because R1 > R2 and G1 > G2.
Because R1 > R2 and AT2 ≥ GC2 are not a consequence of the genetic code, they must be a consequence of the protein code,6,56 which shapes codon usage through tRNAs. The imprint of constraints on proteins in CDSs is RWr. “r” (or Y by symmetry) in the third codon position is more a consequence of A1 ≈ A2 (A1 is slightly lower than A2, on average) and G1 > G2 (G1 is strongly larger than G2, on average), which holds true for the first two positions of codons and compensation for A and G for mutation bias in the third codon position (permitted by tRNA wobble in first position of anti-codon).
In this context, the universal correlation could be observed as a compensation effect for the mutation bias toward AT or GC by T or C, respectively, to maintain RWY.6 This compensation is allowed by the fact that the coding information at the wobble position is “degenerate”. A discussion regarding selective processes potentially acting on DNA at that position is available from Bernardi.2
The amino acids that are encoded by RWY (Ala, Asp, Glu, Gly, Ile, Thr, Val) are compatible with 1) the constraints on protein functionality,7 2) the necessity of a hydrophobic core at the protein center, and 3) the necessity for secondary structures (Ε, H, A). In agreement with the ancestral codon hypothesis, Ala, Asp, Glu, Gly, and Val may have been alternatively used in GC-rich sequences and Ile and Thr may have been used in AT-rich sequences.
The fact that amino acids encoded by RWY have simple lateral chains and were synthesized by Miller31 in a pilot experiment for primordial earth conditions suggests that RWY is a relic of the prebiotic times at the origin of life. Therefore, the 10 amino acids missing from Miller’s list41 may have been unnecessary for producing functional primeval proteins (methionine and phenylalanine were later detected by Parker et al.43 (in 2011) and Johnson et al.42, respectively, in Miller’s extracts). These amino acids probably resulted as a byproduct of metabolism evolution.41 For example, arginine also has relatively high frequency in the secondary structure of Table 2, but it is neither in Miller’s list nor has the RWY pattern. The absence of arginine in the “prebiotic soup” may appear surprising, but is largely accepted60; neither was arginine synthesized through spark discharge nor was it found in meteorites29,61 or hydrothermal vent.29,62 As indicated by Oba et al.22 and McDonald and Storrie-Lombardi,60 the lack of basic amino acids does not prevent peptides or proteins from serving useful structural and biochemical functions. Arginine is encoded by six different codons, ie, SSN (including SSS) or ASR codons. In principle, SSS codons are compatible with an early incorporation of arginine to proteins. Actually, it is surprising to note that in SELEX experiments driven in different independent laboratories using different protocols, aptamers selected to bind arginine are predominantly composed of the modern arginine codons,63 which is reminiscent of the stereochemical hypothesis by Woese et al.64 and extended by Yarus.65 This finding, together with the finding that experiments simulating the early earth’s atmosphere32 yielded as many as 10 different natural amino acids still dominant in modern proteins,34 lays the foundation for a theory that the genetic code evolved under primordial conditions12,26 in two main steps (early and late) of amino acid incorporation to primeval proteins.21 The only reason not to consider arginine codons here is because arginine was likely not present in the prebiotic soup. However, as soon as it became present, it must have been quickly incorporated into the protein synthesis system. The free energy of formation of the amino acids from CO2, NH4+, and H2 in surface seawater at 18 °C and 1 atmosphere66 strongly correlated (r = 0.96) with experimental amino acid concentrations for the 10 early amino acids29 according to the series Gly > Ala > Asp > Glu > Val ~ Ser > Leu ~ Ile ~ Pro > Thr, in which arginine is not included. This finding is confirmed by the fact that class II aminoacyl-tRNA synthetases that match early amino acids do not include aminoacyl-tRNA synthetases for arginine33 and by the consideration that arginine requires several additional metabolic steps for synthesis not needed for early amino acids.21
Ikehara et al.7 proposed that the primitive GNC code encoded Gly, Ala, Asp, and Val. Miller’s experiment indicates that other amino acids with RNN codons (Glu, Ile, Ser, and Thr) together with Ala, Asp, Gly, and Val appeared in primeval earth conditions, satisfying hydropathy and the distribution of secondary structures, such as in modern proteins. The comparison of the Miller41 and Parker et al.43 amino acid doses in extracts from spark discharge samples show the same trend for Gly and Ala (in the range 0.5–1 mM), which are on average three orders of magnitude (four in the case of the dosage by Johnson et al.42) larger than the other amino acids. The concentrations of Gly and Ala were >1,000 times larger than the other amino acids on average, supporting the idea that GSC codons (GGC/GCC) came first15 followed by Val (GTC) and Asp (GAC) according to the primeval GNC code, which would have then evolved to RNY. Interestingly, Gly, Ala, Val, and Asp only need two pairs of complementary codons.16 As shown by Oba et al.22, [GADV]-peptides with catalytic activity could accumulate and participate in the multiplication of [GADV]-proteins by pseudo-replication in a process of repeated drying/heating cycles, thus without the need of an RNA-based translation system. Interestingly, modern peptides are synthesized by a complex and apparently universal protein machinery termed non-ribosomal peptide synthetase,67 whose proteins are more ancient than ribosomal proteins68 and do not involve RNA molecules. Primeval proteins might have possessed the catalytic activity to promote the formation of cyanide and purines from amino acids.69 The route of adenine formation from HCN in aqueous solution by physicochemical means is relatively simple,70 and it is thought to have contributed to the abiotic synthesis of RNA-like polymers via oligomer condensation to trigger the emergence of a replicating system based on RNA.71 At this stage, significant quantities of Gly, Ala, Asp, and Val might have recruited aptamers (tRNA precursors) associated with GNC codons through trial and error. With the other amino acids present in media, GNC codons must have quickly evolved to GNY and successively RNY with the decrease in temperature in the prebiotic environment.72 According to this view, the RNY ancestral codon appears as a fossil of GNC (a subset of RNY). Because the physicochemical constraints acting on the enzyme catalytic activity did not change over time, one may understand why RNY remains statistically significant and conserved in modern proteins. Thus, GC-rich sequences might have favored Ala, Asp, Glu, Gly, and Val, and the progressive increase in AT might have favored Thr and Ile. By consequence, selection could have driven this primeval code toward AT or GC through selective pressures on secondary structures according to the energy available for cell metabolism because Ile and Val are more favorable to E’s, Ala and Glu are more favorable to H’s, and Asp and Gly (and Ser) are more favorable to A’s. Metabolic abilities may have progressively increased thereafter with the codon-amino-acid repertoire.
The average T2 levels found in P. falciparum and C. reinhardtii suggest that mutational bias toward AT or GC might indeed provide a selective advantage according to the environment through the amount of energy available for cell metabolism. P. falciparum and C. reinhardtii are both unicellular eukaryotes that have to thrive in different environments. C. reinhardtii is a free-living green alga (housing a chloroplast) commonly found in soil and fresh water that can grow in simple media consisting of inorganic salts using photosynthesis to provide energy; however, it can also grow in total darkness by producing energy from the catabolism of acetate as a carbon source. With such metabolic ability to grow in environments with limited energy access, C. reinhardtii may have benefited from a mutational bias toward GC to diminish the average cost of protein synthesis by decreasing the E’s contribution to its proteins without affecting the metabolic functionalities. By contrast, P. falciparum is a protozoan parasite that thrives in an environment that is not characterized by limited energy access but extreme aggressiveness due to antibodies in the blood fluid and the acidity of the vacuolar environment of host cells. Thus, a higher energetic cost due to a larger proportion of E’s in proteins as a result of a mutational bias toward AT is not a problem for P. falciparum metabolism. E’s are secondary structures with larger variability and lower potential energy compared to H’s as can be deduced from the broad plateau in the Ramachandran plot73 to which they are associated (centered at φ = −120° and ψ = +135°). Here, we propose that E-rich proteins might offer adaptive benefits for P. falciparum biology74 in the context of a mutational bias toward AT due to a set of specific functions that they encode.75 The large complexity of H. sapiens likely justifies its use of the most protein diversity as deduced from the wide interval of T2 variations in Figure 2B.
It is also interesting to note here that RWY uses 7 (8 if counting Ser in the gray lines of Table 3) of the 10 Miller amino acids. The most abundant (Gly and Ala) in Miller’s experiment were possibly not the most abundant in primeval protein as suggested by the minimal code GWY and even by its generalized form RWY. In actuality, it is not surprising that the amino acid composition of proteins is not correlated to the amino acid concentration in the reaction medium of spark discharge experiments because there is no expected correlation between amino acid concentration in the medium and protein functionality; such a correlation would only address the question of how the genetic system effectively came into existence.
If GWY predated RWY, it is likely that it was for a short period because it does not appear to have any critical difference in the amino acid availability between the codes of GWY and RWY. With regard to amino acid precursor carriers (tRNA analogs), the situation might have been different because they might have needed a time interval for protein complexes with 3 or 4 amino acids to increase to 9 or 10 (see Trifonov16 for a discussion on the chronology of codon evolution). The time necessary to reach the final stage of 20 amino acids might have been greater because it might have needed an additional evolutionary step to increase metabolic complexity, ie, evolution toward a protein system that was likely encapsulated in a proto-cell.
Conclusions
We can conclude from the considerations above that purine bias likely existed from early times when proteins with functional activity were born. Purine bias is observed regardless of the coding DNA base composition; therefore, it is the link between functional constraints on proteins and the use of the genetic code by ribosomes for coding sequences to optimize their average processivity and accuracy. Because of the purine bias, the ancestral codon pattern is RWY and not RNY, which indicates that the amino acids Asp, Val, and Ile (GWY) were most likely essential for the formation of functional proteins under prebiotic conditions.
Acknowledgments
N. Carels thanks J. Pérez Mercader and the research team of Centro de Astrobiología (INTA, Madrid, Spain) for stimulating his interest concerning the origin of life.
Footnotes
ACADEMIC EDITOR: JT Efird, Associate Editor
FUNDING: This work was supported by a grant (INCT-IDN: 573642/2008–7) from Conselho de Desenvolvimento Científico e Tecnológico (CNPq, http://www.cnpq.br/). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: NC. Analyzed the data: NC. Wrote the first draft of the manuscript: NC. Contributed to the writing of the manuscript: NC, MPL. Agree with manuscript results and conclusions: NC, MPL. Jointly developed the structure and arguments for the paper: NC. Made critical revisions and approved final version: NC, MPL. Both authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Kudla G, Lipinski L, Caffin F, Helwak A, Zylicz M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006;4(e180):933–42. doi: 10.1371/journal.pbio.0040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernardi G. The neoselectionist theory of genome evolution. Proc Natl Acad Sci U S A. 2007;104:8385–90. doi: 10.1073/pnas.0701652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shepherd JC. Method to determine the reading frame of a protein from the purine/pyrimidine genome sequence and its possible evolutionary justification. Proc Natl Acad Sci U S A. 1981;78:1596–600. doi: 10.1073/pnas.78.3.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carels N, Vidal R, Frias D. Universal features for the classification of coding and non-coding DNA sequences. Bioinform Biol Insights. 2009;3:1–13. doi: 10.4137/bbi.s2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carels N, Frias D. A statistical method without training step for the classification of coding frame in transcriptome sequences. Bioinform Biol Insights. 2013;7:35–54. doi: 10.4137/BBI.S10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponce de Leon M, de Miranda A, Alvarez-Valin F, Carels N. The purine bias of coding sequences is determined by physicochemical constraints on proteins. Bioinform Biol Insights. 2014;8:93–108. doi: 10.4137/BBI.S13161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikehara K, Omori Y, Arai R, Hirose A. A novel theory on the origin of the genetic code: a GNC-SNS hypothesis. J Mol Evol. 2002;54:530–8. doi: 10.1007/s00239-001-0053-6. [DOI] [PubMed] [Google Scholar]
- 8.Gilbert W. Origin of life: the RNA world. Nature. 1986;319:618. [Google Scholar]
- 9.Wochner A, Attwater J, Coulson A, Holliger P. Ribozyme-catalyzed transcription of an active ribozyme. Science. 2011;332:209–12. doi: 10.1126/science.1200752. [DOI] [PubMed] [Google Scholar]
- 10.Wright MC, Joyce GF. Continuous in vitro evolution of catalytic function. Science. 1997;276:614–7. doi: 10.1126/science.276.5312.614. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu M. Molecular basis for the genetic code. J Mol Evol. 1982;18:297–303. doi: 10.1007/BF01733895. [DOI] [PubMed] [Google Scholar]
- 12.Orgel LE. Self-organizing biochemical cycles. Proc Natl Acad Sci U S A. 2000;97:12503–7. doi: 10.1073/pnas.220406697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eigen M, Schuster P. A principle of natural self-organization part A: emergence of the hypercycle. Naturwissenschaften. 1977;64:541–65. doi: 10.1007/BF00450633. [DOI] [PubMed] [Google Scholar]
- 14.Eigen M, Schuster PA. Principle of natural self-organization: part B: the abstract hypercycle. Naturwissenschaften. 1978;65:7–41. doi: 10.1007/BF00450633. [DOI] [PubMed] [Google Scholar]
- 15.Eigen M, Schuster PA. Principle of natural self-organization part C: the realistic hypercycle. Naturwissenschaften. 1978;65:341–69. doi: 10.1007/BF00450633. [DOI] [PubMed] [Google Scholar]
- 16.Trifonov EN. The triplet code from first principles. J Biomol Struct Dyn. 2004;22:1–11. doi: 10.1080/07391102.2004.10506975. [DOI] [PubMed] [Google Scholar]
- 17.Woese CR. On the evolution of the genetic code. Proc Natl Acad Sci U S A. 1965;54:1546–52. doi: 10.1073/pnas.54.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crick FH. The origin of the genetic code. J Mol Biol. 1968;38:367–79. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 19.Hartman H. Speculations on the evolution of the genetic code. Orig Life. 1975;6:423–7. doi: 10.1007/BF01130344. [DOI] [PubMed] [Google Scholar]
- 20.Hartman H. Speculations on the origin of the genetic code. J Mol Evol. 1995;40:541–4. doi: 10.1007/BF00166623. [DOI] [PubMed] [Google Scholar]
- 21.Hartman H, Smith TF. The evolution of the ribosome and the genetic code. Life. 2014;4:227–49. doi: 10.3390/life4020227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oba T, Fukushima J, Maruyama M, Iwamoto R, Ikehara K. Catalytic activities of [GADV]-peptides. Orig Life Evol Biosph. 2005;34:447–60. doi: 10.1007/s11084-005-3519-5. [DOI] [PubMed] [Google Scholar]
- 23.Di Giulio M. On the origin of the transfer RNA molecule. J Theor Biol. 1992;159:199–214. doi: 10.1016/s0022-5193(05)80702-7. [DOI] [PubMed] [Google Scholar]
- 24.Glaser R, Hodgen B, Farrelly D, McKee E. Adenine synthesis in interstellar space: mechanisms of prebiotic pyrimidine-ring formation of monocyclic HCN-pentamers. Astrobiology. 2007;7:455–70. doi: 10.1089/ast.2006.0112. [DOI] [PubMed] [Google Scholar]
- 25.Powner MW, Gerland B, Sutherland JD. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature. 2009;459:239–42. doi: 10.1038/nature08013. [DOI] [PubMed] [Google Scholar]
- 26.Wächtershäuser G. Before enzymes and templates: theory of surface metabolism. Microb Rev. 1988;52:452–84. doi: 10.1128/mr.52.4.452-484.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivatsan SG. Modeling prebiotic catalysis with nucleic acid-like polymers and its implications for the proposed RNA world. Pure Appl Chem. 2004;76:2085–99. [Google Scholar]
- 28.Benner SA, Kim H-J, Yang Z. Setting the stage: the history, chemistry, and geobiology behind RNA. Cold Spring Harb Perspect Biol. 2012;4:a003541. doi: 10.1101/cshperspect.a003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higgs PG, Pudritz RE. A thermodynamic basis for prebiotic amino acid synthesis and the nature of the first genetic code. Astrobiology. 2009;9:483–90. doi: 10.1089/ast.2008.0280. [DOI] [PubMed] [Google Scholar]
- 30.Oparin AI. The Origin of Life. Moscow: Moscow Worker Publisher; 1924. [Google Scholar]
- 31.Miller SL. A production of amino acids under possible primitive earth conditions. Science. 1953;117:528–9. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 32.Miller SL. Which organic compounds could have occurred on the prebiotic earth? Cold Spring Harb Symp Quant Biol. 1987;52:17–27. doi: 10.1101/sqb.1987.052.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Klipcan L, Safro M. Amino acid biogenesis, evolution of the genetic code and aminoacyl-tRNA synthetases. J Theor Biol. 2004;228:389–96. doi: 10.1016/j.jtbi.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 34.Wong JT, Cedergren R. Natural selection versus primitive gene structure as determinant of codon usage. Eur J Biochem. 1986;159:175–80. doi: 10.1111/j.1432-1033.1986.tb09849.x. [DOI] [PubMed] [Google Scholar]
- 35.Ikehara K, Niihara Y. Origin and evolutionary process of the genetic code. Curr Med Chem. 2007;14:3221–31. doi: 10.2174/092986707782793853. [DOI] [PubMed] [Google Scholar]
- 36.Brooks DJ, Fresco JR. Greater GNN pattern bias in sequence elements encoding conserved residues of ancient proteins may be an indicator of amino acid composition of early proteins. Gene. 2003;303:177–85. doi: 10.1016/s0378-1119(02)01176-9. [DOI] [PubMed] [Google Scholar]
- 37.Tiwary S, Ramchandran S, Bhattacharya A, Bhattacharya S, Ramaswamy R. Prediction of probably genes by Fourrier analysis of genomic sequences. Comput Appl Biosci. 1997;13:263–70. doi: 10.1093/bioinformatics/13.3.263. [DOI] [PubMed] [Google Scholar]
- 38.Grosse I, Herzel H, Buldyrev SV, Stanley HE. Species independence of mutual information in coding and non-coding DNA. Phys Rev E. 2000;61:5624–29. doi: 10.1103/physreve.61.5624. [DOI] [PubMed] [Google Scholar]
- 39.D’Onofrio G, Jabbari K, Musto H, Bernardi G. The correlation of protein hydropathy with the base composition of coding sequences. Gene. 1999;238:3–14. doi: 10.1016/s0378-1119(99)00257-7. [DOI] [PubMed] [Google Scholar]
- 40.Sueoka N. Correlation between base composition of the deoxyribonucleic acid and amino acid and composition of proteins. Proc Natl Acad Sci U S A. 1961;47:1141–9. doi: 10.1073/pnas.47.8.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller ST. The prebiotic synthesis of organic compounds as a step toward the origin of life. In: Schopf JW, editor. Major Events in the History of Life. Boston: Jones & Bartlett; 1992. pp. 1–28. [Google Scholar]
- 42.Johnson AP, Cleaves HJ, Dworkin JP, Glavin DP, Lazcano A, Bada JL. The Miller volcanic spark discharge experiment. Science. 2008;322:444. doi: 10.1126/science.1161527. [DOI] [PubMed] [Google Scholar]
- 43.Parker ET, Cleaves HJ, Dworkin JP, et al. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc Natl Acad Sci U S A. 2011;108:5526–31. doi: 10.1073/pnas.1019191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. doi: 10.1016/s0378-1119(99)00485-0. [DOI] [PubMed] [Google Scholar]
- 45.Carels N, Hatey P, Jabbari K, Bernardi G. Compositional properties of homologous coding sequences from plants. J Mol Evol. 1998;46:45–53. doi: 10.1007/pl00006282. [DOI] [PubMed] [Google Scholar]
- 46.Musto H, Rodriguez-Maseda H, Bernardi G. Compositional properties of nuclear genes from Plasmodium falciparum. Gene. 1995;152:127–32. doi: 10.1016/0378-1119(94)00708-z. [DOI] [PubMed] [Google Scholar]
- 47.Naya H, Romero H, Carels N, Zavala A, Musto H. Translational selection shapes codon usage in the GC-rich genome of Chlamydomonas reinhardtii. FEBS Lett. 2001;501:127–30. doi: 10.1016/s0014-5793(01)02644-8. [DOI] [PubMed] [Google Scholar]
- 48.Saxonov S, Daizadeh I, Fedorov A, Gilbert W. EID: the exon-intron database – an exhaustive database of protein-coding intron-containing genes. Nucleic Acids Res. 2000;28:185–90. doi: 10.1093/nar/28.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shepelev V, Fedorov A. Advances in the exon-intron database. Brief Bioinform. 2006;7:178–85. doi: 10.1093/bib/bbl003. [DOI] [PubMed] [Google Scholar]
- 50.Gouy M, Delmotte S. Remote access to ACNUC nucleotide and protein sequence databases at PBIL. Biochimie. 2008;90:555–62. doi: 10.1016/j.biochi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Merchant SS, Prochnik SE, Vallon O, et al. The Chlamydomonasgenome reveals the evolution of key animal and plant functions. Science. 2007;318(5848):245–50. doi: 10.1126/science.1143609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jolicoeur P. Bivariate allometry: interval estimation of the slopes of the ordinary and standardized normal major axes and structural relationship. J Theor Biol. 1990;144:275–85. [Google Scholar]
- 53.Carels N, Frias D. Classifying coding DNA with nucleotide statistics. Bioinform Biol Insights. 2009;3:141–154. doi: 10.4137/bbi.s3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carels N, Bernardi G. Two classes of genes in plants. Genetics. 2000;154:1819–25. doi: 10.1093/genetics/154.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolf YI, Koonin EV. On the origin of the translation system and the genetic code in the RNA world by means of natural selection, exaptation, and subfunctionalization. Biol Direct. 2007;2:14. doi: 10.1186/1745-6150-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biro JC. The proteomic code: a molecular recognition code for proteins. Theor Biol Med Model. 2007;4:1–44. doi: 10.1186/1742-4682-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berezovsky IN, Trifonov EN. Flowering buds of globular proteins: transpiring simplicity of protein organization. Comp Funct Genomics. 2002;3:525–34. doi: 10.1002/cfg.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frenkel ZM, Trifonov EN. From protein sequence space to elementary protein modules. Gene. 2008;408:64–71. doi: 10.1016/j.gene.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 59.West MW, Hecht MH. Binary patterning of polar and nonpolar amino acids in the sequences and structures of native proteins. Protein Sci. 1995;4:2032–9. doi: 10.1002/pro.5560041008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McDonald GD, Storrie-Lombardi MC. Biochemical constraints in a protobiotic earth devoid of basic amino acids: the “BAA(-) world”. Astrobiology. 2010;10:989–1000. doi: 10.1089/ast.2010.0484. [DOI] [PubMed] [Google Scholar]
- 61.Martins Z, Alexander CMOD, Orzechowska GE, Fogel ML, Ehrenfreund P. Indigenous amino acids in primitive CR meteorites. Meteorit Planet Sci. 2007;42:2125–36. [Google Scholar]
- 62.Hennet RJ-C, Holm NG, Engel MH. Abiotic synthesis of amino acids under hydrothermal conditions and the origin of life: a perpetual phenomenon? Naturwissenschaften. 1992;79:361–5. doi: 10.1007/BF01140180. [DOI] [PubMed] [Google Scholar]
- 63.Knight RD, Landweber LF. Rhyme or reason: RNA-arginine interactions and the genetic code. Chem Biol. 1998;5:R215–20. doi: 10.1016/s1074-5521(98)90001-1. [DOI] [PubMed] [Google Scholar]
- 64.Woese CR, Dugre DH, Dugre SA, Kondo M, Saxinger WC. On the fundamental nature and evolution of the genetic code. Cold Spring Harb Symp Quant Biol. 1966;31:723–36. doi: 10.1101/sqb.1966.031.01.093. [DOI] [PubMed] [Google Scholar]
- 65.Yarus M. Amino acids as RNA ligands: a direct-RNA-template theory for the code’s origin. J Mol Evol. 1998;47:109–17. doi: 10.1007/pl00006357. [DOI] [PubMed] [Google Scholar]
- 66.Amend JP, Shock EL. Energetics of amino acid synthesis in hydrothermal ecosystems. Science. 1998;281:1659–62. doi: 10.1126/science.281.5383.1659. [DOI] [PubMed] [Google Scholar]
- 67.Strieker M, Tanovic A, Marahiel MA. Nonribosomal peptide synthetases: structures and dynamics. Curr Opin Struct Biol. 2010;20:234–40. doi: 10.1016/j.sbi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 68.Bernhardt HS. The RNA world hypothesis: the worst theory of the early evolution of life (except for all the others) Biol Direct. 2012;7:23. doi: 10.1186/1745-6150-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGlynn SE, Beard TE, Broderick JB, Peters JW. Life’s origins: potential for radical mediated cyanide production on the early earth. J Cosmol. 2010;10:3315–24. [Google Scholar]
- 70.Roy D, Najafian K, von Ragué Schleyer P. Chemical evolution: the mechanism of the formation of adenine under prebiotic conditions. Proc Natl Acad Sci U S A. 2007;104:17272–77. doi: 10.1073/pnas.0708434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng LK, Unrau PJ. Closing the circle: replicating RNA with RNA. Cold Spring Harb Perspect Biol. 2012;4:a003566. doi: 10.1101/cshperspect.a002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Di Giulio M. The universal ancestor was a thermophile or a hyperthermophile. Gene. 2001;281:11–7. doi: 10.1016/s0378-1119(01)00781-8. [DOI] [PubMed] [Google Scholar]
- 73.Zhou AQ, O’Hern C, Regan L. Revisiting the Ramachandran plot from a new angle. Protein Sci. 2011;20:1166–71. doi: 10.1002/pro.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hildebrand F, Meyer A, Eyre-Walker A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010;6:e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nowick JS. Exploring β-sheet structure and interactions with chemical model systems. Acc Chem Res. 2008;41:1319–30. doi: 10.1021/ar800064f. [DOI] [PMC free article] [PubMed] [Google Scholar]