

Abstract
Overcoming intrinsic and acquired resistance of cancer stem/progenitor cells to current clinical treatments represents a major challenge in treating and curing the most aggressive and metastatic cancers. This review summarizes recent advances in our understanding of the cellular origin and molecular mechanisms at the basis of cancer initiation and progression as well as the heterogeneity of cancers arising from the malignant transformation of adult stem/progenitor cells. We describe the critical functions provided by several growth factor cascades, including epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), stem cell factor (SCF) receptor (KIT), hedgehog and Wnt/β -catenin signalling pathways that are frequently activated in cancer progenitor cells and are involved in their sustained growth, survival, invasion and drug resistance. Of therapeutic interest, we also discuss recent progress in the development of new drug combinations to treat the highly aggressive and metastatic cancers including refractory/relapsed leukaemias, melanoma and head and neck, brain, lung, breast, ovary, prostate, pancreas and gastrointestinal cancers which remain incurable in the clinics. The emphasis is on new therapeutic strategies consisting of molecular targeting of distinct oncogenic signalling elements activated in the cancer progenitor cells and their local microenvironment during cancer progression. These new targeted therapies should improve the efficacy of current therapeutic treatments against aggressive cancers, and thereby preventing disease relapse and enhancing patient survival.
Keywords: cancer progenitor cells, oncogenic signalling, endothelial progenitor cells, chemotherapeutic treatments, drug resistance mechanisms targeting therapies
Introduction
Major improvements in the treatment of cancer patients have been accomplished in the few last years. The development of more effective diagnostic and prognostic methods is leading to an earlier therapeutic intervention in the clinics [1–17]. Among the clinical treatments, tumour surgical ablation, hormonal therapy, radiotherapy and chemotherapy, alone or in combination, are the most currently used therapies for treating the patients diagnosed with diverse cancers including leukaemias and malignant solid tumours, such as skin, head and neck, brain, lung, kidney, bladder, prostate, breast, ovary, pancreas and gastrointestinal cancers [1–3, 5–9, 12–24]. In general, the patients diagnosed with localized cancers that are treated with these conventional clinical therapies have a high response rate and a good outcome. Although these conventional therapies are effective in initial phase of treatment, the cancer progression to locally invasive and metastatic states is often associated with resistance to treatments and disease relapse, which leads to the death of the patients [1–3, 5–7, 9, 12, 13, 15–17, 21–23, 25–31]. The cancer recurrence phenomenon has been associated with the accumulating genetic and/or epigenic alterations in cancer cells that may contribute to their uncontrolled growth, survival and invasion as well as their intrinsic or acquired resistance to clinical treatments [1–3, 13, 16, 26–28, 31–41]. The altered activation and/or overexpression of numerous growth factors, adenosine 5′ -triphosphate (ATP)-binding cassette (ABC) multidrug efflux transporters, anti-apoptotic factors (MYC, Bcl-2, NF-κ B and survivin) as well as a decreased expression or activity of tumour suppressor genes (p53 and phosphatase and tensin homolog, PTEN) in cancer cells may contribute to the drug resistance and disease relapse [1, 16, 26–28, 32–46].
Recently, new concepts have been proposed on the critical implication of highly leukaemic or tumourigenic cancer progenitor cells also designated as cancer stem cells or cancer-initiating cells, in cancer initiation and progression to metastatic disease states and resistance to conventional therapies [16, 35, 39, 40, 47–53]. These cancer progenitor cell-based concepts may partially explain the recurrence of the most aggressive cancers to current clinical treatments. More specifically, the reactivation of diverse developmental signalling cascades (epidermal growth factor (EGF)/EGFR, stem cell factor (SCF)/KIT, platelet-derived growth factor (PDGF)/PDGFR, sonic hedgehog (SHH/PTCH/GLI transcription factor) and/or Wnt/β -catenin) combined with the increased DNA repair mechanisms and ABC transporter-mediated multi-drug efflux in cancer progenitor cells may be responsible, at least in part, for their resistance to current clinical therapies [13, 16, 30, 31, 35–37, 39–42, 46, 54–61]. Moreover, the changes in the local microenvironment of cancer progenitor cells may also influence their behaviour. In this review, we discuss the importance of considering new concepts on the implication of cancer progenitor cells in cancer development in order to overcoming resistance to conventional cancer therapies. The emphasis is on the oncogenic events occurring frequently in cancer progenitor cells and their local microenvironment during cancer progression and the molecular mechanisms involved in their resistance to current chemotherapeutic drugs. Furthermore, we also discuss the beneficial effects of targeting different intracellular signal transduction pathways in cancer progenitor cells and their microenvironment for the development of more effective therapeutic treatments against the most aggressive and recurrent cancers.
Functions of cancer progenitor cells in the cancer initiation and progression
New model of carcinogenesis based on the cancer progenitor cells
Numerous investigations have provided evidence that the genetic and/or epigenic alterations occurring in the multi-potent tissue-specific adult stem cells and/or their early progenies may lead to their malignant transformation into cancer progenitor cells also designated as cancer stem cells or cancer-initiating cells (Fig. 1 and 2) [3, 13, 37, 39–41, 47–53, 55, 57, 60, 62–65]. A small population of undifferentiated- or poorly differentiated cancer progenitor cells, which possesses the stem cell-like properties including their self-renewal ability and capacity to give rise to the bulk mass of further differentiated malignant cells, appears to represent the principal cancer cells that are responsible for leukaemia or tumour formation [13, 39, 40, 47, 59, 63, 66–76]. Accumulating genetic and/or epigenic alterations in leukaemic or tumourigenic cancer progenitor cells occurring during cancer progression, and more particularly the acquisition of a migratory phenotype by tumour cells during epithelial-mesenchymal transition (EMT) program, may also confer to them the invasive properties that are essential for their migration to distant metastatic sites [39, 40, 49, 64, 72, 73, 77–81]. This cancer progenitor cell model of carcinogenesis is notably supported by the fact that the poorly differentiated and highly-leukaemic or tumourigenic cancer progenitor cells isolated from patients'malignant tissue specimens may give rise to the bulk mass of further differentiated cancer cells ex vitro and in vivo, and thereby be responsible for leukaemia or tumour development (Fig. 1; Table 1, 2) [47, 63, 66–70, 72, 73, 75, 76]. Recent investigations have also revealed that a very small sub-population of cancer progenitor cells with stem cell-like properties may be isolated from well-established cancer cell lines and maintained under an undifferentiated or poorly differentiated state during long years in culture, and this even after multiple passages in medium containing serum [47, 59, 65–69, 72, 73, 75, 76, 82–84]. In regard with this, we describe here the recent investigations that have led to the isolation of cancer progenitor cells from numerous cancer types and the establishment of their functional properties ex vitro and in animal models in vivo.
1.
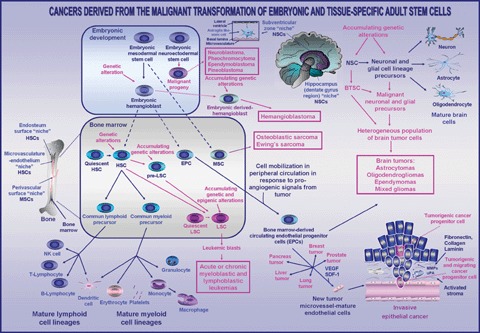
Scheme showing the critical functions assumed by cancer progenitor cells and the bone-marrow derived circulating cells in the development of leukaemias, sarcomas, brain tumours and various epithelial cancers. The accumulating genetic alterations in haematopoietic stem cells (HSCs) and/or lymphoid or myeloid precursors, which may lead to the development of leukaemias, are shown. Moreover, the genetic alteration in the embryonic hemangioblast or neuroectodermal stem cell-derived progeny, which may lead to their persistence in adult life and subsequent hemangioblastoma or neuroectodermal tumour formation, is also indicated. The genetic or epigenic alterations in neural stem cells (NSCs) and/or neuronal and glial cell lineage precursors, whose molecular events may result in their malignant transformation into brain tumour stem cells (BTSCs) and the generation of malignant neuronal and glial cell lineage precursors, are also shown. In addition, the implication of tissue-specific adult stem cells and reactive stromal host cells including the activated fibroblasts, immune cells and bone-marrow-derived endothelial progenitor cells (EPCs) in the tumour neovascularization is also illustrated. Abbreviations: LSC, leukaemic stem cell; MMPs, matrix metalloproteinases; MPS, mesodermal progenitor cells; MSC, mesenchymal stem cell;SDF-1, stromal cell-derived factor-1;uPA, urokinase type plasminogen-activator.
1.
Characterization of specific biomarkers and functional properties of human cancer progenitor cells isolated from diverse malignant tissues and cancer cell lines
| Malignant Tissue/ Established cancer cell line | Specific biomarkers/Stem cell-like properties |
|---|---|
| Hematological malignancy | |
| Acute myeloid leukemia (AML) | CD34+/CD38−, Thy 1−, KIT−, Leukaemic grafts in vivo |
| Multiple myeloma and RPMI 8226 and NCI-H929 myeloma cells | CD138−, Leukaemic grafts in vivo |
| Pediatric brain tumor | |
| Primary medulloblastoma, astrocytoma, glioblastoma multiforme and ependymoma | CD133+/nestin+, Neurospheres, Tumorigenic in vivo |
| Primary gangliogliom | CD133+/nestin+, Neurospheres |
| Adult brain tumor | |
| Primary glioblastoma multiforme | CD133+/nestin+, Neurospheres, Tumorigenic in vivo |
| Melanoma | |
| Metastatic melanoma | CD20+, Melanoma spheroids |
| Primary WM115 melanoma cell line | CD20+, Melanoma spheroids |
| Metastatic WM239A melanoma cell line | CD20+, Melanoma spheroids |
| Breast cancer | |
| Primary and metastatic breast cancers | CD44+/CD24−low, Oct-3/4, Mammospheres, Tumorigenic in vivo |
| MCF-7 breast cancer cell line | CD44+/CD24−low, Mammospheres, Tumorigenic in vivo |
| Ovarian cancer | |
| A2 clone from primary ovarian cancer | CD44+, Oct-3/4, Nanog, EGFR, Vimentin, E-cadherin Tumorigenic in vivo |
| A4-T spontaneously transformed clone | CD44+, Oct-3/4, Nanog, EGFR, Vimentin/Snail, Tumorigenic in vivo, A4-T > A2 clone |
| Prostatic cancer | |
| Primary and metastatic prostatic adenocarcinomas | CD133+/CD44+/α2β1-integrinhigh, Prostatespheres |
| LAPC-4, LAPC-9 and DU 145 prostatic cancer cell lines or xenografts | CD44high/SMO+/β-catenin, Tumorigenic in vivo |
| PC3 prostatic cancer cell line | CD44high/ CD133 |
| Pancreatic cancer | |
| Primary pancreatic adenocarcinoma | CD44+/CD24+/ESA+, Tumorigenic in vivo |
| PancTuI and A81 8–6 pancreatic cancer cell line | CD133+/ABCG2+ |
| Colorectal cancer | |
| Colorectal adenocarcinoma | CD133+, colon spheres, Tumorigenic in vivo |
| Head and neck cancer | |
| Head and neck squamous cell carcinoma | CD44+, Tumorigenic in vivo |
ABCG2/BCRP, breast cancer resistance protein;ESA, epithelial-specific antigen.
2.
Characterization of specific biomarkers and functional properties of Hoechst dye-low side population of cells isolated from diverse malignant tissues and cancer cell lines
| Malignant Tissue/Established cancer cell line | Specific biomarker/ Stem cell-like property |
|---|---|
| Leukemia | |
| Acute myeloidleukemia (AML) cells | CD34−/low |
| from bone marrow | Leukaemic grafts in vivo |
| Lung cancer | |
| Patients' non-small cell lung cancer tissues | |
| Human small-cell carcinoma NCI-H146 | |
| and NCL-H345 cell lines | |
| Human A549*, H460*, H23, HTB58, H441* | ABCG2, MDR1, MPR1, |
| and H-2170 lung carcinoma cell lines | Verapamil-, fumitremorgin C- or reserpine- |
| sensitive, *Tumorigenic in vivo | |
| Brain cancer | |
| Patients' primary neuroblastoma tissues | GD2 ganglioside, KIT+/CD71−/low/CD56−/low |
| CD133−/low, ABCG2, ABCA3 | |
| Human JF, SK-N-SH, IMR32, LAN-1, LAN-5 | ABCG2, ABCA3 (JF/IMR32) |
| and rat B104 neuroblastoma cell lines | |
| Verapamil-sensitive | |
| Human (D54, U87, U251, U373*, HS 683) | *Tumorigenic in vivo |
| glioma cell lines | |
| Rat (C6) glioma cell line | BCRP-1, MDR1, Tumorigenic in vivo |
| Melanoma | |
| Metastatic melanoma DM1N, DM2N, and | Nestin, gp100 |
| DM3N cell lines from lymph node metastases | Verapamil-sensitive |
| Breast cancer | |
| Patients' primary breast cancer tissues | |
| Human breast *MCF-7 and | ABCG2, Notch-1β, -catenin |
| SK-BR-3 cancer cell lines | *Verapamil sensitive*, Tumorigenic in vivo |
| Ovarian cancer | |
| Patients' primary ascite cells | ABCG2, Verapamil-sensitive |
| Human (IGROV-1, SK-OV-3, OVCAR-3, PA-1) | ABCG2/BCRP-1, Verapamil-sensitive*, Tumorigenic in vivo |
| and mouse (*MOVCAR 7 and 4306) | |
| ovarian cancer cell lines | |
| Prostatic cancer | |
| Human LAPC-9 cell-tumor xenograft | Tumorigenic in vivo |
| Human primary RWPE-2 and metastatic | Verapamil sensitive |
| DU145 and PC3 prostate cancer cell lines | |
| Gastrointestinal cancers | |
| Human *HuH7, Hep3B and PLC/PRF/5 | *GATA 6/CK14/CD133, *BMP2, *JAG1 |
| hepatocellular cancer cell lines | *ABCG2, *ABCB1, *CEACAM6, *AREG, |
| Human WiDr, CCK81, Colo201, Colo205, | |
| SW480 and HSC15 colorectal cancer cell lines, | |
| Human esophageal TE1, TE2 and TE13 and | |
| gastric NUGC3, MKNI, MKN7 and MKN28 | |
| cancer cell lines | |
| Pancreatic cancer | |
| Human CD18 pancreatic cancer cell line | Verapamil sensitive |
| Human PK9 and PK45H pancreatic cancer cell lines | |
| Retinal cancer | |
| Mouse retinoblastoma | ABCG2, Verapamil-sensitive |
| Human WERI-Rb27 retinoblastoma cell line | ABCG2, Verapamil-sensitive |
| Cervix cancer | |
| HeLa cancer cell line | |
| Thyroid cancer | |
| Human anaplastic (*ARO and FRO) and | Verapamil-sensitive, |
| papillary (NPA) thyroid cancer cell lines | *Tumorigenic in vivo |
| WRO follicular carcinoma cell line | |
| Head and neck sqamous cell carcinoma | |
| Human metastatic UMSCC10B and | Verapamil-sensitive |
| HN12 cell lines | |
| Nasopharyngeal carcinoma | |
| Human CNE-1, CNE-2*, SUNE-1, | CK19+, SMO+, Verapamil-sensitive |
| HONE-1 and C-666-1 cell lines | *Tumorigenic in vivo |
| Hepatocellular carcinoma | |
| Human *Huh7 and *PLC/PRF/5 | AFP+/CK19+,*ABCG1 and ABCF2 or |
| liver cancer cell lines | *ABCB2, ABCC7, ABCA5, ABCB1, |
| *Wnt ligands and FZD7, | |
| Verapamil-sensitive, Tumorigenic in vivo | |
BCRP-1/ABCG2, breast cancer resistance protein-1;BMP2, bone morphogenetic protein 2;CEACAM6, carcinoembryonic antigen-related cell adhesion molecule; MDR1, multidrug resistance 1 gene, MPR1, multidrug resistant associated protein 1.
Isolation and ex vitro and in vivo characterization of functional properties of cancer progenitor cells
Significant advancements have been made in the identification of the specific biomarkers of multi-potent tissue-specific adult stem cells. Researchers have been able to isolate these adult stem cells as well as their malignant counterparts, cancer progen-itor cells from total cell mass in cancer patients’ malignant tissue specimens and well-established cancer cell lines for their ex vitro and in vivo functional characterization (Table 1, 2) [47, 59, 65–69, 71–73, 75, 76, 82–89]. Among the methods that are frequently used for the enrichment and isolation of very small population of cancer progenitor cells with stem cell-like properties, there are the fluorescence-activated cell sorting (FACS), using the specific antibodies directed against one or several stem cell-like surface markers, such as CD34, CD138, CD20, CD133 and/or CD44 and the Hoechst dye efflux technique [47, 59, 65–69, 71–73, 75, 76, 82–89]. Hence, the isolated small sub-population of cancer progenitor cells may be subsequently expanded ex vivo in serum-free medium and further characterized by the non-adherent spheroid generation and clonogenicity assays for establishing their self-renewal and multi-lineage capacities in vitro. The implantation and serial transplantations assays may also be carried out with the isolated cancer progenitor cells in animal models for estimating their leukaemic or tumouri-genic potential and self-renewal ability in vivo [47, 59, 65–69, 71–73, 75, 76, 82–87]. More particularly, a very small sub-population of human cancer progenitor cells expressing the specific stem cell-like surface markers has been successfully isolated from malignant tissues and/or well-established cancer cell lines. Among the cancer types harbouring a sub-population of cancer progenitor cells, there are the acute myeloid leukaemia, multiple myeloma, melanoma, head and neck, brain, breast, ovary, prostate, pancreas and colorectal cancers (Table 1) [47, 66–69, 71–73, 75, 76, 82–87, 90–92]. It has been shown that all these cancer progenitor cells, which possess a self-renewal capacity, are able to give rise in vitro and/or in vivo to the bulk mass of further differentiated cancer cells that recapitulates the cellular heterogeneity and morphological characteristics of cancer tissues from which they originate [66–69, 71–73, 75, 76, 82–84, 86]. The fact that the engrafted leukaemic or tumourigenic cells could be serially transplanted into other mice has also provided further experimental evidence of the self-renewal capacity of these cancer-initiating cells [67–69,73,76]. Particularly, a small number of these poorly differentiated cancer progenitor cells showed a higher leukaemic or tumourigenic potential in animal models in vivo as compared to their further differentiated progenies [67–69, 72, 75, 76, 83, 84, 86, 87]. For instance, a sub-population of non-adherent melanoma spheroid cells expressing CD20+ antigen has been isolated from human metastatic melanoma tissues and established primary WM115 and metastatic WM239A melanoma cell lines derived from a same patient [70]. The multi-potent individual cells within these non-adherent melanoma spheres established from metastatic melanoma tissues were able to give rise to multiple mesenchymal cell lineages including melanocytes, adipocytes, osteocytes and chondrocytes ex vitro and in vivo (Table 1) [70]. These cells forming non-adherent melanoma spheres were also more tumourigenic than their adherent melanoma cell counterpart in severe combined immunodeficient (SCID) mice in vivo [70]. Similarly, a single clone (A2) expressing different markers, such as CD44, Oct-3/4, Nanog, EGFR, vimentin and E-cadherin and able to form the multi-layered spheroids ex vivo, has been isolated from the total cancer cell population of the ascites of a patient with advanced ovarian cancer [63]. Additionally, another clone A4-T derived from multi-layered spheroids that underwent a spontaneous transformation in culture has also been isolat-ed. A4-T was characterized by an expression marker profile comparable to that of A2 clone with the exception that it expressed a detectable level of the transcriptional repressor of E-cadherin, Snail but not E-cadherin suggesting that this clone may have to undergo a more complete EMT program [63]. Importantly, the A4-T clone was more potential than the A2 clone to form the tumours with an architecture resembling to the patients’ original tumours, and undergo metastases in nude mice in vivo (Table 1) [63]. On the other hand, by using a specific antibody directed against the embryonic stem cell-like marker Oct-3/4, which is a POU family transcription factor, combined with the analyses of its expression level by RT-PCR, it has been observed that the human breast, liver, pancreas, kidney, mesenchymal and gastric adult stem cells, and a few keratinocytes in the basal layer of human skin epidermis express significant levels of Oct-3/4 [93]. Moreover, the human, dog and rat tumours and HeLa and MCF-7 cancer cell lines also expressed a detectable level of its stem cell-related marker Oct-3/4, while the differentiated cells did not, supporting thereby the cancer progenitor cell concept of tumourigenesis [93].
In addition, the enrichment of undifferentiated or poorly differentiated cancer progenitor cells expressing the stemness genes from patient's cancer samples or well-established cancer cell lines has also been performed by the Hoechst dye efflux technique, which is particularly useful when the tissue-specific stem cell markers are not well established. Hence, a very small fraction of cancer cells designated as a ‘side population’ (SP), and which possesses a high ability to actively efflux the fluorescent DNA-binding dye, Hoechst 33342 due to elevated expression levels of ABC multi-drug efflux pumps, has been identified in several mammalian malignant tumour tissues and well-established cancer cell lines (Table 2) [54, 74, 75, 86–89, 94–105]. It has been observed that a small number of SP cells may generate both SP and non-SP cells in culture ex vivo or in vitro by asymmetric division and are able to induce the leukaemia-like disease or tumour formation in animal models in vivo resembling to the patients’ original cancers [54, 74–76, 80, 86–89, 94, 96–98, 101, 103]. For instance, the FACS sorted SP fraction from the C6 glioma cell line may differentiate in vitro and in vivo into neurons and glia expressing the neuronal (low molecular weight neurofilament [NF-L] or βIII-tubulin) and glial (glial fibrillary acidic protein, [GFAP]) markers, respectively, and form the metastatic tumours when injected intraperitonally in nude mice in vivo [96,97]. Importantly, it has been noted that the number of cancer cells detected in the SP population versus the non-SP fraction may be significantly influenced by the experimental conditions including the source of the patients'malignant tissues (untreated, treated and/or relapsed patients) and the culture conditions used (cell density and growth factors) [54, 97, 102, 105].
In light of these observations, it appears that several types of malignant tissues and well-established cancer cell lines contain a very small population of undifferentiated or poorly differentiated cancer progenitor cells that may be responsible for the generation of leukaemia or tumour formation in animal models in vivo. Hence, the most aggressive human cancers may originate from this small highly leukaemic or tumourigenic cancer progenitor cell sub-population possessing stem cell-like properties and an aberrant differentiating ability. Based on these observations, it appears important to consider the presence of these putative cancer progenitor cells in certain cancer cell lines which have been initially established from patients with leukaemia and malignant primary or secondary neoplasms. More particularly, this small sub-population of cancer progenitor cells isolated from patients'malignant tissue specimens or well-established cancer cell lines may represent a more appropriate experimental model for the basic cancer research and drug testing in vitro and in vivo.
Functions of cancer progenitor cells in the cancer development
Numerous recent investigations indicated that the most cancers may arise from the malignant transformation of multi-potent tissue-specific adult stem cells and/or their early progenitors into cancer progenitor cells. Furthermore, the accumulation of different genetic and/or epigenic alterations in cancer progenitor cells during cancer progression also seems to be associated with the occurrence of highly aggressive cancer subtypes. However, one of the major challenges now is to determine more precisely whether the cancers may originate in the majority of cases from accumulating oncogenic events occurring in the cancer progenitor cells that may lead to the generation of all the further differentiated cancer cells or whether the more committed and differentiated cell lineage precursors with the stem-cell-like properties could be responsible of the initiation of certain cancer subtypes. The available experimental lines of evidence appear to corroborate the possibility that these two processes are not exclusive and may be dependent on the cancer subtypes [69, 90, 106–113]. In this matter, the familial genetic alterations, such as germinal mutations may notably promote the incidence of certain cancers derived from the embryonic or adult stem cells [114–123]. For instance, it has been proposed that the deregulation of the hedgehog signalling pathway associated with the germinal mutations in PTCH receptor gene may lead to the developmental defects and a pre-disposition to develop certain tumours, such as basal cell carcinoma, medulloblastoma, meningioma, foetal rhabdomyoma as well as embryonal rhabdomyosarcoma, which appears to originate in myogenic satellite cells [118, 119]. Intriguingly, it has also been suggested that the transformed cells possessing the biomarkers and differentiating properties like embryonic hemangioblasts, which represent the putative common embryonic precursor to both haematopoietic and endothelial cell lineages (Fig. 1), may contribute to the development of certain malignancies in adult life [123, 124]. Among them, the ocular and central nervous system (CNS) hemangioblastomas, which are highly vascular tumours that may occur in patients with inherited von Hippel-Lindau (VHL) disease, have been proposed to likely originate from a haematopoi-etic/vascular cell lineage [123, 125]. More specifically, it has been proposed that the malignant cells that are at the origin of the hemangioblastoma formation within CNS in patients with VHL syndrome could represent the mesoderm-derived embryologically arrested hemangioblasts that have persisted in adults, and which may be further transformed under specific conditions (Fig. 1) [123]. In addition, the primitive neuroectodermal tumours (PNETs) including neuroblastoma, pheochromocytoma, ependymoblastoma and pineoblastoma may also originate during the embryonic development from neuroecto-dermal stem cells such as neural crest stem cells (Fig. 1) [10, 11, 121, 122, 126, 127]. For instance, it has been reported that the immature and tumouri-genic human neuroblastoma stem cells termed as intermediate type (I-type) cells isolated from neuroblastoma tissues or cell lines were able to give rise to the neuronal cells, melanocytes and Schwann cell precursors that constitute the tumours [128–130]. Although further genetic studies are necessary to confirm the possible implication of embryonic cells in the development of certain cancers, it is noteworthy that several cancer progenitor cells derived from adult stem cells also may re-express the markers associated with the more primitive pluripotent embryonic stem cells. In addition to these oncogenic changes occurring in cancer progenitor cells, the local tumour microenvironment may also influence their behaviour during the multiple steps of carcinogenesis.
Influence of local tumour microenvironment on the behaviour of cancer progenitor cells
The functional properties of cancer progenitor cells may be influenced through external signals mediated by other further differentiated cancer cells and host stromal cells including activated fibroblasts and infiltrating immune cells, such as macrophages and endothelial cells [131–134]. Among the diverse growth factors, chemokines and angiogenic substances released by stromal cells, there are matrix-degrading enzymes, matrix metalloproteinases (MMPs) and urokinase-type plasminogen activator (uPA), stromal cell-derived factor-1 (SDF-1) acting as CXC chemokine receptor 4 (CXCR4) ligand and vascular endothelial growth factor (VEGF) [132, 133, 135, 136]. All these soluble factors can influence, of autocrine or paracrine manner, the tumour cell behaviour and neovascularization process during cancer progression. In fact, the tumour and stromal cells may actively collaborate during each step of the tumour formation at primary and secondary sites in mediating the pro-angiogenic signals (VEGF and SDF-1) that contribute to the mobilization and recruitment of the bone-marrow (BM)-derived cells into peripheral circulation, and their subsequent migration to distant tumoural sites (Fig. 1) [134, 136]. Among the cells that may be attracted from BM and peripheral circulation into primary or secondary tumours, there are the circulating endothelial progenitor cells (EPCs) characterized by different biomarkers, including CD34+ or CD34−, CD133, VEGF receptor-2 (VEGFR-2), also designated as Flk-1 (foetal liver kinase-1), SCF receptor (KIT) and CXCR4. Moreover, the haematopoietic progenitor cells expressing VEGFR-1 (Flt-1, fms-like tyrosine kinase) also can participate to the new vessel formation within tumour [131, 133–135, 137–139]. More specifically,the multi-potent BM-derived EPCs possessing the stem cell-like properties may migrate into peripheral circulation and reach the tumoural sites where they can contribute to the tumour neovascularization process (Fig. 1). The circulating EPCs can promote the sprouting of pre-existent endothelial cells within tumours and/or participate in the formation of new microvasculature in differentiating into mature endothelial cells with the CD34+/CD133−/VEGFR-2+/KIT+/CXCR4+ phenotype that physically incorporate to new tumour microvasculature (Fig. 1) [131, 134, 137, 138, 140–142]. In regard with this, it has been observed that the BM-derived EPCs were recruited into the tumour site and incorporated in lymphoma, melanoma, brain, lung, liver, pancreas, breast and prostate tumour vessels in the mouse models in vivo (Fig. 1) [135, 142–144]. It has been observed that a small number of EPCs may be detected in circulation and within tumours in the patients diagnosed with Hodgkin's lymphoma and non-small cell lung cancer (NSCLC) as well as head and neck, thyroid, mucoepidermoid, osteogenic and breast carcinomas [138, 145]. Although it appears that the number of circulating EPCs that migrate to tumour sites and contribute to tumour vascularization is low, an enhanced number of EPCs in peripheral circulation in NSCLC patients has been associated with a weak response rate to antitumoural therapy and a poor clinical behaviour and overall survival [141]. Moreover, the possibility that a part of the EPCs that migrates to tumour sites can give rise to further differentiated endothelial cells within neo-plasms, and thereby may be undetectable, in peripheral circulation should also be considered. Further investigations by the engineering of specific modification in EPCs are therefore necessary to estimate more precisely the number of EPCs that can migrate to the tumour site as well as the amount of EPC-derived-endothelial cells that are incorporated in new tumour microvessels in diverse human cancer types. In addition, recent studies has also revealed that the EPCs including CD133+ EPCs with mesenchymal stem cell (MSC)-like characteristics and MSCs could also reside within the specific niches in certain normal human tissues [146–148]. Interestingly, the resident CD133+ renal progenitor cells, which are committed to differentiate into endothelial cells, were able to form the new vessels in the presence of tumour-derived growth factors, and thereby can promote the tumour growth established by co-transplanted renal carcinoma cells in SCID mice in vivo [148]. Moreover, the injection of BM-derived human Flk+/CD34− MSCs as well as the differentiated human endothelial cells into severe SCID mice engrafted with human malignant melanoma, resulted in their incorporation into new tumour vessels as confirmed by an immunohistochemical analysis of the walls of murine tumour vessels [149]. On the other hand, the circulating VEGFR-1 positive haematopoietic progenitor cells that migrate to tumour sites may release the growth factors that promote the neoangiogenic process, and thereby contribute to the stabilization of the microenvironment that favours the incorporation of EPC-derived endothelial cells in tumour microvasculature [134, 138, 139]. The changes within the local tumour microenvironment combined with different oncogenic events occurring in cancer progenitor cells during cancer progression may also lead to the occurrence of distinct cancer subtypes.
Cancer types originating from cancer progenitor cells
New concepts of the heterogeneity of cancers derived from distinct cancer progenitor cells
According to the hierarchical organization model of carcinogenesis, the leukaemic or tumourigenic cancer progenitor cells with unlimited potential for self-renewal and altered differentiating capacity are considered to be able to generate the total cancer cell mass of further differentiated progenies that constitutes the leukaemia or tumour [16, 39, 40, 48, 57, 65, 69, 112, 150–154]. Based on this cancer progenitor cell concept, it is likely that the differences between the phenotypic and functional properties of cancer progenitor cells from which a cancer originates may be at the basis of the heterogeneity of cancers [13, 16, 39, 40, 42, 49, 64, 73, 77, 155]. More specifically, the differentiating potential of cancer progenitor cells from which a cancer originates may corroborate the differentiation status of the resulting cancer sub-type. For instance, certain undifferentiated- to poorly-differentiated cancer subtypes could arise from the malignant transformation of primitive multi-potent stem/progenitor cells that acquire an aberrant differentiating potential resulting in a maturation arrest at an early stage of cell lineage differentiation [39, 152, 156]. In contrast, other moderately- to well-differentiated cancer subtypes could derive either from multi-potent cancer progenitor cells with a high differentiating capacity that are able to give rise to further differentiated cancer cells or more committed differentiated cancer progenitor cells [39, 106, 112, 113, 152, 156]. Furthermore, the differences between the specific oncogenic events activated in cancer progenitor cells, such as a gain of new oncogenic product expression and/or repression of tumour suppressor genes combined with the changes in their microenvironment during the progression from localized cancers into invasive forms may lead to the development of distinct cancer subtypes or be responsible for the intratumoural heterogeneity noticed for a certain cancer type [39, 40, 42, 64, 69, 72, 73, 77–81, 111, 157, 158]. More particularly, the acquisition of a more malignant behaviour by poorly- or moderately differentiated cancer progenitor cells during the EMT program including a migratory potential may result in highly aggressive cancer forms [39, 40, 42, 64, 72, 73, 77–81]. In support with these new concepts on the heterogeneity of cancers, several recent lines of evidence revealed that distinct poorly- to moderately differentiated cancer progenitor cells with different stem cell-like properties may be at the origin of cancer subtypes. In this regard, we describe here the recent observations supporting the heterogeneity of cancers derived from distinct cancer progenitor cells with a particular emphasis on the cellular origin of leukaemia, brain and breast cancer subtypes.
Implication of cancer progenitor cells in bone marrow-derived cancers
Leukaemias
Leukaemias are a type of cancer that arise in the blood-forming tissue, the BM and result in the production of a large number of immature blood cells termed as leukaemic blasts that accumulate in the BM and within the bloodstream, and which did not provide the functions of normal blood cells (Fig. 1). Several experimental lines of evidence suggested that the leukaemias, including acute myeloid/myelogenous leukaemia (AML), acute promyelocytic leukaemia (APL), chronic myeloid/myelogenous leukaemia (CML), acute lymphoblastic leukaemia (ALL) and chronic lymphocytic leukaemia (ALL) may originate from the malignant transformation of multi-potent haematopoietic stem cells (HSCs) or their early progenies in BM into leukaemic stem cells (LSCs) also designated as leukaemia-initiating cells (Fig. 1) [21, 22, 69, 90, 94, 95, 108, 109, 112, 154, 159, 160]. Among the frequent malignant transforming events occurring in HSCs or their more committed progenitors, there are the gene re-arrangements leading to the generation of chimeric fusion oncoproteins and aberrant activation of diverse haematopoietic growth factor and cytokine signalling cascades involved in the stringent regulation of self-renewal and/or differentiation of HSCs, such as hedgehog, Wnt/β -catenin, Notch, KIT and/or FMS-like tyrosine kinase 3 (FLT3) pathways [48, 58, 108, 109, 150, 161–165]. Hence, the generation of LSCs, which possess a self-renewal capacity and aberrant differentiating potential, can give rise to a heterogeneous population of leukaemic blasts containing different haematopoietic cell lineage precursors and further differentiated cells that vary with the leukaemia subtype (Fig. 1) [65, 90, 108, 112]. Especially, the accumulating genetic alterations including gene translocations and/or mutations in CD34− or CD34+/CD38−/LIN− primitive HSCs or their early progenitors may result in the cell sub-populations that display strong repopulating activity and leukaemic-initiating-capacity in non-obese diabetic (NOD)/SCID mice in vivo [90, 94, 166]. In this matter, since it has been observed that the primitive CD34 HSCs can give rise to CD34+ cells in animal models in vivo, it is therefore likely that these two HSC sub-populations may initiate certain types of leukaemia under specific conditions [94, 95, 167].
The specific chromosomal translocations generating different fusion oncoproteins, such as TEL-AML1, MLL-ENL, MN1-TEL and AML1-ETO and mutations in the genes encoding transcriptional factors such as Spi-1/PU.1 that are involved in regulation of the differentiation of haematopoietic progenitor cells may notably inhibit their differentiation into mature myeloid or lymphoid cells, and hence contribute to the induction and progression of certain AML or ALL subtypes [1, 153, 154, 168–172]. Among the initiating events associated with APL which is an AML subtype, there is a gene re-arrangement involving the retinoic acid receptor-α gene (RAR-α) with the promyelocytic leukaemia gene (PML), t(15;17) chromosomal translocation that generates the PML-RAR-fusion protein. PML-RAR-α oncoprotein may induce an arrest of granulopoiesis concomitant with the accumulation of immature granulocytes designated as promyelocytes [1]. Additional molecular oncogenic events that frequently occur during leukaemogenesis include the activating mutations in K- and N-Ras and the members of class III receptor tyrosine kinase family, such as FLT3, KIT and PDGFR as well as tumour suppressor gene p53 mutations and enhanced expression of Bcl-2 and Wilms tumour gene product (WT1) [1, 161, 170, 172]. These events may enhance the proliferation and/or inhibit the apoptosis in AML or ALL cells, and thereby promote the risk of relapse [1, 161, 170, 172]. Importantly, it has also been noted that the occurrence of one transforming event that leads to an arrest of the differentiation combined with another event inducing a proliferative effect in haematopoietic progenitor cells may co-operate to induce the more malignant leukaemiasubtypes [1, 170, 173].
Accumulating evidence has indicated that certain leukaemia subtypes could originate from additional genetic and/or epigenic alterations occurring in the intermediate haematopoietic progenitor cells, the pre-cancerous-LSCs (pre-LSCs) also designated as pre-cancerous stem cells (p-CSCs) (Fig. 1) [106, 112, 113]. The pre-LSCs, which may derive from more primitive HSCs, could possess a more limited self-renewal and multi-potent differentiating ability. In support with this, it has been noted that the CD34+/CD38−/LIN− LSCs in the most AMLs did not express the Thy-1 surface marker like normal Thy-1+ HSCs, suggesting that these leukaemia types could be accompanied either by the mutations leading to the down-regulation of its expression during the malignant transformation of primitive Thy-1+ HSCs, or derived from more committed leukaemic progenitor cells (Fig. 1) [90, 106, 112]. For instance, the analyses of BM cells from AML patients showing long-term remission after chemotherapeutic treatment, alone or combined with autologous mobilized blood transplantation (MBT), have revealed the persistence of AML-ETO chromosomal translocation in Thy-1+ HSCs [106]. This suggests that this genetic alteration may be insufficient to cause the cancer initiation. In contrast, the same patient harboured a population of Thy-1− LSCs at diagnosis corroborating the fact that this cancer subtype may have arisen from the additional oncogenic events in more differentiated leukaemic progenitor cells [106]. The single clones of p-CSCs (pre-LSCs) with CD45−/KIT−/Sca-1−/LIN−/CD44high phenotype, which have been established from the spleen of a mouse with dendritic cell-like leukaemia, also retained an incomplete multi-potency and can differentiate into non-malignant or malignant cells depending on their microenvironment [113]. The malignant transformation of pre-LSCs was associated with the up-regulation of KIT and Sca-1, suggesting that pre-LSCs may represent a cell population with an intermediate phenotype between the normal HSCs and LSCs (Fig. 1) [113]. Additionally, further oncogenic events in LSCs during cancer progression, as observed for the cancer progenitor cells during EMT in solid tumours, may also be accompanied by the acquisition of more malignant phenotypes [174]. For instance, during the initial chronic phase of CML, a t(9;22) BCR-ABL chromosomal translocation resulting in a shortened chromosome 22, designated the Philadelphia (Ph+) chromosome occurs in the HSCs and generates the chimeric BCR-ABL fusion oncoprotein with an enhanced constitutive tyrosine kinase activity in CML-LSCs [108, 175]. Moreover, the initial phase of CML may be accompanied by the overexpression of Bcl-2 and/or decreased Jun B expression [108, 175]. These molecular events may enhance the rate of growth factor-independent proliferation and reduce apoptotic signals in haematopoietic progenitor cells [174]. This initial chronic phase is followed by an accelerated phase driven by the occurrence of further oncogenic transformation of LSCs leading ultimately to the Ph+ B cell-ALL and the terminal stage of the disease designated as blast-crisis (Bc)-CML [108, 164, 175]. More specifically, the late-stage Bc-CML is accompanied by an expansion of granulo-cyte-macrophage progenitors (GMPs) expressing an enhanced level of β-catenin whose oncogenic event appears to be associated with an enhanced self-renewal capacity and leukaemic potential of human GMPs [108, 109]. On the other hand, it has also been observed that the BCR-ABL transduction in mouse committed myeloid progenitors does not induce their malignant transformation in vitro whereas MOZ-TIF2 or MLL-ENL fusion proteins were transforming for both mouse HSCs and their more committed myeloid progenitors [107, 108, 110]. BCR-ABL-transduced mouse committed myeloid progenitors also did not generate an AML in animal model in vivo while MOZ-TIF2 or MLL-ENL-transduced-mouse myeloid progenitors were able to give rise to an AML in irradiated mouse in vivo after serial transplantations [107, 108, 110]. This suggests then that only certain types of genetic alterations may contribute to the malignant transformation of HSCs and/or the more committed haematopoietic cells during leukaemia initiation and progression.
Despite the initial implication of the primitive HSCs and/or their early progeny (pre-LSCs) in the occurrence of the most the leukaemias, these observations suggest that the loss of the multi-potent or bipotent differentiating potential of LSCs may occur at an early or later stage of the disease depending on the oncogenic events occurring in the malignant cells during cancer development. Particularly, the fact that certain more committed haematopoietic cells may acquire a more malignant phenotype, including an enhanced self-renewal capacity during the cancer progression, underlines the importance of also considering their implication for a more appropriate therapeutic intervention. Further investigations on the genetic/epigenic alterations occurring in primitive human HSCs and their early progenies versus the more committed progenitor cells should establish more precisely the molecular oncogenic events associated with each particular case of leukaemias.
Sarcomas
Adult BM also constitutes a homing site for several other types of non-haematopoietic multi-potent stem cells including EPCs and mesenchymal stem/progenitor cells (MSCs also designated MPCs) which provide the specific functions (Fig. 1) [176]. For instance, MSCs can give rise to diverse mature cell lineages, such as adipocytes, chondrocytes and osteocytes. Several lines of evidence have revealed that the bone sarcomas, which are among the most aggressive mesenchymal malignancies in childhood and young adults, may originate from the malignant transformation of primitive stem cell-like MSCs [114, 115]. More specifically, the osteoblastic sarcoma, which is the most common type of primary bone cancer, may result in distant metastases to lungs [114, 115]. The germinal mutations in retinoblastoma (Rb) and p53 tumour suppressor genes may notably promote the incidence of osteoblastic sarcoma [114–117]. Since MSCs are the bone-forming osteogenic progenitor cells, they may then represent the putative BM adult stem cells that may be at the origin of osteoblastic sarcoma initiation. In regard with this, it has been reported that a small population of bone sarcoma cells that are able to form the sarcospheres in culture expressed several markers of MSCs (Stro-1, CD105 and CD4) as well as the pluripotent embryonic cells (Oct-3/4 and Nanog) [177]. This supports the potential implication of MSCs in bone sarcoma development. More recently, it has been observed that the cultured murine MSC co-infused with BM into irradiated allogeneic host could drive the development of foci of sarcoma in the lungs and extremities in certain mice in vivo [178]. The infusion of MSC-derived sarcoma cells having a cytogenetic profile comparable to human sarcoma also resulted in the malignant lesions in secondary recipients [178]. Additionally, Ewing's sarcoma, which is a member of Ewing's family tumours (EFTs), and the second most common solid bone and soft tissue malignancy of childhood and young adults, also appears to originate from MSCs [179]. The EFTs are generally associated with the occurrence of a chromosomal translocation that involves the fusion of a 5′ segment of the EWS gene with a 3′ segment of the ETS transcription factor family gene FLI-1, given the EWS-FLI-1 fusion protein. EWS-FLI-1 oncoprotein acts as an aberrant transcriptional activator and contributes to tumour development. In support with this, it has been shown that EWS-FLI-1 can transform the primary BM MSCs and generate tumours in animal models in vivo showing the phenotypic characteristics that resemble Ewing's sarcoma [179].
Implication of cancer progenitor cells in paediatric and adult brain tumours
Primary malignant CNS tumours are the most frequent forms of solid malignant tumours diagnosed in children and the glioblastoma multiformes (GBMs) represent one of the more frequent aggressive tumour types in adults [7, 11, 15, 29]. The cellular origin of paediatric and adult brain tumour types has not yet been precisely established. The paediatric brain tumours constitute a diverse group in terms of their localization, prognosis and response to various therapeutic treatments relative to adult brain tumours (Fig. 1) [10, 11, 29, 126, 127]. Gliomas, which are the most common malignant primary brain tumours, include astrocytomas (grade I slow-growing pilocytic astrocytomas, grade II astrocytomas, grade III anaplastic astrocytomas and grade IV GBMs), oligo-dendrogliomas, ependymomas and mixed gliomas, which may contain the astrocytes, oligodendrocytes and/or ependymal cell-like cells in different proportions (Fig. 1) [7,15]. Several recent lines of evidence support the concept that certain brain cancers including medulloblastomas, astrocytomas, GBMs, oligodendrogliomas and ependymomas could derive from the malignant transformation of neural stem cells (NSCs) or more restricted-neuronal and glial cell lineage precursors into brain cancer progenitor cells also designated as brain tumour stem cells (BTSCs) [15, 41, 52, 66, 67, 72, 73, 82, 180–182]. BTSCs could possess a self-renewal capacity but showed an abnormal ability to differentiate (Fig. 1). More specifically, the infratentorial medulloblastomas, which are the most common brain tumours diagnosed in childhood, may originate either from NSCs within the ventricular zone germinal layer or neuronal progenitors designated as cerebellar granule cell precursors (GCPs) located in the external granular layer within the cerebellum (Fig. 1) [37, 127, 157, 158]. Aberrant activation of SHH pathway (SHH/PTCH/GLI), which is recognized to triggering normal growth of the cerebellum in the foetus, induced through the mutations in patched receptor (PTCH), smoothened (SMO) or Sfu, may notably contribute to uncontrolled growth of GCPs in childhood and young children, and thereby induce the medulloblastoma formation [37, 41, 62, 111, 127, 157,158, 183]. The p53 inactivation or sustained activation of Wnt/β -catenin pathway may also result in an increased expression of downstream oncogenic gene products that contribute to development of certain human medulloblastoma subtypes [41, 111, 157, 158]. In addition, the data from a recent study have revealed that the pilocytic astrocytomas and ependymomas arising in the supratentorial (cortex and sub-cortex) versus infratentorial (cerebellum and brainstem) regions of the brain show specific genetic signatures, which were also detected in NSCs localized in these regions [184]. This suggests then that these brain tumours may derive from the malignant transformation of a common precursor that possesses different gene-expression patterns depending on its brain localization. Interestingly the tumourigenic CD133+/nestin+ BTSCs isolated from ependymomas display a phenotype resembling to radial glia-like cells, which are the neuronal precursor cells that may give rise to mature ependymal cells lining membrane of brain ventricles (Fig. 1) [92]. This observation supports the fact that certain ependymoma subtypes may derive from the malignant transformation of radial glia-like cells. Similarly, the secondary or progressive GBMs may also arise from the malignant transformation of NSCs into BTSCs that give rise to a heterogeneous population of further differentiated cancer cells (Fig. 1) [41, 64]. The secondary GBMs, which develop slowly from low-grade tumours, are often accompanied by the p53 tumour suppressor gene mutations. In this matter, it has been observed that the inactivation both of p53 and neurofibromatosis type 1 (NF1), which is a tumour suppressor gene that is involved in negative regulation of Ras pathway, may co-operate to induce the development of malignant astrocytomas from early pre-symptomatic lesions within sub-ventricular zone containing NSCs in animal model in vivo [185]. In contrast, other subgroup of aggressive primary GBM tumours, which progress rapidly without evidence of a transitory step of lower grade tumour, are frequently accompanied by the overexpression of EGFR and acquisition of mesenchymal properties by cancer progenitor cells [41, 64, 72, 73]. The results from a recent study have also revealed that an inhibition of the nuclear co-repressor (N-CoR) of astroglial differentiation may contribute to de novo GBM development in mice suggesting that this molecular event may occur in certain GBM tumours in human [182].
In addition, the isolation of CD133/nestin positive-tumour cells from patients’ malignant tissues has revealed that these primitive cancer progenitor cells can give rise in vitro and in vivo to different neural cell lineages, including neuron and glial cell-like cells (astrocytes, oligodendrocytes and ependymal cells), but in different proportion in respect to the normal NSCs [15, 41, 52, 66, 67, 72, 73, 82]. For instance, it has been observed that the neurospheres established from the paediatric medulloblastomas, astrocytomas and GBMs were able to differentiate majoritarly ex vitro and in vivo into cells expressing the neural markers of original tumour cell phenotype, including the neuronal (β-III tubulin) and astrocytic GFAP markers [66, 67, 82]. Similarly, the multi-potent neurosphere-like clusters isolated from adult primary GBMs can also give rise to further differentiated progenitors expressing the specific markers to three neural cell lineages including neurons (β-III tubulin), astrocytes (GFAP) and/or oligodendrocytes (myelin or galactocerebroside) ex vitro and in vivo resembling to the cellular heterogeneity characterizing the initial GBMs [72, 73]. An immunohistochemical analysis of brain tissue specimens from astrocytomas of different grades has also revealed that the number of tumour cells expressing a NSC-like phenotype (doublecortin, β-III-tubulin, collapsing response-mediated protein-4 [CRMP-4/TUC-4] and nestin) and proliferation marker (Ki67 antigen), was increased in high-grade astrocytomas including GBMs relative to low-grade neoplasms [186]. This suggests then that a high proliferative capacity of BTSCs could be related with the occurrence of highly aggressive tumours. In regard with this, it has been observed that the co-injection of primary human endothelial cells with CD133+ BTSCs from primary medulloblastomas in the cerebral cortex of immuno-compromised mice, markedly increased the tumour growth derived from CD133+ cells in vivo (Fig. 1) [187]. Based on this observation, it has been proposed that the perivascular niche of self-renewing BTSCs could assume an important role for their expansion during cancer development [187]. In addition, the results from a recent study revealed that certain primary glioblastoma subtypes may also derived from a small sub-population of CD133− tumour cells with stem cell-like properties and showing a distinct gene expression pattern relative to CD133+ cancer stem/progenitor cells [188]. Hence, since these observations suggest that different cancer stem/progenitor cells may be at origin from certain brain cancer subtypes, further investigations are then necessary to establish more precisely their specific properties and sensitivity to therapeutic treatments.
Implication of cancer progenitor cells in other cancer types
The isolation of cancer progenitor cells with the stem cell-like properties from diverse solid tumour specimens and cancer cell lines also supports the fact that melanoma, skin, head and neck, thyroid, cervix, lung, liver, breast, ovary, prostate, pancreas, gastrointestinal and retinal cancers may arise from the malignant transformation of tissue-specific adult stem cells and/or their early progenies (Table 1, 2) [39, 40,47, 49, 50, 54, 59, 61, 65, 68, 70, 71, 75, 76, 80, 83–87, 96, 98–105, 189]. Particularly, the re-activation of distinct growth factor signalling including EGFR, hedgehog, Wnt/β -catenin, Notch and/or integrin pathways, which frequently occurs in cancer progenitor cells during the cancer initiation and EMT program, may lead to different cancer subtypes (Fig. 1, 2) [13, 31, 35–37, 39, 40, 42, 49, 54–57, 59–62, 64, 102, 190, 191]. For instance, the occurrence of different malignant transforming events in breast stem cells during cancer initiation and/or accumulating of distinct genetic/epigenic alterations in breast cancer progenitor cells and/or their early progenies during cancer progression may result in the formation of different breast cancer subtypes [39, 42, 47, 56, 60, 68, 75, 77, 190, 192–196]. It has been observed that the targeted expression of stabilized -catenin in the basal epithelial cells of mouse mammary epithelium resulted in an enhanced proliferation of basal-type cell-like progenitor cells possessing an abnormal differentiation potential, whose oncogenic event led to development of invasive basal-type carcinomas [192]. Based on the gene expression signatures detected in distinct breast cancers including the expression levels of cytokeratin 5/6 (CK5/6), erbB2/HER-2/Neu, estrogen receptor (ERα) and/or progesterone receptor (PR), the invasive breast cancers have been classified at least five subtypes [8, 14, 156, 197–201]. Among them, there are the basal-like (CK5/6+, ERα−, PR−, erbB2−/low, EGFR+, vimentin+ and KIT+); erbB2+ overexpressing (ERα− and PR−; luminal A (ERα+ and/or PR+ and erbB2−); luminal B (ER + and/or PR+ and erbB2+) and normal breast-cancer subtype (high expression of normal epithelium genes and low expression of luminal epithelial gene products) [8, 14, 152, 197–201]. It has been observed that the ERα -negative breast cancer subtypes including the basal-like breast cancers and erbB2-overexpressing breast cancers, which are among the most aggressive breast cancer forms diagnosed, are generally associated with a poor prognosis and survival of patients to current clinical therapies [197–200, 202, 203]. Despite the activation of estrogen-ERα signalling cascade may contribute to ERα+-epithelial cell proliferation, it has been observed that the ovariectomy had no effect on the size of the mouse mammary EGFR+ stem cell-enriched population, which did not express ERα, PR or erbB2 [204, 205]. Moreover, the mouse ERα−breast cancer cells may express a lower level of E-cadherin than the ERα+ breast cancer cells, and therefore they can display a higher migratory capacity [206]. Although further studies are essential to confirm the implication of estrogens/ERα+ cascade for the proliferation of human ER − breast cancer progenitor cells, it appears likely that the different breast cancer subtypes may respond differently to the therapies consisting to targeting ERα, erbB2 and/or EGFR [202, 203, 207]. In this matter, we are reporting the specific functional properties of cancer progenitor cells that may contribute to their resistance to current therapies.
2.

Scheme showing the possible oncogenic cascades involved in the stimulation of sustained growth, survival, migration and drug resistance of cancer progenitor cells. The intracellular elements induced through the activation of EGF-EGFR, PDGF/PDGFR, SCF/KIT, hedgehog (SHH/PTCH/GLI), Notch and Wnt/β -catenin signalling and possible cross-talks between these cascades are shown. The changes in the expression levels of numerous target gene products, including down-regulated E-cadherin and up-regulated matrix metalloproteinases (MMPs), urokinase plasminogen activator (uPA) and vascular endothelial growth factor (VEGF), which can contribute both to the malignant transformation of cancer progenitor cells during cancer progression and angiogenesis, are also indicated. Furthermore, the effects of pharmacological agents acting as the potent inhibitors of the oncogenic cascades including the selective inhibitors of EGF-EGFR system (gefitinib and erlotinib), smoothened hedgehog signalling element (cyclopamine), Notch (γ-secretase inhibitor) as well as Wnt/β -catenin cascades (monoclonal anti-Wnt antibody ‘mAb’) on the cancer cells are also indicated. Abbreviations: APC, adenomatous polyposis coli; ABCG2/BCRP-1, brain cancer resistence protein-1; CDK, cyclin-dependent kinase; CoA, co-activators; COX-2, cyclooxygenase-2; Dsh, Dishevelled; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; Fzd, Frizzled receptor, GSKβ, glycogen synthase kinaseβ; ICN, intracellular domain of Notch; Iκ Bα, inhibitor of nuclear factor- κBβ; KIT, stem cell factor receptor; LEF, lymphocyte enhancer factor;LPR, lipoprotein co-receptor;MAPKs, mitogen-activated protein kinases;MEK, extracel-lular signal-related kinase kinase;NF-κ B, nuclear factor-kB;PI3K, phosphatidylinositol-3’kinase; PTEN, tensin homo-logue deleted on chromosome 10;PDGF, platelet-derived growth factor;PDGFR, platelet-derived growth factor-receptor; PLC-γ, phospholipase C-γ; PTCH, hedgehog-patched receptor; SCF, stem cell factor; SHH, sonic hedgehog ligand;SMO, smoothened;TCL, T-cell factor;WIF-1, Wnt-inhibitory factor-1;Wnt, Wingless ligand.
Novel cancer therapies by molecular targeting of cancer progenitor cells and their microenvironment
New concepts on the functions of cancer progenitor cells in the resistance to current cancer therapies
Accumulating evidence revealed that the intrinsic or acquired resistance of poorly differentiated and leukaemic or tumourigenic cancer progenitor cells to current clinical therapies may lead to their persistence in primary and secondary neoplasms after treatments, and thereby contribute to cancer recurrence [13, 16, 35, 39, 40, 55, 56, 59, 62, 64, 71, 78, 81, 88, 89, 155]. In this regard, certain ABC multi-drug efflux transporters including P-glycoprotein (P-gp) encoded by the MDR1 (ABCB1) gene, the multi-drug resistance-associated protein 1 (MRP1) and breast cancer resistance protein-1 (mouse BCRP-1)/human ABCG2/MXR gene products are frequently overex-pressed in cancer progenitor cells [16, 35, 46, 88, 89, 95, 208]. Since the SP cells, which possess the stem cell-like properties, generally express higher levels of ABC multi-drug efflux pump(s) than the non-SP cells, it is likely that this phenotype may be also related to the intrinsic resistance of certain cancer progenitor cells to chemotherapeutic treatments. In support with this, several data have revealed that the SP cells from malignant tissues or cancer cell lines were more resistant than the non-SP cells to the chemotherapeutic drugs, and the enhanced drug efflux in the SP cells was associated with a high rate of survival [54, 80, 88, 89, 209]. For instance, the SP cells isolated from HuH7 hepatocellular cancer cells, which expressed the high levels of ABCG2 and ABCB1 multi-drug efflux pumps as well as carcinoembryonic antigen-related cell adhesion molecule (CEACAM6), which may contribute to gemcitabine chemoresis-tance, displayed a greater resistance to doxorubicin, 5-fluorouracil (5-Fu) and gemcitabine treatment than the non-SP fraction [80, 210]. Similarly, the SP cells from the mouse ovarian MOVCAR 7 cancer cell line, which express the BCRP-1 gene product, were also more resistant to the anti-proliferative effect of doxorubicin which acts as a substrate of BCRP-1 efflux pump than the non-SP fraction [98]. The ABCG2 gene product overexpression in mitoxantrone-resistant MCF-7/MitoR breast cancer cells also resulted in a substantial decrease of intracellular accumulation of Hoechst 33342 dye and enhanced the number of SP cells with an acquired multi-drug resistance phenotype as compared to parental MCF-7 cell line [208]. Thus, the selection of multi-drug-resistant cancer progenitor cells by continuous drug treatment may result in an increase of their proportion in total cancer cell mass, and thereby contribute to the cancer progression to highly aggressive cancers and disease relapse [54]. In addition, the enhanced expression/activation of many growth factors and anti-apoptotic signalling elements may also contribute to the drug resistance and survival of cancer progenitor cells [13, 16, 30, 31, 35–37, 39–42, 46, 54–61, 78, 89]. For instance, many changes in the apoptotic signalling elements have been observed in the chemotherapy resistant ex vivo selected ‘survivor cell’ population which was enriched for CD34+ LSCs [211]. Similarly, CD133+ BTSCs isolated from primary cell lines established from glioblastoma patients, which expressed higher levels of CD90, CD44, CXCR4, Nestin, Msi1 and MELK relative to CD133− cancer cells, were also resistant to diverse chemotherapeutic drugs such as temo-zolomide, carboplatin, etoposide and paclitaxel [78].
On the other hand, the cancer stem/progenitor cell model of carcinogenesis may also explain the differences of response of distinct cancer subtypes to current therapies as well as the dormancy phenomenon and disease relapse, which may be associated with a higher resistance of cancer progenitor cells to conventional therapies under specific conditions prevalent in primary and/or secondary neoplasms relative to their further differentiated progeny [14, 25, 39, 40, 46, 79, 207, 212]. Importantly, it has been observed that the CD44/CD24−/low mammosphere cell cultures established from human MCF-7 breast cancer cells were more resistant to radiation than the adherent breast cancer cell fraction and that their number was enhanced after radiotherapy [213]. Similarly, Sca-1+ multi-potent progenitor cells from an immortalized COMMA-Dβ -geo murine mammary gland cell line expressing a high Wnt/β -catenin level were also resistant to radiation at a clinically relevant dose [212]. Moreover, the CD133+ breast cancer stem cell population from glioblastomas was also enriched after ionizing radiation treatment whose radioresistance phenomenon may be associated with a preferential activation of the DNA damage checkpoint response concomitant with an increase in DNA repair capacity relative to the CD133 fraction [79]. Additionally, since most of the solid tumours are molecularly heterogeneous and distinct cancer cells may possess specific hyper-activated intracellular signalling cascades, it appears that the targeting of distinct oncogenic pathways may represent a more effective strategy to eradicate the total cancer cell population in certain aggressive and recurrent cancers. Consistent with this, it has been observed that the undifferentiated-type areas of heterogeneous gastric tumours display a higher GLI1 nuclear immunostaining than the differentiated-type areas suggesting that the undifferentiated gastric cancer subtype could be more sensitive to the agents targeting hedgehog cascade [214]. In analogy with the BM-resident HSCs which possess long-term in vivo re-populating abilities and are mainly under a quiescent state, the primitive quiescent LSCs may also be more resistant than diving LSCs to the agents, such as 5-FU or imatinib mesylate that principally act on the proliferative cells (Fig. 1) [78, 81]. Hence, all these stem cell-like properties attributed to cancer progenitor cells may provide them with a higher resistance to current cancer therapies, and thereby constitute a substantial obstacle to the successful treatment of cancer patients. Together, these observations underline then the critical importance of targeting the cancer progenitor cells and their early progenies as well as their local microenvironment in the earlier stages of cancer treatment to counteract the rapid progression of certain cancer types and prevent the metastatic spread at distant sites. In regard with this, we describe here the new cancer therapeutic strategies based on targeting of different oncogenic cascades activated in highly leukaemic or tumourigenic cancer progenitor cells, and which must now be considered for improving the current therapeutic treatments.
New combination therapies against the aggressive and recurrent cancers
Targeting cancer progenitor cells
The simultaneous blockade of several oncogenic cascades activated in cancer progenitor cells during cancer progression may be essential for improving the current clinical treatments against high-risk, metastatic or relapsed leukaemias, multiple myelomas and numerous solid cancers, including neuroblastomas, medulloblastomas, GBMs and skin, lung, head and neck, breast, ovary, prostate, liver, pancreas and gastrointestinal cancers [1, 12, 13, 15–17, 30, 31, 35–42, 55–57, 59–62, 163, 164, 190, 191, 214, 215]. Among them, the molecular targeting of diverse developmental cascades including hedgehog, Wnt/β -catenin, Notch, EGFR, PDGFR and KIT pathways and/or oncogenic signalling elements (telomerase, Src, ABL, PI3K/Akt, MYC, NF-κ B and survivin) which assume a critical function in regulating the self-renewal, survival and invasion of cancer progenitor cells as well as in drug resistance and disease relapse, is of therapeutic interest [1, 12, 13, 16, 17, 26, 30, 31, 35–45, 49, 55–59, 61, 62, 64, 78, 102, 164, 190, 191, 214, 216, 217]. In this matter, one of the major significant advances in cancer therapeutics over the few last years has been the development of a number of novel anti-carcinogenic agents, including the antibodies and small chemical molecules such as the protein tyrosine kinase inhibitors (TKIs) (Table 3) [12, 13, 16, 17, 24, 26, 30, 36, 37, 39, 40, 57, 216, 218–220]. These pharmacological agents, which target one or several oncogenic products involved in the malignant transformation of human cells, may be used alone or in combination with conventional therapeutic treatments against certain aggressive cancers. For instance, certain anti-carcinogenic agents may induce the apoptotic death and/or a differentiating effect on primitive cancer-initiating cells, and thereby may constitute the useful tools for the development of more effective cancer therapies [13, 16, 26, 30, 35, 37, 39–41, 48, 55, 57, 59, 150, 215, 221]. The combination of high-dose chemotherapy or radiation with autologous or allogenic stem cell transplantation also represent another alternative for the patients diagnosed with advanced and refractory/relapsed leukaemias, multiple myeloma, lymphomas and sarcoma as well as brain, lung, breast, kidney, ovary and colorectal cancers [13, 16, 222–224].
3.
Specific inhibitors of growth factor cascades and signaling elements involved in sustained growth, survival and/or drug resistance of cancer progenitor cells and angiogenic process
| Targeted signaling element | Name of inhibitor |
|---|---|
| EGFR family member inhibitor | |
| Anti-EGFR (erbB1) antibody | mAb-C225, IMC-C225 |
| Anti-EGF antibody | ABX-EGF |
| EGFR-TKI | Gefitinib, erlotinib, AG1478, EKB-569 |
| Anti-erbB2 antibody | Trastuzumab |
| EGFR-erbB2-TKI | PKI-166, TAK165, GW572017 (lapatinib) |
| erbB1/erbB2/erbB3/erbB4-TKI | CI1033 |
| Other growth factor signaling inhibitor | |
| Hedgehog | SMO inhibitor cyclopamine, anti-SHH antibody |
| Anti-Wnt antibody, WIF-1 | |
| Wnt/ -catenin | |
| Notch | -secretase inhibitor DAPT, GSI-18 |
| PDGFRβ/KIT/ABL-TKI | Imatinib mesylate (STI571) |
| Sorafenib | |
| PDGFR /FLT3 | |
| VEGF | Anti-VEGF antibody (bevacizumab), As-VEGF |
| VEGFR | Anti-VEGFR antibody |
| VEGFR1,2 and 3 | CEP-7055, AZD2171 |
| AMG 706, Vatalanib (PTK787/ZK 222584) | |
| VEGFR1,2,3 /PDGFRβ/KIT | |
| Signaling element inhibitor | |
| Telomerase | Telomerase template antagonist |
| MYC | As-MYC |
| Bcl-2 | ABT-737 |
| PI3K | LY294002, rapamycin, CCI-779 |
| NF-κ B inhibitor | IkBβ inhibitor, sulfasalazine, bortezomib (PS-341) |
| Src-family and ABL-TKI | PD180970 |
| Src-family-TKI | CGP-76030 |
| Fusion oncoprotein inhibitor | |
| BCR-ABL | Imatinib mesylate (STI571), dasatinib (BMS-354825), nilotinib |
| Imatinib mesylate (STI571) | |
| FIP1L1-PDGFRα | |
| All-trans retinoic acid | |
| PML-RAR-α | |
| Drug transporter inhibitor | |
| ABC multidrug efflux transporter | |
| MDR1/ABCB1/P-gp | MS-209, gefitinib, CI1033, tamoxifen, cyclopamine |
| MRP1/ABCC1 | MS-209 |
| ABCG2/BCRP | Gefitinib, CI1033, tamoxifen derivatives, cyclopamine |
| Organic cation intracellular transporter Oct-1 | Prazosin |
DAPT, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester;EGFR, epidermal growth factor receptor; KIT, stem cell factor (SCF) receptor;GSI-18, [11-endo]-N-(5,6,7,8,9,10-hexahydro-6,9-methanobenzo[a][8]annulen-11-ylthiophene-2-sulfonamide;MDR1, multidrug resistance 1;MRP1, multidrug resistance-associated protein 1;MRP2, multidrug resistance-associated protein 2;NF-kB, nuclear factor-kB;PDGFR, platelet-derived growth factor receptor;P-gp, P-glycoprotein;PI3K, phosphatidylinositide-3'-kinase;TKI, tyrosine kinase inhibitor;VEGF, vascular epithelial growth factor;VEGFR, vascular epithelial growth factor receptor;WIF-1, Wingless inhibitory factor-1;Wnt, Wingless ligand
Among the current clinical targeted therapies, the use of imatinib mesylate, which is a TKI targeting BCR-ABL+ oncoprotein, has provoked great interest based on its high efficacy and safe profile and successful use for the first-line clinical treatment of patients with BCR-ABL+ (Ph+) CMLs in initial chronic phase [16, 24, 27, 28, 33]. Moreover, imatinib mesylate may also act as a potential therapeutic agent for the treatment of Ph+ AML, FIP1L1-PDGFRA positive chronic eosinophilic leukaemia and gastrointestinal stromal tumours which frequently harbour the activating mutations in KIT and PDGFRA tyrosine kinase receptors [24, 27, 28, 33, 225]. Particularly, the inhibition of the tyrosine kinase activity of chimeric BCR-ABL fusion oncoprotein by imatinib mesylate may lead to a complete cytogenetic remission after initiation treatment in the most patients diagnosed with BCR-ABL+ (Ph+) CMLs [27, 28, 33]. However, the persistence of quiescent CML-LSCs resistant to ima-tinib treatment as well as the progression to an advanced and late-stage CML disease, accelerated phase or terminal blast crisis phase, is often accompanied by acquisition of a more malignant transformed phenotype by LSCs and disease relapse [27, 28, 32–34, 81, 226–228]. The mutations in BCL-ABL and other independent BCR-ABL mechanisms, such as the up-regulation of Scr-related LYN kinase, SHH, Wnt/β -catenin, anti-apoptotic factor Bcl-2, MDR1/ABCB1 and ABCG2/BCRP muli-drug efflux transporters and/or cellular organic cation transporter Oct-1 involved in the uptake of imatinib may contribute to the CML recurrence [16, 27, 28, 32–34, 81, 164, 226–228]. Therefore, the combined use of distinct anti-leukaemic agents that are able to inhibit diverse oncogenic cascades that are activated in LSCs is of particular interest for overcoming imatinib resistance in the refractory/relapsed CML patients. Among them, there are the inhibitors of BCR-ABL (dasatinib and nilotinib), Scr-related LYN kinase (PD180970), anti-apoptotic factor Bcl-2 (ABT-737) or Oct-1 transporter (prazosin) which may be used alone or incombination with imatinib mesylate and/or other cytotoxic drugs, such as interferon-α[27, 28, 34, 175, 227, 229–232]. Recently, it has also been observed that the exposure of quiescent BCR-ABL+ CML-LSCs to the growth factors such as granulo-cyte-colony stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF) may decrease the number of non-dividing BCR-ABL+ CML-LSCs, and thereby could also constitute an alternative strategy for improving the ima-tinib mesylate-based therapies [233, 234]. Similarly, the current treatment for APL, which consists of the combined use of all-trans retinoic acid (ATRA) and anthracycline-based chemotherapy, generally results in a high rate of complete cytogenetic remission (about 90%) and an overall survival rate of 80% in the initial phase. Certain patients with high-risk APL may, however, relapse [21, 23]. In these cases, the efficacy of clinical treatment of high-risk and relapsed PML-RAR+ APL may also be improved by the combined used of a differentiating agent, ATRA plus cytotoxic drugs such as arsenic trioxide that may co-operate to eradicate the total population of PML-RAR+ APL-LSCs [21–23]. Additionally, it has also been observed that the use of an inhibitor of γ-secretase (DAPT), a protease that is involved in the activation of Notch receptor-mediated intracellular signalling by its lig-and Jagged-2, which is overexpressed in the LSCs from AML samples, may inhibit the growth of CD34+ CD38− AML-LSC fraction in colony formation assays in vitro [235]. The molecular mechanisms underlying the cytotoxic effect of DAPT on LSCs appears to be due in an alteration of the AML-LSC fate and the interference of this agent with the transportation of LSCs into the stem cell-supportive microenvironmen-tal niche [45]. Interestingly, a recent study has also indicated that the targeting of LSCs at the origin of certain AML subtypes by using an activating mono-clonal antibody directed against the cell surface-adhesion molecule CD44 results in a reduction of leukaemic population in human AML-LSC cells transplanted NOD-SCID mice in vivo as well as in the absence of leukaemia development after subsequent serial transplantations in other mice [236]. The data from another study carried out on a murine leukaemia model have also revealed that CALM/AF10-positive AML can be propagated by a transformed leukaemic progenitor cells, with lym-phoid characteristics including CD45RA (B220) expression and Ig DH-JH re-arrangement, which can be targeted by antibodies that do not cross-react with normal HSCs [237].
Interestingly, recent studies have also revealed the possibility of using the agents targeting the EGFR, hedgehog, Wnt/β -catenin and/or Notch cascades to inhibit the ABC muli-drug efflux transporters and/or eliminate the cancer progenitor cells (Fig. 2;Table 3) [57, 89, 102, 191, 215, 238–240]. For instance, it has been observed that the EGF treatment of human metastatic UMSCC10B and HN12 head and neck squamous carcinoma cell lines was accompanied by an increase of number of the SP cells and the add of gefitinib reduced the SP population [102]. The cyclopamine treatment of poorly differentiated and highly-tumourigenic CD44+ PC3 metastatic prostate cancer cells, which possess an intermediate phenotype (androgen receptor negative, CK5/18), was also accompanied by a decrease of the expression levels of MDR1 and BCRP/ABCG2 suggesting that the activation of hedgehog signalling may contribute to MDR in these cancer cells [239]. Importantly, the continuous cyclopamine treatment of PC3 cell-xenografts established in nude mice in vivo has also been observed to result in apoptotic cell death and tumour regression [191]. Also, no sign of the tumour recurrence and secondary effects on normal cells was detected 72 hrs after the cessation of treatment, suggesting that the cancer-initiating cells may have been eliminated [191]. Similarly, it has also been reported that the hedgehog ligand induces a clonal expansion of multiple myeloma (MM) stem cells while cyclopamine treatment counteracts the expansion of purified MM stem cells but has little effect on the growth of their further differentiated malignant plasma cells [238]. The number of SP cells detected in the NCL-H929 MM cell line was also decreased after the cyclopamine treatment [238]. Of particular therapeutic interest, cyclopamine alone also inhibited the self-renewal ability of CD133+ glioma cancer stem cell cultures established from human GBM tumour samples in vitro, and induced an additive or synergistic anti-proliferative and apoptotic effect at a lower concentration in combination with the current therapeutic drug, temozolomide on the gliomasphere cells [240]. Long-term treatment of gliomasphere cells expressing a high expression of the stemness genes (GLI1, PTCH1, Nanog, Oct-4, SOX2, BMI1 and PCNA) with the cyclopamine alone also killed all these cancer stem cells in culture, and induced the regression of glioma tumours established from the gliomasphere cells in nude mice in vivo, without detectable secondary effects [240]. Moreover, the radioresistant property of CD133+ glioblastoma stem cells could also be reversed by using a specific inhibitor of the CDK1 and CDK2 checkpoint kinases [79]. Additionally, the suppressing of β-catenin expression in Sca-1+ multi-potent progenitor cells from an immortalized COMMA-D β-geo murine mammary gland cell line has been observed to decrease their self-renewal capacity [212]. The results from a recent investigation have also indicated that the Notch signalling blockade by using an inhibitor of γ-secretase (GSI-18) significantly reduced the CD133+ cell fraction and eliminated the SP cells detected in medulloblastoma cell mass, which express a high Notch signalling levels [215]. Since the medulloblas-toma cells treated with Notch inhibitor did not effectively form the tumour xenografts in vivo, it has been suggested that the cancer-initiating cells have been eradicated [215].
Targeting the local microenvironment of cancer progenitor cells
The development of more effective antitumoural strategies also implicates the inhibition of the neoangiogenic process which is necessary for the tumour vascularization and growth. Many anti-angiogenic agents that are able to interfere with the VEGF-VEGFR transduction system, including the anti-VEGF or VEGFR antibody, VEGFR antagonists and the soluble truncated form of VEGFR have been designed and observed to effectively counteract the tumour growth in animal models in vivo (Table 3) [30, 131, 132, 134–136, 140, 187, 241]. Particularly, it has been observed that the targeting of VEGFRs may be more effective than the inhibition of the single receptor to prevent and/or counteract the tumour formation in animal models in vivo[135, 137]. Interestingly, the treatment of the mice bearing ortho-topic U87 glioma cell xenografts with anti-VEGF monoclonal antibody, bevacizumab markedly reduced the microvasculature density and tumour growth and this anti-angiogenic effect was also accompanied by a decrease of the number of vessel-associated self-renewing CD133+/nestin+ tumour cells [187]. Based on these observations, it has been suggested that the bevacizumab may act, at least in part, by disrupting the vascular microenvironment ‘niche’ of BTSCs, and thereby impairs their self-renewal capacity (Fig. 1) [187, 242]. In addition, several lines of evidence have revealed the potential benefit of targeting the tumour-specific vascular precursor cells including the circulating VEGFR-2+-EPCs and VEGFR-1+-PMC which may be recruited within the activated stromal compartment in the primary and secondary neoplasms for preventing and/or counteracting the neovascularization process associated with tumour development (Fig. 1) [134, 136, 137, 140]. More particularly, it has been shown that the use of BM-derived EPCs engineered to express an anti-angiogenic gene product, a soluble truncated form of VEGFR-2, may impair tumour growth in vivo[140]. The identification of further specific biomarkers to the tumour-specific EPCs should therefore help to develop the more effective anti-angio-genic treatments based on their molecular targeting.
Conclusions
Altogether, these recent investigations have revealed that the most aggressive cancers may originate from the malignant transformation of embryonic or adult stem/progenitor cells into cancer progenitor cells. The cancer progenitor cells can provide critical functions in cancer initiation and progression into metastatic and recurrent disease states. Based on these observations, the molecular targeting of highly leukaemic or tumourigenic cancer progenitor cells must be considered for improving the efficacy of the current cancer therapies. New drug combinations targeting cancer progenitor cells have been developed and shown to be effective against certain aggressive cancers in the clinical settings. Additional investigations to identify the specific biomarkers and resistance mechanisms of cancer progenitor cells versus their further differentiated malignant progenies and their normal counterparts, adult stem cells should lead to more effective and safe therapeutic treatments for the most the aggressive and incurable cancers.
Future directions and perspectives
The technical advancements in the adult stem cell biology have provided the methods to isolate and characterize the ex vitro and in vivo properties of cancer stem/progenitor cells. However, the evaluation of their functional properties ex vivo must be considered with caution since multiple extrinsic factors may influence their behaviour, and more particularly, the experimental culture conditions did not necessarily reflect their local microenvironment within the tumours. Further investigations of cancer progenitor cells in more vigorous experimental conditions are required, and thereby should help to elucidate the molecular mechanisms that provide a critical role for their uncontrolled self-renewal and/or aberrant differentiation capacity. The differences between the specific biomarkers and functional properties of cancer progenitor cells versus their normal counterpart, adult stem cells should notably be exploited for the specific targeting of cancer progenitor cells, and thereby preserve the integrity of normal tissue-specific adult stem cells. The cancer progenitor cells also exhibit a differential gene expression pattern, as compared to the bulk mass of further cancer cells, and therefore a re-analyse of the oncogenic gene products activated specifically in cancer progenitor cells appears to be necessary. Further investigations by using the cancer progenitor cells isolated from primary cancer patient's malignant tissues at different stages during the cancer progression and metastatic disease should help to identify new biomarkers for the development of more effective diagnostic and prognostic methods and targeted therapies against the aggressive and metastatic cancers. The development of new effective and safe targeted therapies by eradicating the total population of cancer progenitor cells and their further differentiated progenies at the primary and secondary neoplasms should allow us to improve the current cancer treatments, prevent the disease relapse and thereby induce a complete cytogenetic remission and cure of cancer patients in the clinics.
Acknowledgments
We apologize to researchers that have contributed to the advancements in cancer stem/progenitor cell research and whose works have not been cited due to space limitations. The authors are supported by the grants from the U. S. Department of Defense (PC04502, OC04110) and the National Institutes of Health (CA78590, CA72712, CA111294). We thank Ms. Kristi L. Berger for editing the manuscript.
References
- 1.Lowenberg B, Griffin JD, Tallman MS. Acute myeloid leukemia and acute promyelocytic leukemia. Hematology Am Soc Hematol Educ. Program. 2003:82–101. [PubMed] [Google Scholar]
- 2.Mimeault M, Brand RE, Sasson AA, Batra SK. Recent advances on the molecular mechanisms involved in pancreatic cancer progression and therapies. Pancreas. 2005;31:301–16. doi: 10.1097/01.mpa.0000175893.04660.1b. [DOI] [PubMed] [Google Scholar]
- 3.Mimeault M, Batra SK. Recent advances on multiple tumorigenic cascades involved in prostatic cancer progression and targeting therapies. Carcinogenesis. 2006;27:1–22. doi: 10.1093/carcin/bgi229. [DOI] [PubMed] [Google Scholar]
- 4.Lock-Andersen J, Horn J, Sjostrand H. Prognosis after sentinel node biopsy in malignant melanoma. Ugeskr Laeger. 2006;168:2457–62. [PubMed] [Google Scholar]
- 5.Shiraishi J, Abe H, Li F, Engelmann R, MacMahon H, Doi K. Computer-aided diagnosis for the detection and classification of lung cancers on chest radiographs ROC analysis of radiologists performance. Acad Radiol. 2006;13:995–1003. doi: 10.1016/j.acra.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Chatterton K, Ray E, O'Brien TS. Fluorescence diagnosis of bladder cancer. Br J Nurs. 2006;8(15):595–7. doi: 10.12968/bjon.2006.15.11.21226. [DOI] [PubMed] [Google Scholar]
- 7.Alexander A, Murtha A, Abdulkarim B, Mehta V, Wheatley M, Murray B, Riauka T, Hanson J, Fulton D, McEwan A, Roa W. Prognostic significance of serial magnetic resonance spectroscopies over the course of radiation therapy for patients with malignant glioma. Clin Invest Med. 2006;29:301–11. [PubMed] [Google Scholar]
- 8.Calza S, Hall P, Auer G, Bjohle J, Klaar S, Kronenwett U, Liu ET, Miller L, Ploner A, Smeds J, Bergh J, Pawitan Y. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 2006;8:R34. doi: 10.1186/bcr1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petignat P, Du Bois A. Bruchim I, Fink D, Provencher DM. Should intraperitoneal chemotherapy be considered as standard first-line treatment in advanced stage ovarian cancer? Crit Rev Oncol Hematol. 2006;62:137–47. doi: 10.1016/j.critrevonc.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 10.MacDonald TJ, Rood BR, Santi MR, Vezina G, Bingaman K, Cogen PH, Packer RJ. Advances in the diagnosis, molecular genetics, and treatment of pediatric embryonal CNS tumors. Oncologist. 2003;8:174–86. doi: 10.1634/theoncologist.8-2-174. [DOI] [PubMed] [Google Scholar]
- 11.Takei H, Bhattacharjee MB, Rivera A, Dancer Y, Powell SZ. New immunohistochemical markers in the evaluation of central nervous system tumors: a review of 7 selected adult and pediatric brain tumors. Arch Pathol Lab Med. 2007;131:234–41. doi: 10.5858/2007-131-234-NIMITE. [DOI] [PubMed] [Google Scholar]
- 12.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–7. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 13.Mimeault M, Batra SK. Recent advances on the significance of stem cells in tissue regeneration and cancer therapies. Stem Cells. 2006;24:2319–45. doi: 10.1634/stemcells.2006-0066. [DOI] [PubMed] [Google Scholar]
- 14.Lacroix M. Significance, detection and markers of disseminated breast cancer cells. Endocr Relat Cancer. 2006;13:1033–67. doi: 10.1677/ERC-06-0001. [DOI] [PubMed] [Google Scholar]
- 15.Baehring JM. An update on oligodendroglial neo-plasms. Curr Opin Neurol. 2005;18:639–44. doi: 10.1097/01.wco.0000189877.31637.74. [DOI] [PubMed] [Google Scholar]
- 16.Mimeault M, Hauke R, Batra SK. Recent advances on the molecular mechanisms involved in drug-resistance of cancer cells and novel targeting therapies. Clin Pharmacol Ther. 2007;82:252–64. doi: 10.1038/sj.clpt.6100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorscher SM. Biological therapy update in colorectal cancer. Expert Opin Biol Ther. 2007;7:509–19. doi: 10.1517/14712598.7.4.509. [DOI] [PubMed] [Google Scholar]
- 18.Brawley OW, Kramer BS. Cancer screening in theory and in practice. J Clin Oncol. 2005;23:293–300. doi: 10.1200/JCO.2005.06.107. [DOI] [PubMed] [Google Scholar]
- 19.Van Der Merwe DE, Oikonomopoulou K, Marshall J, Diamandis EP. Mass spectrometry: uncovering the cancer proteome for diagnostics. Adv Cancer Res. 2006;96:23–50. doi: 10.1016/S0065-230X(06)96002-3. [DOI] [PubMed] [Google Scholar]
- 20.Clarke NW. Management of the spectrum of hormone refractory prostate cancer. Eur Urol. 2006;50:428–38. doi: 10.1016/j.eururo.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Tallman MS. Treatment of relapsed or refractory acute promyelocytic leukemia. Best Pract Res Clin Haematol. 2007;20:57–65. doi: 10.1016/j.beha.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Zhou GB, Zhang J, Wang ZY, Chen SJ, Chen Z. Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide:a paradigm of synergistic molecular targeting therapy. Philos Trans R Soc Lond B Biol Sci. 2007;362:959–71. doi: 10.1098/rstb.2007.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X, Seshire A, Ruster B, Bug G, Beissert T, Puccetti E, Hoelzer D, Henschler R, Ruthardt M. Arsenic but not all-trans retinoic acid overcomes the aberrant stem cell capacity of PML/RARalpha-positive leukaemic stem cells. Haematologica. 2007;92:323–31. doi: 10.3324/haematol.10541. [DOI] [PubMed] [Google Scholar]
- 24.Jovanovic JV, Score J, Waghorn K, Cilloni D, Gottardi E, Metzgeroth G, Erben P, Popp H, Walz C, Hochhaus A, Roche-Lestienne C, Preudhomme C, Solomon E, Apperley J, Rondoni M, Ottaviani E, Martinelli G, Brito-Babapulle F, Saglio G, Hehlmann R, Cross NC, Reiter A, Grimwade D. Low-dose imatinib mesylate leads to rapid induction of major molecular responses and achievement of complete molecular remission in FIP1L1-PDGFRA positive chronic eosinophilic leukemia. Blood. 2007;109:4635–40. doi: 10.1182/blood-2006-10-050054. [DOI] [PubMed] [Google Scholar]
- 25.Pantel K, Woelfle U. Micrometastasis in breast cancer and other solid tumors. J Biol Regul Homeost Agents. 2004;18:120–5. [PubMed] [Google Scholar]
- 26.Milas L, Raju U, Liao Z, Ajani J. Targeting molecular determinants of tumor chemo-radioresistance. Semin Oncol. 2005;32:S78–81. doi: 10.1053/j.seminoncol.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 27.Mauro MJ. Defining and managing imatinib resistance. Hematology Am Soc Hematol Educ Program. 2006:219–25. doi: 10.1182/asheducation-2006.1.219. [DOI] [PubMed] [Google Scholar]
- 28.Mughal T, Goldman JM. Optimal management of patients with newly diagnosed chronic phase chronic myeloid leukemia in 2007. Clin Lymphoma Myeloma. 2007;7:S95–101. doi: 10.3816/clm.2007.s.008. [DOI] [PubMed] [Google Scholar]
- 29.Ceschel S, Casotto V, Valsecchi MG, Tamaro P, Jankovic M, Hanau G, Fossati F, Pillon M, Rondelli R, Sandri A, Silvestri D, Haupt R, Cuttini M. Survival after relapse in children with solid tumors:a follow-up study from the Italian off-therapy registry. Pediatr Blood Cancer. 2006;47:560–6. doi: 10.1002/pbc.20726. [DOI] [PubMed] [Google Scholar]
- 30.Rosell R, Cecere F, Cognetti F, Cuello M, Sanchez JM, Taron M, Reguart N, Jablons D. Future directions in the second-line treatment of non-small cell lung cancer. Semin Oncol. 2006;33:S45–51. doi: 10.1053/j.seminoncol.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Sato M, Shames DS, Gazdar AF, Minna JD. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol. 2007;2:327–43. doi: 10.1097/01.JTO.0000263718.69320.4c. [DOI] [PubMed] [Google Scholar]
- 32.Goldman J, Gordon M. Why do chronic myelogenous leukemia stem cells survive allogeneic stem cell transplantation or imatinib: does it really matter? Leuk Lymphoma. 2006;47:1–7. doi: 10.1080/10428190500407996. [DOI] [PubMed] [Google Scholar]
- 33.Copland M, Jorgensen HG, Holyoake TL. Evolving molecular therapy for chronic myeloid leukaemia–are we on target? Hematology. 2005;10:349–59. doi: 10.1080/10245330500234195. [DOI] [PubMed] [Google Scholar]
- 34.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, Barow M, Mountford JC, Holyoake TL. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107:4532–9. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 35.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 36.Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 37.Rubin LL, De Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5:1026–33. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 38.Roberg K, Jonsson AC, Grenman R, Norberg-Spaak L. Radiotherapy response in oral squamous carcinoma cell lines:Evaluation of apoptotic proteins as prognostic factors. Head Neck. 2007;29:325–34. doi: 10.1002/hed.20520. [DOI] [PubMed] [Google Scholar]
- 39.Mimeault M, Batra S. Interplay of distinct growth factors during epithelial-mesenchymal transition of cancer progenitor cells and molecular targeting as novel cancer therapies. Ann Oncol. 2007;18:1605–19. doi: 10.1093/annonc/mdm070. [DOI] [PubMed] [Google Scholar]
- 40.Mimeault M, Batra SK. Functions of tumorigenic and migrating cancer progenitor cells in cancer progression and metastasis and their therapeutic implications. Cancer Metastasis Rev. 2007;26:203–14. doi: 10.1007/s10555-007-9052-4. [DOI] [PubMed] [Google Scholar]
- 41.Nicolis SK. Cancer stem cells and “stemness” genes in neuro-oncology. Neurobiol Dis. 2007;25:217–29. doi: 10.1016/j.nbd.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 42.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–8. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 43.Li XN, Shu Q, Su JM, Adesina AM, Wong KK, Perlaky L, Antalffy BA, Blaney SM, Lau CC. Differential expression of survivin splice isoforms in medulloblastomas. Neuropathol Appl Neurobiol. 2007;33:67–76. doi: 10.1111/j.1365-2990.2006.00782.x. [DOI] [PubMed] [Google Scholar]
- 44.Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pahway. Carcinogenesis. 2007;28:1379–86. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- 45.Fuster JJ, Sanz-Gonzalez SM, Moll UM, Andres V. Classic and novel roles of p53: prospects for anticancer therapy. Trends Mol Med. 2007;13:192–9. doi: 10.1016/j.molmed.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 46.De Jonge-Peeters SD, Kuipers F, De Vries EG, Vellenga E. ABC transporter expression in hematopoietic stem cells and the role in AML drug resistance. Crit Rev Oncol Hematol. 2007;62:214–26. doi: 10.1016/j.critrevonc.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 47.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene. 2004;23:7274–82. doi: 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 48.Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. 2004;51:1–28. doi: 10.1016/j.critrevonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion:migrating cancer stem cells – an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–9. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 50.Miller SJ, Lavker RM, Sun TT. Interpreting epithelial cancer biology in the context of stem cells: tumor properties and therapeutic implications. Biochim Biophys Acta. 2005;1756:25–52. doi: 10.1016/j.bbcan.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Li L, Neaves WB. Normal stem cells and cancer stem cells:the niche matters. Cancer Res. 2006;66:4553–7. doi: 10.1158/0008-5472.CAN-05-3986. [DOI] [PubMed] [Google Scholar]
- 52.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–36. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 53.Massard C, Deutsch E, Soria JC. Tumour stem cell-targeted treatment:elimination or differentiation. Ann Oncol. 2006;17:1620–4. doi: 10.1093/annonc/mdl074. [DOI] [PubMed] [Google Scholar]
- 54.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 56.Woodward WA, Chen MS, Behbod F, Rosen JM. On mammary stem cells. J Cell Sci. 2005;118:3585–94. doi: 10.1242/jcs.02532. [DOI] [PubMed] [Google Scholar]
- 57.Galmozzi E, Facchetti F, La Porta CA. Cancer stem cells and therapeutic perspectives. Curr Med Chem. 2006;13:603–7. doi: 10.2174/092986706776055661. [DOI] [PubMed] [Google Scholar]
- 58.Rizo A, Vellenga E, De HG, Schuringa JJ. Signaling pathways in self-renewing hematopoietic and leukaemic stem cells:do all stem cells need a niche. Hum Mol Genet. 2006;15:R210–9. doi: 10.1093/hmg/ddl175. [DOI] [PubMed] [Google Scholar]
- 59.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea–a paradigm shift. Cancer Res. 2006;66:1883–90. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 60.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog sig-nalling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepa-tocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 62.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–31. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 63.Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025–9. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- 64.Tso CL, Shintaku P, Chen J, Liu Q, Liu J, Chen Z, Yoshimoto K, Mischel PS, Cloughesy TF, Liau LM, Nelson SF. Primary glioblastomas express mes-enchymal stem-like properties. Mol Cancer Res. 2006;4:607–19. doi: 10.1158/1541-7786.MCR-06-0005. [DOI] [PubMed] [Google Scholar]
- 65.Tan BT, Park CY, Ailles LE, Weissman IL. The cancer stem cell hypothesis: a work in progress. Lab Invest. 2006;86:1203–7. doi: 10.1038/labinvest.3700488. [DOI] [PubMed] [Google Scholar]
- 66.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 67.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 68.Al-Hajj M, Wicha MS, Ito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukaemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–43. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 70.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–37. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 71.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 72.Yuan X, Curtin J, Xiong Y, Liu G, Waschsmann-Hogiu S, Farkas DL, Black KL, Yu JS. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 73.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De VS, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 74.Hadnagy A, Gaboury L, Beaulieu R, Balicki D. SP analysis may be used to identify cancer stem cell populations. Exp Cell Res. 2006;312:3701–10. doi: 10.1016/j.yexcr.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 75.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 76.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De MR. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 77.Dontu G, El-Ashry D, Wicha MS. Breast cancer, stem/progenitor cells and the estrogen receptor. Trends Endocrinol Metab. 2004;15:193–7. doi: 10.1016/j.tem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 78.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 80.Haraguchi N, Utsunomiya T, Inoue H, Tanaka F, Mimori K, Barnard GF, Mori M. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006;24:506–13. doi: 10.1634/stemcells.2005-0282. [DOI] [PubMed] [Google Scholar]
- 81.Ravandi F, Estrov Z. Eradication of leukemia stem cells as a new goal of therapy in leukemia. Clin Cancer Res. 2006;12:340–4. doi: 10.1158/1078-0432.CCR-05-1879. [DOI] [PubMed] [Google Scholar]
- 82.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L, Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 84.Tang DG, Patrawala L, Calhoun T, Bhatia B, Choy G, Schneider-Broussard R, Jeter C. Prostate cancer stem/progenitor cells:identification, characterization, and implications. Mol Carcinog. 2007;46:1–14. doi: 10.1002/mc.20255. [DOI] [PubMed] [Google Scholar]
- 85.Collins AT, Maitland NJ. Prostate cancer stem cells. Eur J Cancer. 2006;42:1213–8. doi: 10.1016/j.ejca.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 86.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 87.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104:973–8. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827–33. doi: 10.1158/0008-5472.CAN-06-3557. [DOI] [PubMed] [Google Scholar]
- 89.Wang J, Guo LP, Chen LZ, Zeng YX, Lu SH. Identification of cancer stem cell-like side population cells in human nasopharyngeal carcinoma cell line. Cancer Res. 2007;67:3716–24. doi: 10.1158/0008-5472.CAN-06-4343. [DOI] [PubMed] [Google Scholar]
- 90.Blair A, Hogge DE, Ailles LE, Lansdorp PM, Sutherland HJ. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood. 1997;89:3104–12. [PubMed] [Google Scholar]
- 91.Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, Smith BD, Civin CI, Jones RJ. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332–6. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, Macdonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–35. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 93.Tai MH, Chang CC, Kiupel M, Webster JD, Olson LK, Trosko JE. Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis. 2005;26:495–502. doi: 10.1093/carcin/bgh321. [DOI] [PubMed] [Google Scholar]
- 94.Wulf GG, Wang RY, Kuehnle I, Weidner D, Marini F, Brenner MK, Andreeff M, Goodell MA. A leukaemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood. 2001;98:1166–73. doi: 10.1182/blood.v98.4.1166. [DOI] [PubMed] [Google Scholar]
- 95.Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99:507–12. doi: 10.1182/blood.v99.2.507. [DOI] [PubMed] [Google Scholar]
- 96.Setoguchi T, Taga T, Kondo T. Cancer stem cells persist in many cancer cell lines. Cell Cycle. 2004;3:414–5. doi: 10.4161/cc.3.4.799. [DOI] [PubMed] [Google Scholar]
- 97.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci USA. 2004;101:781–6. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, Dombkowski D, Preffer F, Maclaughlin DT, Donahoe PK. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian inhibiting substance responsiveness. Proc Natl Acad Sci USA. 2006;103:11154–9. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haraguchi N, Inoue H, Tanaka F, Mimori K, Utsunomiya T, Sasaki A, Mori M. Cancer stem cells in human gastrointestinal cancers. Hum Cell. 2006;19:24–9. doi: 10.1111/j.1749-0774.2005.00004.x. [DOI] [PubMed] [Google Scholar]
- 100.Seigel GM, Campbell LM, Narayan M, Gonzalez-Fernandez F. Cancer stem cell characteristics in retinoblastoma. Mol Vis. 2005;11:729–37. [PubMed] [Google Scholar]
- 101.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2 +and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–19. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 102.Chen JS, Pardo FS, Wang-Rodriguez J, Chu TS, Lopez JP, Aguilera J, Altuna X, Weisman RA, Ongkeko WM. EGFR regulates the side population in head and neck squamous cell carcinoma. Laryngoscope. 2006;116:401–6. doi: 10.1097/01.mlg.0000195075.14093.fb. [DOI] [PubMed] [Google Scholar]
- 103.Grichnik JM, Burch JA, Schulteis RD, Shan S, Liu J, Darrow TL, Vervaert CE, Seigler HF. Melanoma, a tumor based on a mutant stem cell? J Invest Dermatol. 2006;126:142–53. doi: 10.1038/sj.jid.5700017. [DOI] [PubMed] [Google Scholar]
- 104.Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, Nakauchi H, Taniguchi H. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006;44:240–51. doi: 10.1002/hep.21227. [DOI] [PubMed] [Google Scholar]
- 105.Mitsutake N, Iwao A, Nagai K, Namba H, Ohtsuru A, Saenko V, Yamashita S. Characterization of side population in thyroid cancer cell lines:cancer stem-like cells are enriched partly but not exclusively. Endocrinology. 2007;148:1797–803. doi: 10.1210/en.2006-1553. [DOI] [PubMed] [Google Scholar]
- 106.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myeloge-nous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci USA. 2000;97:7521–6. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–35. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jamieson CH, Weissman IL, Passegue E. Chronic versus acute myelogenous leukemia: a question of self-renewal. Cancer Cell. 2004;6:531–3. doi: 10.1016/j.ccr.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 109.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. Granulocyte-macrophage progenitors as candidate leukaemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 110.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K, Gilliland DG. MOZ-TIF2, but not BCR-ABL, confers properties of leukaemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 111.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ. Genomics identifies medul-loblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–31. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 112.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–84. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 113.Chen L, Shen R, Ye Y, Pu XA, Liu X, Duan W, Wen J, Zimmerer J, Wang Y, Liu Y, Lasky LC, Heerema NA, Perrotti D, Ozato K, Kuramochi-Miyagawa S, Nakano T, Yates AJ, Carson Iii WE, Lin H, Barsky SH, Gao JX. Precancerous stem cells have the potential for both benign and malignant differentiation. PLoS ONE. 2007;2:e293. doi: 10.1371/journal.pone.0000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer. 2003;3:685–94. doi: 10.1038/nrc1168. [DOI] [PubMed] [Google Scholar]
- 115.Mackall CL, Meltzer PS, Helman LJ. Focus on sarcomas. Cancer Cell. 2002;2:175–8. doi: 10.1016/s1535-6108(02)00132-0. [DOI] [PubMed] [Google Scholar]
- 116.Tataria M, Quarto N, Longaker MT, Sylvester KG. Absence of the p53 tumor suppressor gene promotes osteogenesis in mesenchymal stem cells. J Pediatr Surg. 2006;41:624–32. doi: 10.1016/j.jpedsurg.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 117.Thomas D, Kansara M. Epigenetic modifications in osteogenic differentiation and transformation. J Cell Biochem. 2006;98:757–69. doi: 10.1002/jcb.20850. [DOI] [PubMed] [Google Scholar]
- 118.Tiffin N, Williams RD, Shipley J, Pritchard-Jones K. PAX7 expression in embryonal rhabdomyosarco-ma suggests an origin in muscle satellite cells. Br J Cancer. 2003;89:327–32. doi: 10.1038/sj.bjc.6601040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tostar U, Malm J, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208:17–25. doi: 10.1002/path.1882. [DOI] [PubMed] [Google Scholar]
- 120.McDonald SA, Preston SL, Lovell MJ, Wright NA, Jankowski JA. Mechanisms of disease: from stem cells to colorectal cancer. Nat Clin Pract Gastroenterol Hepatol. 2006;3:267–74. doi: 10.1038/ncpgasthep0473. [DOI] [PubMed] [Google Scholar]
- 121.Hoebeeck J, Vandesompele J, Nilsson H, De PK, Van RN, De SE, Yigit N, De PA, Laureys G, Pahlman S, Speleman F. The von Hippel-Lindau tumor suppressor gene expression level has prognostic value in neuroblastoma. Int J Cancer. 2006;119:624–9. doi: 10.1002/ijc.21888. [DOI] [PubMed] [Google Scholar]
- 122.Mannelli M, Simi L, Gagliano MS, Opocher G, Ercolino T, Becherini L, Parenti G. Genetics and biology of pheochromocytoma. Exp Clin Endocrinol Diabetes. 2007;115:160–5. doi: 10.1055/s-2007-970407. [DOI] [PubMed] [Google Scholar]
- 123.Park DM, Zhuang Z, Chen L, Szerlip N, Maric I, Li J, Sohn T, Kim SH, Lubensky IA, Vortmeyer AO, Rodgers GP, Oldfield EH, Lonser RR. von Hippel-Lindau disease-associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med. 2007;4:e60. doi: 10.1371/journal.pmed.0040060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lugus JJ, Park C, Choi K. Developmental relationship between hematopoietic and endothelial cells. Immunol Res. 2005;32:57–74. doi: 10.1385/IR:32:1-3:057. [DOI] [PubMed] [Google Scholar]
- 125.Chan CC, Collins AB, Chew EY. Molecular pathology of eyes with von Hippel-Lindau (VHL) Disease: a review. Retina. 2007;27:1–7. doi: 10.1097/01.iae.0000244659.62202.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sklar CA. Childhood brain tumors. J Pediatr Endocrinol Metab. 2002;2:669–73. doi: 10.1515/jpem.2002.15.s2.669. [DOI] [PubMed] [Google Scholar]
- 127.Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, Golub TR. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 128.Nakagawara A, Ohira M. Comprehensive genomics linking between neural development and cancer: neu-roblastoma as a model. Cancer Lett. 2004;204:213–24. doi: 10.1016/S0304-3835(03)00457-9. [DOI] [PubMed] [Google Scholar]
- 129.Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, Biedler JL, Cheung NK, Ross RA. Characteristics of stem cells from human neuroblas-toma cell lines and in tumors. Neoplasia. 2004;6:838–45. doi: 10.1593/neo.04310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ross RA, Spengler BA. Human neuroblastoma stem cells. Semin Cancer Biol. 2007;17:241–7. doi: 10.1016/j.semcancer.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 131.Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002;2:826–35. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]
- 132.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9:777–94. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 134.Kopp HG, Ramos CA, Rafii S. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol. 2006;13:175–81. doi: 10.1097/01.moh.0000219664.26528.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ganss R. Tumor stroma fosters neovascularization by recruitment of progenitor cells into the tumor bed. J Cell Mol Med. 2006;10:857–65. doi: 10.1111/j.1582-4934.2006.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 136.Moreira IS, Fernandes PA, Ramos MJ. Vascular endothelial growth factor (VEGF) inhibition–a critical review. Anticancer Agents Med Chem. 2007;7:223–45. doi: 10.2174/187152007780058687. [DOI] [PubMed] [Google Scholar]
- 137.Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu Z, Hackett NR, Crystal RG, Moore MA, Hajjar KA, Manova K, Benezra R, Rafii S. Impaired recruitment of bone-marrow-derived endothe-lial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 138.Peters BA, Diaz LA, Polyak K, Meszler L, Romans K, Guinan EC, Antin JH, Myerson D, Hamilton SR, Vogelstein B, Kinzler KW, Lengauer C. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med. 2005;11:261–2. doi: 10.1038/nm1200. [DOI] [PubMed] [Google Scholar]
- 139.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Davidoff AM, Ng CY, Brown P, Leary MA, Spurbeck WW, Zhou J, Horwitz E, Vanin EF, Nienhuis AW. Bone marrow-derived cells contribute to tumor neovasculature and, when modified to express an angiogenesis inhibitor, can restrict tumor growth in mice. Clin Cancer Res. 2001;7:2870–9. [PubMed] [Google Scholar]
- 141.Dome B, Timar J, Dobos J, Meszaros L, Raso E, Paku S, Kenessey I, Ostoros G, Magyar M, Ladanyi A, Bogos K, Tovari J. Identification and clinical significance of circulating endothelial progenitor cells in human non-small cell lung cancer. Cancer Res. 2006;66:7341–7. doi: 10.1158/0008-5472.CAN-05-4654. [DOI] [PubMed] [Google Scholar]
- 142.Hammerling GJ, Ganss R. Vascular integration of endothelial progenitors during multistep tumor progression. Cell Cycle. 2006;5:509–11. doi: 10.4161/cc.5.5.2517. [DOI] [PubMed] [Google Scholar]
- 143.Spring H, Schuler T, Arnold B, Hammerling GJ, Ganss R. Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci USA. 2005;102:18111–6. doi: 10.1073/pnas.0507158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Santarelli JG, Udani V, Yung YC, Cheshier S, Wagers A, Brekken RA, Weissman I, Tse V. Incorporation of bone marrow-derived Flk-1-expressing CD34+ cells in the endothelium of tumor vessels in the mouse brain. Neurosurgery. 2006;59:374–82. doi: 10.1227/01.NEU.0000222658.66878.CC. [DOI] [PubMed] [Google Scholar]
- 145.Hilbe W, Dirnhofer S, Oberwasserlechner F, Schmid T, Gunsilius E, Hilbe G, Woll E, Kahler CM. CD133 positive endothelial progenitor cells contribute to the tumour vasculature in non-small cell lung cancer. J Clin Pathol. 2004;57:965–9. doi: 10.1136/jcp.2004.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Da Silva ML, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all postnatal organs and tissues. J Cell Sci. 2006;119:2204–13. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 147.Zengin E, Chalajour F, Gehling UM, Ito WD, Treede H, Lauke H, Weil J, Reichenspurner H, Kilic N, Ergun S. Vascular wall resident progenitor cells: a source for postnatal vasculogenesis. Development. 2006;133:1543–51. doi: 10.1242/dev.02315. [DOI] [PubMed] [Google Scholar]
- 148.Bruno S, Bussolati B, Grange C, Collino F, Graziano ME, Ferrando U, Camussi G. CD133 +renal progenitor cells contribute to tumor angiogene-sis. Am J Pathol. 2006;169:2223–35. doi: 10.2353/ajpath.2006.060498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sun B, Zhang S, Ni C, Zhang D, Liu Y, Zhang W, Zhao X, Zhao C, Shi M. Correlation between melanoma angiogenesis and the mesenchymal stem cells and endothelial progenitor cells derived from bone marrow. Stem Cells Dev. 2005;14:292–8. doi: 10.1089/scd.2005.14.292. [DOI] [PubMed] [Google Scholar]
- 150.Sell S. Leukemia:stem cells, maturation arrest, and differentiation therapy. Stem Cell Rev. 2005;1:197–205. doi: 10.1385/SCR:1:3:197. [DOI] [PubMed] [Google Scholar]
- 151.Tokar EJ, Ancrile BB, Cunha GR, Webber MM. Stem/progenitor and intermediate cell types and the origin of human prostate cancer. Differentiation. 2005;73:463–73. doi: 10.1111/j.1432-0436.2005.00047.x. [DOI] [PubMed] [Google Scholar]
- 152.Villadsen R. In search of a stem cell hierarchy in the human breast and its relevance to breast cancer evolution. APMIS. 2005;113:903–21. doi: 10.1111/j.1600-0463.2005.apm_344.x. [DOI] [PubMed] [Google Scholar]
- 153.Morrow M, Samanta A, Kioussis D, Brady HJ, Williams O. TEL-AML1 preleukemic activity requires the DNA binding domain of AML1 and the dimerization and corepressor binding domains of TEL. Oncogene. 2007;26:4404–14. doi: 10.1038/sj.onc.1210227. [DOI] [PubMed] [Google Scholar]
- 154.Martinez-Jaramillo G, Vela-Ojeda J, Sanchez-Valle E, Montesinos JJ, Mayani H. in vitro functional alterations in the hematopoietic system of adult patients with acute lymphoblastic leukemia. Leuk Res. 2007;31:83–9. doi: 10.1016/j.leukres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 155.Zhang M, Rosen JM. Stem cells in the etiology and treatment of cancer. Curr Opin Genet Dev. 2006;16:60–4. doi: 10.1016/j.gde.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 156.Birnbaum D, Bertucci F, Ginestier C, Tagett R, Jacquemier J, Charafe-Jauffret E. Basal and lumi-nal breast cancers:basic or luminous? (review) Int J Oncol. 2004;25:249–58. [PubMed] [Google Scholar]
- 157.Eberhart CG. In search of the medulloblast: neural stem cells and embryonal brain tumors. Neurosurg. Clin N Am. 2007;18:59–ix. doi: 10.1016/j.nec.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 158.Zindy F, Uziel T, Ayrault O, Calabrese C, Valentine M, Rehg JE, Gilbertson RJ, Sherr CJ, Roussel MF. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebel-lar granule neuron precursors. Cancer Res. 2007;67:2676–84. doi: 10.1158/0008-5472.CAN-06-3418. [DOI] [PubMed] [Google Scholar]
- 159.Cobaleda C, Gutierrez-Cianca N, Perez-Losada J, Flores T, Garcia-Sanz R, Gonzalez M, Sanchez-Garcia I. A primitive hematopoietic cell is the target for the leukaemic transformation in human philadelphia-positive acute lymphoblastic leukemia. Blood. 2000;95:1007–13. [PubMed] [Google Scholar]
- 160.Nakagawa R, Soh JW, Michie AM. Subversion of protein kinase C alpha signaling in hematopoietic progenitor cells results in the generation of a B-cell chronic lymphocytic leukemia-like population in vivo. Cancer Res. 2006;66:527–34. doi: 10.1158/0008-5472.CAN-05-0841. [DOI] [PubMed] [Google Scholar]
- 161.Reilly JT. Class III receptor tyrosine kinases: role in leukaemogenesis. Br J Haematol. 2002;116:744–57. doi: 10.1046/j.0007-1048.2001.03294.x. [DOI] [PubMed] [Google Scholar]
- 162.Moore MA. Converging pathways in leukemogenesis and stem cell self-renewal. Exp Hematol. 2005;33:719–37. doi: 10.1016/j.exphem.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 163.Ysebaert L, Chicanne G, Demur C, De TF, Prade-Houdellier N, Ruidavets JB, Mansat-De M, Rigal-Huguet F, Laurent G, Payrastre B, Manenti S, Racaud-Sultan C. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia. 2006;20:1211–6. doi: 10.1038/sj.leu.2404239. [DOI] [PubMed] [Google Scholar]
- 164.Sengupta A, Banerjee D, Chandra S, Banerji SK, Ghosh R, Roy R, Banerjee S. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and Notch signaling in chronic myeloid leukemia progression. Leukemia. 2007;21:949–55. doi: 10.1038/sj.leu.2404657. [DOI] [PubMed] [Google Scholar]
- 165.Frankfurt O, Tallman MS. Growth factors in leukemia. J Natl Compr Canc Netw. 2007;5:203–15. doi: 10.6004/jnccn.2007.0020. [DOI] [PubMed] [Google Scholar]
- 166.Bhatia M, Bonnet D, Murdoch B, Gan OI, Dick JE. A newly discovered class of human hematopoietic cells with SCID-repopulating activity. Nat Med. 1998;4:1038–45. doi: 10.1038/2023. [DOI] [PubMed] [Google Scholar]
- 167.Zanjani ED, Meida-Porada G, Livingston AG, Zeng H, Ogawa M. Reversible expression of CD34 by adult human bone marrow long-term engrafting hematopoi-etic stem cells. Exp Hematol. 2003;31:406–12. doi: 10.1016/s0301-472x(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 168.Gupta P, Gurudutta GU, Verma YK, Kishore V, Gulati S, Sharma RK, Chandra R, Saluja D. PU.1: An ETS family transcription factor that regulates leukemogenesis besides normal hematopoiesis. Stem Cells Dev. 2006;15:609–17. doi: 10.1089/scd.2006.15.609. [DOI] [PubMed] [Google Scholar]
- 169.Carella C, Bonten J, Rehg J, Grosveld GC. MN1-TEL, the product of the t(12;22) in human myeloid leukemia, immortalizes murine myeloid cells and causes myeloid malignancy in mice. Leukemia. 2006;20:1582–92. doi: 10.1038/sj.leu.2404298. [DOI] [PubMed] [Google Scholar]
- 170.Nishida S, Hosen N, Shirakata T, Kanato K, Yanagihara M, Nakatsuka S, Hoshida Y, Nakazawa T, Harada Y, Tatsumi N, Tsuboi A, Kawakami M, Oka Y, Oji Y, Aozasa K, Kawase I, Sugiyama H. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood. 2006;107:3303–12. doi: 10.1182/blood-2005-04-1656. [DOI] [PubMed] [Google Scholar]
- 171.Somervaille TC, Cleary ML. PU.1 and Junb: suppressing the formation of acute myeloid leukemia stem cells. Cancer Cell. 2006;10:456–7. doi: 10.1016/j.ccr.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 172.Moore MA, Dorn DC, Schuringa JJ, Chung KY, Morrone G. Constitutive activation of Flt3 and STAT5A enhances self-renewal and alters differentiation of hematopoietic stem cells. Exp Hematol. 2007;35:105–16. doi: 10.1016/j.exphem.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 173.Kosmider O, Denis N, Lacout C, Vainchenker W, Dubreuil P, Moreau-Gachelin F. Kit-activating mutations cooperate with Spi-1/PU.1 overexpression to promote tumorigenic progression during ery-throleukemia in mice. Cancer Cell. 2005;8:467–78. doi: 10.1016/j.ccr.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 174.Primo D, Flores J, Quijano S, Sanchez ML, Sarasquete ME, Del Pino-Montes J, Gaarder PI, Gonzalez M, Orfao A. Impact of BCR/ABL gene expression on the proliferative rate of different sub-populations of haematopoietic cells in chronic myeloid leukaemia. Br J Haematol. 2006;135:43–51. doi: 10.1111/j.1365-2141.2006.06265.x. [DOI] [PubMed] [Google Scholar]
- 175.Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukaemic progenitors and stem cells is essential for improved treatment of Ph +leukemia in mice. Proc Natl Acad Sci USA. 2006;103:16870–5. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Asahara T, Kawamoto A. Endothelial progenitor cells for postnatal vasculogenesis. Am J Physiol Cell Physiol. 2004;287:C572–9. doi: 10.1152/ajpcell.00330.2003. [DOI] [PubMed] [Google Scholar]
- 177.Gibbs CP, Kukekov VG, Reith JD, Tchigrinova O, Suslov ON, Scott EW, Ghivizzani SC, Ignatova TN, Steindler DA. Stem-like cells in bone sarcomas:impli-cations for tumorigenesis. Neoplasia. 2005;7:967–76. doi: 10.1593/neo.05394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tolar J, Nauta AJ, Osborn MJ, Panoskaltsis MA, McElmurry RT, Bell S, Xia L, Zhou N, Riddle M, Schroeder TM, Westendorf JJ, McIvor RS, Hogendoorn PC, Szuhai K, Oseth L, Hirsch B, Yant SR, Kay MA, Peister A, Prockop DJ, Fibbe WE, Blazar BR. Sarcoma derived from cultured mesenchymal stem cells. Stem Cells. 2007;25:371–9. doi: 10.1634/stemcells.2005-0620. [DOI] [PubMed] [Google Scholar]
- 179.Riggi N, Cironi L, Provero P, Suva ML, Kaloulis K, Garcia-Echeverria C, Hoffmann F, Trumpp A, Stamenkovic I. Development of Ewing's sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005;65:11459–68. doi: 10.1158/0008-5472.CAN-05-1696. [DOI] [PubMed] [Google Scholar]
- 180.Holland EC, Li Y, Celestino J, Dai C, Schaefer L, Sawaya RA, Fuller GN. Astrocytes give rise to oligo-dendrogliomas and astrocytomas after gene transfer of polyoma virus middle T antigen in vivo. Am J Pathol. 2000;157:1031–7. doi: 10.1016/S0002-9440(10)64615-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sanai N, Varez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–22. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- 182.Lubensky IA, Vortmeyer AO, Kim S, Lonser RR, Park DM, Ikejiri B, Li J, Okamoto H, Walbridge S, Ryschkewitsch C, Major E, Oldfield EH, Zhuang Z. Identification of tumor precursor cells in the brains of primates with radiation-induced de novo glioblastoma multiforme. Cell Cycle. 2006;5:452–6. doi: 10.4161/cc.5.4.2482. [DOI] [PubMed] [Google Scholar]
- 183.Dellovade T, Romer JT, Curran T, Rubin LL. The hedgehog pathway and neurological disorders. Annu Rev Neurosci. 2006;29:539–63. doi: 10.1146/annurev.neuro.29.051605.112858. [DOI] [PubMed] [Google Scholar]
- 184.Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD, Albin MG, Emnett RJ, Loeser S, Watson MA, Nagarajan R, Gutmann DH. Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res. 2007;67:890–900. doi: 10.1158/0008-5472.CAN-06-0973. [DOI] [PubMed] [Google Scholar]
- 185.Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–30. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mao Y, Zhou L, Zhu W, Wang X, Yang G, Xie L, Mao X, Jin K. Proliferative status of tumor stem cells may be correlated with malignancy grade of human astro-cytomas. Front Biosci. 2007;12:2252–9. doi: 10.2741/2227. [DOI] [PubMed] [Google Scholar]
- 187.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 188.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–5. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 189.Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007;67:4807–15. doi: 10.1158/0008-5472.CAN-06-4608. [DOI] [PubMed] [Google Scholar]
- 190.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 192.Teuliere J, Faraldo MM, Deugnier MA, Shtutman M, Ben-Ze'ev A, Thiery JP, Glukhova MA. Targeted activation of beta-catenin signaling in basal mammary epithelial cells affects mammary development and leads to hyperplasia. Development. 2005;132:267–77. doi: 10.1242/dev.01583. [DOI] [PubMed] [Google Scholar]
- 193.Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol. 2005;277:443–56. doi: 10.1016/j.ydbio.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 194.Du Z, Podsypanina K, Huang S, McGrath A, Toneff MJ, Bogoslovskaia E, Zhang X, Moraes RC, Fluck M, Allred DC, Lewis MT, Varmus HE, Li Y. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proc Natl Acad Sci USA. 2006;103:17396–401. doi: 10.1073/pnas.0608607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tang P, Wang X, Schiffhauer L, Wang J, Bourne P, Yang Q, Quinn A, Hajdu S. Expression patterns of ER-alpha, PR, HER-2/neu, and EGFR in different cell origin subtypes of high grade and non-high grade ductal carcinoma in situ. Ann Clin Lab Sci. 2006;36:137–43. [PubMed] [Google Scholar]
- 196.Moraes RC, Zhang X, Harrington N, Fung JY, Wu MF, Hilsenbeck SG, Allred DC, Lewis MT. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development. 2007;134:1231–42. doi: 10.1242/dev.02797. [DOI] [PubMed] [Google Scholar]
- 197.Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, Gilks CB, Van De Rijn M, Perou CM. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–74. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 198.Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, Perou CM. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19:264–71. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 199.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492–02. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 200.Jumppanen M, Gruvberger-Saal S, Kauraniemi P, Tanner M, Bendahl PO, Lundin M, Krogh M, Kataja P, Borg A, Ferno M, Isola J. Basal-like phenotype is not associated with patient survival in estrogen receptor negative breast cancers. Breast Cancer Res. 2007;9:R16. doi: 10.1186/bcr1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sorlie T, Wang Y, Xiao C, Johnsen H, Naume B, Samaha RR, Borresen-Dale AL. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics. 2006;7:127. doi: 10.1186/1471-2164-7-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Potemski P, Kusinska R, Watala C, Pluciennik E, Bednarek AK, Kordek R. Prognostic relevance of basal cytokeratin expression in operable breast cancer. Oncology. 2005;69:478–85. doi: 10.1159/000090986. [DOI] [PubMed] [Google Scholar]
- 203.Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109:25–32. doi: 10.1002/cncr.22381. [DOI] [PubMed] [Google Scholar]
- 204.Asselin-Labat ML, Shackleton M, Stingl J, Vaillant F, Forrest NC, Eaves CJ, Visvader JE, Lindeman GJ. Steroid hormone receptor status of mouse mammary stem cells. J Natl Cancer Inst. 2006;98:1011–4. doi: 10.1093/jnci/djj267. [DOI] [PubMed] [Google Scholar]
- 205.Sleeman KE, Kendrick H, Robertson D, Isacke CM, Ashworth A, Smalley MJ. Dissociation of estrogen receptor expression and in vivo stem cell activity in the mammary gland. J Cell Biol. 2007;176:19–26. doi: 10.1083/jcb.200604065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–19. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 207.Kurbel S. Selective reduction of estrogen receptor (ER) positive breast cancer occurrence by estrogen receptor modulators supports etiological distinction between ER positive and ER negative breast cancers. Med Hypotheses. 2005;64:1182–7. doi: 10.1016/j.mehy.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 208.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kim M, Turnquist H, Jackson J, Sgagias M, Yan Y, Gong M, Dean M, Sharp JG, Cowan K. The mul-tidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin Cancer Res. 2002;8:22–8. [PubMed] [Google Scholar]
- 210.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. CEACAM6 gene silencing impairs anoikis resistance and in vivo metastatic ability of pancreatic ade-nocarcinoma cells. Oncogene. 2004;23:465–73. doi: 10.1038/sj.onc.1207036. [DOI] [PubMed] [Google Scholar]
- 211.Kornblau SM, Qiu YH, Bekele BN, Cade JS, Zhou X, Harris D, Jackson CE, Estrov Z, Andreeff M. Studying the right cell in acute myelogenous leukemia:dynamic changes of apoptosis and signal transduction pathway protein expression in chemotherapy resistant ex-vivo selected “survivor cells”. Cell Cycle. 2006;5:2769–77. doi: 10.4161/cc.5.23.3507. [DOI] [PubMed] [Google Scholar]
- 212.Chen MS, Woodward WA, Behbod F, Peddibhotla S, Alfaro MP, Buchholz TA, Rosen JM. Wnt/beta-catenin mediates radiation resistance of Sca1+ pro-genitors in an immortalized mammary gland cell line. J Cell Sci. 2007;120:468–77. doi: 10.1242/jcs.03348. [DOI] [PubMed] [Google Scholar]
- 213.Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44 +breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–85. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 214.Yanai K, Nagai S, Wada J, Yamanaka N, Nakamura M, Torata N, Noshiro H, Tsuneyoshi M, Tanaka M, Katano M. Hedgehog signaling pathway is a possible therapeutic target for gastric cancer. J Surg Oncol. 2007;95:55–62. doi: 10.1002/jso.20606. [DOI] [PubMed] [Google Scholar]
- 215.Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–52. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 216.Mimeault M, Johansson SL, Venkatraman G, Moore E, Henichart JP, Depreux P, Lin MF, Batra SK. Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Mol Cancer Ther. 2007;6:967–78. doi: 10.1158/1535-7163.MCT-06-0648. [DOI] [PubMed] [Google Scholar]
- 217.Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene. 2007;26:1357–60. doi: 10.1038/sj.onc.1210200. [DOI] [PubMed] [Google Scholar]
- 218.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–84. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 219.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, Cameron D, Dowsett M, Barrios CH, Steger G, Huang CS, Andersson M, Inbar M, Lichinitser M, Lang I, Nitz U, Iwata H, Thomssen C, Lohrisch C, Suter TM, Ruschoff J, Suto T, Greatorex V, Ward C, Straehle C, McFadden E, Dolci MS, Gelber RD Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–72. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 220.Zhang Q, Chen G, Liu X, Qian Q. Monoclonal antibodies as therapeutic agents in oncology and antibody gene therapy. Cell Res. 2007;17:89–99. doi: 10.1038/sj.cr.7310143. [DOI] [PubMed] [Google Scholar]
- 221.Sano T, Kagawa M, Okuno M, Ishibashi N, Hashimoto M, Yamamoto M, Suzuki R, Kohno H, Matsushima-Nishiwaki R, Takano Y, Tsurumi H, Kojima S, Friedman SL, Moriwaki H, Tanaka T. Prevention of rat hepatocarcinogenesis by acyclic retinoid is accompanied by reduction in emergence of both TGF-alpha-expressing oval-like cells and activated hepatic stellate cells. Nutr Cancer. 2005;51:197–206. doi: 10.1207/s15327914nc5102_10. [DOI] [PubMed] [Google Scholar]
- 222.Ringden O. Immunotherapy by allogeneic stem cell transplantation. Adv Cancer Res. 2007;97C:25–60. doi: 10.1016/S0065-230X(06)97002-X. [DOI] [PubMed] [Google Scholar]
- 223.Avramova B, Jordanova M, Michailov G, Konstantinov D, Christosova I, Bobev D. Myeloablative chemotherapy with autologous peripheral blood stem cell transplantation in patients with poor-prognosis solid tumors – Bulgarian experience. J BUON. 2006;11:433–48. [PubMed] [Google Scholar]
- 224.Mimeault M, Hauke R, Batra SK. Stem cells:A revolution in therapeutics–Recent advances on the stem cell biology and their therapeutic applications in regenerative medicine and cancer therapies. Clin Pharm Ther. 2007;82:252–64. doi: 10.1038/sj.clpt.6100301. [DOI] [PubMed] [Google Scholar]
- 225.Bickenbach K, Wilcox R, Veerapong J, Kindler HL, Posner MC, Noffsinger A, Roggin KK. A review of resistance patterns and phenotypic changes in gastrointestinal stromal tumors following imatinib mesylate therapy. J Gastrointest Surg. 2007;11:758–66. doi: 10.1007/s11605-007-0150-y. [DOI] [PubMed] [Google Scholar]
- 226.Burger H, Nooter K. Pharmacokinetic resistance to imatinib mesylate: role of the ABC drug pumps ABCG2 (BCRP) and ABCB1 (MDR1) in the oral bioavailability of imatinib. Cell Cycle. 2004;3:1502–05. doi: 10.4161/cc.3.12.1331. [DOI] [PubMed] [Google Scholar]
- 227.Schenone S, Manetti F, Botta M. Last findings on dual inhibitors of abl and SRC tyrosine-kinases. Mini Rev Med Chem. 2007;7:191–201. doi: 10.2174/138955707779802598. [DOI] [PubMed] [Google Scholar]
- 228.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, Eaves C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–35. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- 229.Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myeloge-nous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–8. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- 230.Angstreich GR, Matsui W, Huff CA, Vala MS, Barber J, Hawkins AL, Griffin CA, Smith BD, Jones RJ. Effects of imatinib and interferon on primitive chronic myeloid leukaemia progenitors. Br J Haematol. 2005;130:373–81. doi: 10.1111/j.1365-2141.2005.05606.x. [DOI] [PubMed] [Google Scholar]
- 231.White DL, Saunders VA, Dang P, Engler J, Zannettino AC, Cambareri AC, Quinn SR, Manley PW, Hughes TP. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107):reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108:697–704. doi: 10.1182/blood-2005-11-4687. [DOI] [PubMed] [Google Scholar]
- 232.Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, Roberts AW, Strasser A. Bim and Bad mediate imatinib-induced killing of Bcr/Abl +leukaemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci USA. 2006;103:14907–12. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Jorgensen HG, Copland M, Allan EK, Jiang X, Eaves A, Eaves C, Holyoake TL. Intermittent exposure of primitive quiescent chronic myeloid leukemia cells to granulocyte-colony stimulating factor in vitro promotes their elimination by imatinib mesylate. Clin Cancer Res. 2006;12:626–33. doi: 10.1158/1078-0432.CCR-05-0429. [DOI] [PubMed] [Google Scholar]
- 234.Holtz M, Forman SJ, Bhatia R. Growth factor stimulation reduces residual quiescent chronic myeloge-nous leukemia progenitors remaining after imatinib treatment. Cancer Res. 2007;67:1113–20. doi: 10.1158/0008-5472.CAN-06-2014. [DOI] [PubMed] [Google Scholar]
- 235.Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, Nagler A, Tavor S, Ein-Dor L, Lapidot T, Domany E, Rechavi G, Givol D. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia. 2006;20:2147–54. doi: 10.1038/sj.leu.2404401. [DOI] [PubMed] [Google Scholar]
- 236.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukaemic stem cells. Nat Med. 2006;12:1167–74. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 237.Deshpande AJ, Cusan M, Rawat VP, Reuter H, Krause A, Pott C, Quintanilla-Martinez L, Kakadia P, Kuchenbauer F, Ahmed F, Delabesse E, Hahn M, Lichter P, Kneba M, Hiddemann W, Macintyre E, Mecucci C, Ludwig WD, Humphries RK, Bohlander SK, Feuring-Buske M, Buske C. Acute myeloid leukemia is propagated by a leukaemic stem cell with lymphoid characteristics in a mouse model of CALM/AF10-positive leukemia. Cancer Cell. 2006;10:363–74. doi: 10.1016/j.ccr.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 238.Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA, Watkins DN, Matsui W. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. 2007;104:4048–53. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007;26:5674–9. doi: 10.1038/sj.onc.1210356. [DOI] [PubMed] [Google Scholar]
- 240.Clement V, Sanchez P, De TN, Radovanovic I, Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–72. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antian-giogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67:3560–4. doi: 10.1158/0008-5472.CAN-06-4238. [DOI] [PubMed] [Google Scholar]
- 242.Yang ZJ, Wechsler-Reya RJ. Hit ‘em where they live: targeting the cancer stem cell niche. Cancer Cell. 2007;11:3–5. doi: 10.1016/j.ccr.2006.12.007. [DOI] [PubMed] [Google Scholar]