

Abstract
There is now accumulating evidence that bone marrow-derived mesenchymal stem cells (MSCs) make an important contribution to postnatal vasculogenesis, especially during tissue ischaemia and tumour vascularization. Identifying mechanisms which regulate the role of MSCs in vasculogenesis is a key therapeutic objective, since while increased neovascularization can be advantageous during tissue ischaemia, it is deleterious during tumourigenesis. The potent angiogenic stimulant vascular endothelial growth factor (VEGF) is known to regulate MSC mobilization and recruitment to sites of neovascularization, as well as directing the differentiation of MSCs to a vascular cell fate. Despite the fact that MSCs did not express VEGF receptors, we have recently identified that VEGF-A can stimulate platelet-derived growth factor (PDGF) receptors, which regulates MSC migration and proliferation. This review focuses on the role of PDGF receptors in regulating the vascular cell fate of MSCs, with emphasis on the function of the novel VEGF-A/PDGF receptor signalling mechanism.
Keywords: mesenchymal stem cell, platelet-derived growth factor receptor, vascular endothelial growth factor, neovascularization, vasculogenesis
Introduction
The differentiation of adult bone marrow-derived mesenchymal stem cells (MSCs) towards chondrocytes, osteocytes or adipocytes is reasonably well characterized and documented [1]. In comparison, MSC differentiation to a vascular cell fate, such as endothelial cells or vascular smooth muscle cells (SMCs), is less well defined and more controversial. However, recent studies both in vitro and in vivo utilizing animal models, clearly identify bone marrowderived MSCs in making an important contribution to the formation of new blood vessels in the adult [2–4], a process known as neovasculogenesis. Local microenvironmental factors, such as growth factor expression and concentration, the ECM composition, the level of oxygen tension and mechanical strain, all contribute to modulating and directing the MSC fate. Two structurally related vascular growth factors, vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), both play crucial roles in regulating vascular cells during blood vessel wall assembly [5–8], but their involvement in regulating MSCs during vasculogenesis is less well understood and likely to be more complex.
We have been studying the role of MSCs during vasculogenesis and factors that may be important in determining their differentiation. One intriguing finding is that VEGF-A can induce VEGF receptor-negative MSCs to migrate and proliferate, by binding to and activating PDGF receptors [9]. MSCs in vitro have a high cell surface PDGFR : PDGFR ratio [10], with abundant PDGFR appearing to be a characteristic of undifferentiated MSCs. Thus cell surface PDGF receptor expression will be an important determinant in mediating the regulation of MSCs. In this review, we first introduce the VEGF/PDGF super-family of ligands and receptors, then focus on the role of PDGF receptors in directing MSCs towards a vascular cell fate, with emphasis on the role of the novel VEGF-A/PDGF receptor signalling mechanism. Finally, the contributions of MSCs to vascular repair and neovascularization are discussed.
The vascular endothelial growth factor/platelet-derived growth factor super-family of ligands and receptors
VEGF and PDGF belong to the VEGF/PDGF superfamily of ligands and receptors, grouped together on the basis of their close sequence homology and evolutionary relationship. Each of the ligands contains eight conserved cysteine residues, forming a typical cystine-knot motif [11]. Sequence analysis predicts that PDGF and VEGF evolved from a common ancestral gene [5]. PDGF/VEGF-like factors (PVFs) containing a cystine-knot motif, have been identified in invertebrates. Drosophila melanogaster contains three different PVFs which function through a single receptor (PVR). These play an essential role in the guidance of cell migration during oogenesis and haematopoiesis [12, 13] and also are important in mediating haemocyte cell survival [14]. In Caenorhabditis elegans, a PVF and four possible PDGF/VEGF receptors (VER-1–VER-4) have been identified. Interestingly, the PVF was able to bind specifically to the human VEGF receptors, VEGFR1 and VEGFR2, which induced receptor phosphorylation and promoted angiogenesis [15].
Vascular endothelial growth factor ligands
The large number of VEGF family members highlights their structural and functional diversity. The most abundant and biologically active VEGF ligand is VEGF-A [5, 6], containing eight exons which undergo alternative splicing to produce several different isoforms. Exons 6 and 7 are important since these contain heparin binding domains. The most prevalent human VEGF-A isoforms are VEGF-A165 which lacks exon 6 but can bind heparin, and VEGF-A121 which lacks exons 6 and 7, therefore cannot bind heparin, but is freely diffusible [16]. Both isoforms are secreted as homodimers. A variety of different cell types express VEGF-A, including macrophages, vascular SMC, numerous tumour cells and MSC [5, 17, 18]. Several factors can up-regulate VEGF-A expression, including exposure to cytokines or growth factors, such as transforming growth factor-, fibroblast growth factor-2β (FGF-2) or PDGF [16]. A synergistic relationship exists between VEGF-A and FGF-2, which potently stimulates both in vitro and in vivo neovascular formation [19]. VEGF-A was shown to enhance endothelial cell PDGF-B expression, while FGF-2 promoted mural cell PDGFRβ expression [20].
Another important physiological and pathological stimulator of VEGF-A expression is the local environmental oxygen concentration. While VEGF-A expression is normally low in most adult tissues, atmospheres with reduced oxygen tension potently stimulate VEGF-A secretion [21]. Increased VEGF-A expression plays a critical role during neovascularization, wound healing and tissue repair processes. Hypoxia-induced transcription of VEGF-A is mediated by the binding of a hypoxia-inducible transcription factor-1 (HIF-1α), a heterodimer composed of subunits HIF-1α and HIF-1β, to hypoxia responsive enhancer elements (HREs) within the VEGF-A promoter [22], resulting in transcriptional activation. During normal oxygen concentrations, intracellular HIF-1α levels are low, due to rapid degradation mediated by oxygen-dependent prolyl hydroxylase-2 (PHD-2) [23]. However, hypoxia suppresses PHD-2 activity, resulting in increase levels of HIF-1α which associate with HIF-1β to form a heterodimer, resulting in transcriptional activation of VEGF-A.
Platelet-derived growth factor ligands
The PDGF signalling system has a versatile composition, consisting of four ligands, PDGF-A, PDGF-B, PDGF-C and PDGF-D [24, 25]. All four ligands, which are inactive in their monomeric form, assemble intracellularly to form disulfide-linked homodimers, but only PDGF-A and PDGF-B form a heterodimer. It is assumed that regulation of PDGF isoform expression is largely due to transcriptional control. Thus if both PDGF-A and PDGF-B are expressed, a PDGF-AB heterodimer is just as likely to form as a PDGF homodimer. Human PDGF-A and PDGF-D transcripts may also undergo alternative splicing. A short form of PDGF-A can be generated by exclusion of exon 6, while a short sequence can be alternatively spliced in PDGF-D [25]. PDGF-C and PDGF-D are distinct in having a novel N-terminal CUB domain and are structurally more similar to the VEGF family than the PDGFs [25]. Both isoforms are controlled by post-translational processing, since they are secreted as latent inactive forms, which require extracellu-lar proteolytic removal of the CUB domain to allow receptor binding [25].
Vascular endothelial growth factor receptors
VEGF and PDGF receptors are members of the class III receptor tyrosine kinase sub-family, which have extracellular immunoglobulin-like (Ig) domains, a transmembrane region and split intracellular kinase domains. VEGF receptors have seven Ig domains, while PDGF receptors have five extracellular Ig domains [26]. These and other structural similarities suggest a close evolutionary relationship. Three human VEGF receptors (VEGFR1, VEGFR2 and VEGFR3) which each form homodimers on ligand binding have been identified [5, 6]. VEGF-A binds to VEGFR1 and VEGFR2, but not VEGFR3, however, the majority of signalling is mediated by VEGFR2 [5, 6]. VEGF-A165 but not VEGF-A121 interacts with neuropilin-1 (NRP-1) and neuropilin-2 (NRP-2), which are cell surface transmembrane glycoprotein receptors [27]. Since NRPs have a short cytoplasmic domain, it is presumed they do not signal independently, but function only as co-receptors [27]. NRP-1 can associate and form a complex with VEGFR2 [28], resulting in enhanced VEGF-A165 induced VEGFR2 signalling, which was assumed to be mediated by VEGF-A165 binding to NRP-1. However, recently NRP-1 and NRP-2 have been shown to enhance VEGF-A121 induced VEGFR2 signal transduction [29], even though VEGF-A121 does not bind to NRPs.
Platelet-derived growth factor receptors
Active PDGF isoforms stimulate cells by binding to and activating the signalling of two distinct, but structurally related, membrane bound receptor tyrosine kinases, PDGF receptor-α (PDGFRα) and PDGF receptor-β (PDGFRβ) [24, 25]. Ligand binding to a PDGF receptor induces either receptor homodimerization or heterodimerization. PDGF-BB has a greater binding affinity for PDGFRβ, but can also bind PDGFRα, thus can induce all three receptor homodimer and heterodimer combinations. PDGF-AA can only interact with PDGFR, forming a homodimer, but with higher affinity than PDGF-BB, while PDGF-AB may initiate both PDGFRα homod-imer and heterodimer formation. Similarly, while PDGF-CC specifically interacts with PDGFRα, it can induce the formation of both homodimer and heterodimer, thereby resembling PDGF-AB binding affinities [30], while PDGF-DD preferentially binds to PDGFR to form a homodimer, but may induce low levels of heterodimer [31].
Ligand induced PDGF receptor homodimerization or heterodimerization, results in autophosphorylation of specific tyrosine residues within the cytoplasmic domain, referred to as receptor activation. Phosphotyrosine residues provide docking sites for SH2-domain containing signalling molecules such as phosphatidylinositol 3 kinase (PI3K), phospholipase C-γ(PLCγ), Ras GTPase-activating protein (Ras-GAP) and Src family kinases, which initiate signal transduction.
During development, PDGF-A/PDGFRα signalling has been shown to mediate a wide spectrum of critical functions during embryogenesis and organogenesis, affecting a broad range of mesenchymal tissues [7, 32]. In comparison, PDGF-B/PDGFRβ signalling has a narrower range of tissues and functions, primarily essential for the recruitment and differentiation of cells during vascular development [7, 32]. The importance of PDGF signalling during development is clearly demonstrated by knockout studies in mice. PDGF-A and PDGFRα knockouts are both lethal [33, 34], but PDGFRα knockouts are more severe, resulting in death during late gestation caused by a range of defects in several tissues, including vascular abnormalities. Both PDGF-B and PDGFRβ knockouts are severe [35, 36], inducing defective vascular development, resulting in death during the late embryonic stage.
Role of platelet-derived growth factor receptors in regulating the MSC fate
Prenatal and postnatal development studies have identified PDGF signalling as being crucial to the selective expansion andrecruitment of undifferentiated mesenchymal cell and progenitor cell populations, which generate distinct differentiated cell types [8, 32]. PDGF-A/PDGFRα signalling appears to regulate a broad range of progenitor cells, in several different development processes. During mammalian central nervous system development, PDGF-AA expressed by astrocytes and neurons in the spinal cord, plays a central role in stimulating PDGFRα -positive oligodendrocyte progenitor (O2A) cells to proliferate and migrate [37]. PDGF-A/PDGFRα signalling has also been shown to be important for the proliferation and recruitment of lung alveolar SMC progenitor cells [38], mesenchymal stem or progenitor cells during skin development [39] and progenitor cells which form villus clusters during intestine morphogenesis [40].
In comparison, PDGF-B/PDGFRβ signalling is primarily involved in regulating mural (vascular SMC, pericyte) progenitor cells. Endothelial-derived PDGF-BB induces the migration and proliferation of PDGFRβ -positive mural progenitor cells during vascular development [41]. Similarly, pericyte-like mesangial progenitor cells require PDGF-B/PDGFRβ signalling for their proliferation and migration to the developing glomerular capillary tuft, during kidney glomerulus formation [42].
Our studies have shown that MSCs in vitro are defined by abundant cell surface PDGFRα and a high PDGFRα :PDGFRβ ratio [10]. Thus the cell surface PDGFR expression will be an important determinant in mediating the fate of MSCs.
MSCs utilize a novel vascular endothelial growth factor/platelet-derived growth factor receptor signalling mechanism
It was previously assumed that VEGF could only signal by binding to and activating VEGF receptors. Human MSCs derived from normal bone marrow of five different donors, ranging in age from 18 to 26 years old, were shown not to express VEGF receptors. Using these cells, our studies identified a novel VEGF-A signalling mechanism, whereby VEGF-A can directly stimulate PDGF receptor activation [9]. VEGF-A interacted with and stimulated both PDGFRα and PDGFRβ tyrosine phosphorylation activation, which regulated MSC migration and proliferation. This was demonstrated by neutralizing cell surface PDGFRα or PDGFRβ using specific blocking antibodies, or expression knockdown using siRNA oligonucleotides, which significantly attenuated VEGF-A165 induced MSC migration and proliferation. Since VEGF-A165 stimulation resulted in similar tyrosine phosphorylation levels in both PDGFRα and PDGFR, our data suggested that, at least in part, VEGF-A165 induced and activated PDGFR(αβ) het-erodimer formation and signalling.
MSCs express high levels of both PDGF receptors [10], which creates the basis for abundant PDGFR() heterodimer formation, however, a functional role for this receptor combination has not been clearly defined. Double PDGFRα and PDGFRβ knockout studies demonstrate a greater phenotypic severity than individual PDGF receptor knockout phenotypes [8]. PDGF-AB, which initiates the formation and activation of a PDGFRα homodimer and heterodimer, is the most potent PDGF mitogen [43], which suggests that PDGF-AB may specifically activate a high level of PDGF receptor heterodimer phosphorylation. In this respect, both in vitro and in vivo studies demonstrated that PDGF-AB stimulation promoted the differentiation of adult bone marrow-derived cells to cardiomyocytes [44]. In addition, PDGF-AB has also recently been shown to potently stimulate MSC migration [45]. Furthermore, PDGF-C a potent angiogenic factor similar to VEGF [46], which is more similar in structure to VEGFs than PDGFs [25], has been shown to induce the formation and activation of the PDGF receptor heterodimer [46]. Taken together, these studies clearly demonstrate the signalling capacity of the PDGF receptor heterodimer. Since MSCs possess a high potential for abundant PDGF receptor heterodimer formation, which may be a characteristic of MSCs, this suggests PDGFR(αβ) will play an important role in mediating the MSC fate.
VEGF-A165 isoform interactivity and biological function can be regulated by NRP-1 and NRP-2 coreceptors, in a heparin-dependent manner [6, 16, 27]. Co-expression of NRP-1 with VEGFR2 in endothelial cells, significantly facilitates VEGF-A165 induced VEGFR2 signal activation, resulting in enhanced endothelial cell migration [28]. While our studies demonstrated that MSCs clearly expressed NRP-1 and NRP-2 transcripts [9], because both VEGF-A isoforms, VEGF-A165 or VEGF-A121, were shown to equally induced PDGF receptor activation and MSC migration, this suggested that neither heparin binding nor NRPs played an important role. However, recently both NRP-1 and NRP-2 have now been shown to enhance VEGF-A121 induced VEGFR2 signalling [29], thus it remains to be established whether NRPs may function to enhance VEGF-A induced PDGF receptor signalling.
In addition to VEGF-A induced PDGF receptor activation, our analysis also revealed that VEGF-A165 stimulated the phosphorylation of other receptor tyrosine kinases, EGFR, EphA7 and Axl, on the cell surface of MSCs [9]. While PDGF-BB stimulation also resulted in EGFR activation, EphA7 or Axl receptors were not phosphorylated, suggesting the induced activation of EphA7 and Axl receptors were VEGF-A165 specific. The EphA7 receptor has an important role during neural tissue development, whereas, the Axl receptor has been shown to regulate several essential processes during vascular injury and neovascularization [47]. While Axl receptor expression in endothelial cells and vascular SMCs is involved in regulating their migration, proliferation and apoptosis [47], the role of Axl receptor expression in MSCs is not defined. Interestingly, cross-talk between Axl and other membrane bound receptors has recently been reported. While Axl can be transactivated by the IL-15 receptor [48], Axl can inhibit VEGF activation of VEGFR2 [49]. Thus, as discussed later, formation of a membrane complex containing both PDGF and Axl receptors may lead to receptor transmodulation, which could regulate VEGF-A stimulated PDGF receptor activation or Axl receptor tyrosine phosphorylation.
Besides VEGF-A165 being able to induce MSC migration, we also demonstrated that when VEGF-A165 was localized at the cell surface, it could inhibit PDGF-induced MSC migration, presumably by competing for PDGF receptor occupancy [9]. Since MSCs can express and secrete high levels of VEGF-A, especially in atmospheres of reduced oxygen, this suggests an intriguing possibility that autocrine VEGF-A production could regulate paracrine PDGF responses. A schematic diagram representing this proposed mechanism is shown in Figure 1A.
1.
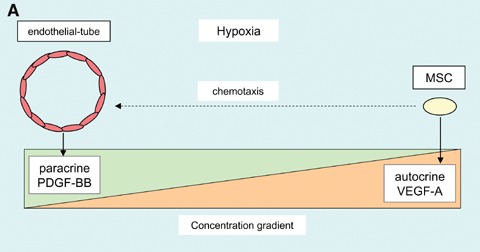
Schematic diagrams highlighting potential mechanisms regulating PDGF receptor signalling.(A) MSC autocrine VEGF-A expression may regulate paracrine PDGF ligand stimulation of PDGF receptors, by VEGF-A competitively binding to PDGF receptors and competing with PDGF concentration gradients. (B) Modulation of VEGF-A induced PDGF receptor signalling specificity is likely to be multifactorial, depending in part on quantitative and qualitative differences, such as; (1) local oxygen concentration regulating VEGF-A expression, (2) matrix sequestration and retention of VEGF-A, (3) soluble VEGF-A level, (4) concentration of PDGF ligands, (5) integrin-matrix interactions, (6) local membrane-associated proteins, (7) recruitment of signalling molecules.
Regulation of vascular endothelial growth factor/platelet-derived growth factor receptor signalling
The finding that VEGF may bind and activate both PDGFRα and PDGFRβ extends the repertoire of ligands which can potentially stimulate individual PDGF receptors. However, this raises the crucial question of how different ligands stimulating the same receptor tyrosine kinase can induce distinct biological responses. Signalling specificity will be reliant on both qualitative and quantitative differences in the stimulation perceived by a particular receptor, which will be dependent on both ligand and receptor expression profiles in a particular microenvironment. Vascular growth factor expression is normally very low in adult tissues, but VEGF and PDGF expression are both significantly up-regulated during vascular repair and regeneration situations. Similarly, while the MSC cell surface PDGF receptor profile may adjust in response to local microenvironmental factors, abundant PDGFRα and the high PDGFRα : PDGFRβ ratio appears to be a characteristic of undifferentiated MSCs.
Crystal structures and mutation analyses have revealed how VEGF-A and PDGFs bind their respective receptors and highlighted similarities in their docking sites. Extracellular immunoglobulin-like (Ig) domains 2 and 3 in both PDGF receptors provide high-affinity binding sites for the PDGF ligands [50]. Similarly, VEGF-A binding to VEGFR2 is also mediated by Ig domains 2 and 3 [51]. Electron microscopy demonstrated that VEGF-A binding to VEGFR2 extracellular Ig domains 2 and 3 induced receptor dimerization [52]. This dimeric association was further stabilized by interactions through membrane-proximal Ig domain 7, which facilitated the positioning and activation of intracellular kinase domains. Since PDGF and VEGF-A ligands utilize similar docking sites on their respective receptors, VEGF-A binding to a PDGF receptor would be expected to be mediated by an analogous mechanism involving subtle differences. Identifying critical growth factor specific binding residues within the PDGF receptors will form the basis for developing ligand specific inhibitors, which potentially may be exploited therapeutically.
While a particular ligand can differ in receptor binding affinities, it may also have distinct activation thresholds. VEGF-A interacts with VEGFR1 with at least a tenfold higher binding affinity than VEGFR2, however, VEGF-A binding to VEGFR1 results in a significantly lower tyrosine phosphorylation activity compared to VEGF-A induced VEGFR2 activation [53]. Ligand signal strength has been shown to regulate PDGFR -induced migration and proliferation, by activating different signalling pathways [54]. A low ligand concentration induced PDGFRα activation of PI3K and migration, whereas a higher ligand concentration was required for activation of PLCγ and proliferation. Thus PDGFRα signalling would be predicted to play an important role in mediating distinct MSC biological responses, following either PDGF or VEGF-A stimulation.
The bioavailability of ligands may also be an important determinant in modulating the strength and duration of PDGF receptor activation. In this regard, the extracellular environment plays a crucial role. Heparin binding motifs present in the C-terminal regions of PDGF-A, PDGF-B [55] and PDGF-C [56] and also in exon 7 of VEGF-A165, can interact with heparan sulphate proteoglycans within the ECM and at the cell surface [57]. This may result in the ECM sequestrating and retaining a significant fraction of the secreted growth factors. In comparison, PDGF-D binding to heparin has not been shown, while VEGF-A121 does not contain a heparin-binding domain and is freely diffusible. Since heparin-binding motifs are important in regulating growth factor diffusion, PDGF-D and VEGF-A121 are likely to have distinct signalling functions compared to the more common heparin binding VEGF/PDGF family members. The deposition of secreted growth factors within the ECM is thought to promote the formation of short-range concentration gradients, which facilitates paracrine signalling and chemotaxis. PDGF-A and PDGF-B homodimers and heterodimer were shown to interact with collagen types I–VI, while retaining their biological activity [58]. VEGF-A165 bound to fibronectin resulted in enhanced and sustained VEGF-A165 stimulated biological activity, which was mediated by an integrin α5 β1/VEGFR2 complex [59, 60]. In contrast, while VEGF-A165 bound to vitronectin, this association resulted in only a transient increase in VEGF-A165 stimulated biological activity [59], highlighting the role of the ECM in modulating the strength and duration of receptor tyrosine kinase signalling activity. Thus, the local matrix composition can regulate the retention and presentation of bioactive ligands at the pericellular interface. Consequently, this may modulate the intensity and/or duration of PDGF or VEGF induced PDGF receptor signalling, resulting in distinct biological responses.
Integrins are an important class of receptors mediating cell–matrix interactions, conveying both inside-out and outside-in signals, which can also modulate receptor tyrosine kinase signalling events. The level of integrin-mediated PDGF receptor regulation will depend on the local ECM composition, together with the number and type of cell surface integrins expressed. Studies have demonstrated that specific integrin/matrix engagement induces integrin clustering and formation of an integrin/PDGF receptor complex. Vitronectin induced αvβ3/PDGFRβ and αvβ3/PDGFRα complexes [61, 62], while collagen type I induced a α2β1/PDGFRβ complex [63], all of which synergistically increased PDGF stimulated responses. Importantly, other membrane-associated proteins have also been shown to transmodulate PDGF/PDGF receptor signalling, including low-density lipoprotein receptor-related protein-1 [64], sphin-gosine 1-phosphate [65] and membrane-type1 matrix metalloproteinase [66]. The formation of membrane complexes or modules, containing receptor tyrosine kinases localized together with other trans-membrane proteins has previously been suggested [67]. A picture therefore emerges of a particular PDGF receptor forming different complexes with local membrane associated proteins, the spatial organization and composition of which, might transmodulate PDGF receptor signalling. In this regard, it is unknown whether the transmembrane NRP receptors, NRP-1 and NRP-2 expressed by MSCs, can transmodulate PDGF receptor activity. Thus it is possible that VEGF or PDGF stimulation of the same PDGF receptor may generate different localized biological responses, dependent upon permissive integrin/matrix interactions and transmem-brane protein dynamics.
Extracellular ligand stimulation of either PDGFRα or PDGFRβ can induce the recruitment, activation and interaction with, slightly different compliments of intracellular signalling molecules. For example, the Crk family of signalling relay molecules only interact with activated PDGFRα, while Ras-GAP only binds to activated PDGFRβ[68]. Distinct integrin/matrix interactions may also regulate the composition of signalling relay molecules which associate with activated PDGF receptors. Fibronectin was shown to mediate the reduced recruitment and association of RasGAP to PDGFRβ, a negative regulator of PDGFR but not PDGFRα activity [69]. In addition, modulation of the intracellular cytoskeleton by integrin outside-in signals, may regulate the activity of PDGF effectors, such as intracellular enzymes and transcription factors, resulting in distinct cellular responses. A schematic diagram illustrating potential mechanisms regulating PDGF receptor signalling is shown in Figure 1B.
MSC and the vasculature
Differentiation of MSC towards endothelial cells
While there is compelling evidence that haematopoietic stem cells (HSC) are a source of endothelial cell progenitors, or angioblasts [70, 71], MSC-derived endothelial cells have been more difficult to establish, due in part, to potential HSC contamination. However, several recent studies have now shown that bone marrow-derived MSCs can be induced to differentiate towards endothelial cells, following exposure to VEGF [72–74]. Human bone marrow-derived MSCs, positive for CD105, CD73, CD166, CD90 and CD44, but negative for CD34, CD133, VEGFR1, VEGFR2, VE-cadherin, VCAM-1 and von Willebrand factor (vWF), when cultured in 2% foetal calf serum (FCS) supplemented with VEGF, differentiated towards endothelial-like cells after seven days [72]. The MSCs acquired several distinct characteristics of mature endothelial cells, such as VEGFR1, VEGFR2, VE-cadherin, VCAM-1 and vWF expression. Similarly, human bone marrow-derived MSCs, which were positive for CD105, CD166, but negative for CD34, VEGFR2 and vWF, following 5 days culture in 10% FCS supplemented with VEGF, expressed the endothelial cell marker vWF [73]. Using an in vivo murine matrigel model, VEGFR2-negative MSCs implants formed vascular tubes, which were inhibited by VEGF antisera, suggesting their formation was VEGF-dependent. Moreover, MSCs which incorporated into the vessel walls were reported to have differentiated towards CD31-positive endothelial-like cells [74].
A reduction in oxygen level (hypoxia) has also been shown to induce murine MSCs to form in vitro capillary-like structures [75]. Our studies have also demonstrated human MSCs have an inherent capacity to rapidly form in vitro capillary-like structures, when cultured on three-dimensional matrigels or fibrin gels, under either normoxic or hypoxic conditions, as shown in Figure 2. Human MSCs co-cultured with mature endothelial cells also differentiated towards endothelial-like cells, which were inhibited using VEGF antisera [73], highlighting the principal role of VEGF in this differentiation event. While these studies clearly demonstrate that VEGF can stimulate VEGF receptor-negative MSCs, inducing differentiation towards endothelial cells, the mechanism has not been defined, but may well involve VEGF induced PDGF receptor signalling. Our studies using VEGF receptor-negative MSCs, demonstrated that 5 hrs VEGF-A stimulation did not induce VEGF receptor transcript expression [9], indicating a more sustained exposure is required to induce differentiation towards an endothelial fate. Thus, the role of VEGF-A induced PDGF receptor activation and signalling in directing MSC differentiation to endothelial cells requires further investigation.
2.
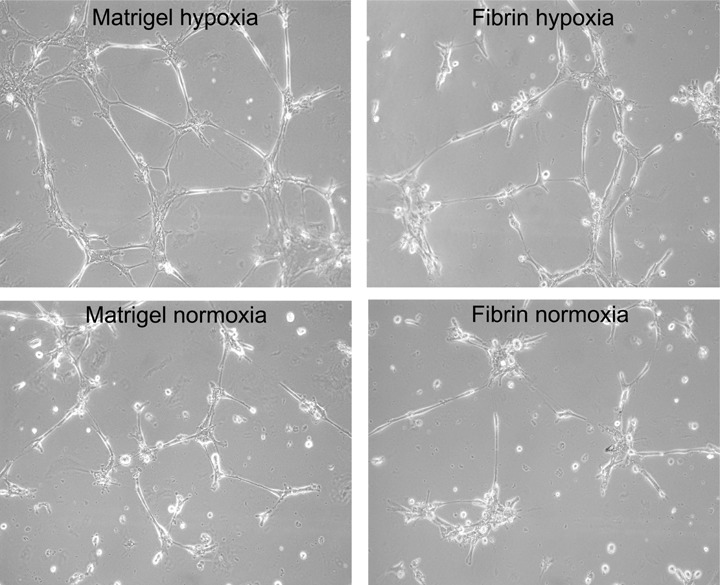
MSCs have inherent capacity to rapidly form capillary-like structures. The ability for MSCs to form in vitro tube-like structures was determined by culturing MSCs on three-dimensional Matrigels or fibrin gels, under either normoxic (20%) or hypoxic (1%) oxygen levels for 4 hrs. Both Matrigel and fibrin induced capillary-like structure formation, with Matrigel and hypoxia promoting increased tube length and branching points. However, after 24 hrs (data not shown), normoxic or hypoxic conditions resulted in similar capillary-like structure organization. Images were taken using phase-contrast microscopy with a 10x objective lens.
An important caveat particularly pertaining to MSC biology, is the difficulty in comparing data between individual studies. Obvious caution needs to be exercised when comparing investigations utilizing MSC from different species. In addition, when evaluating human MSC studies, variables such as isolation and culture protocols, passage number and donor age also require careful consideration.
Differentiation of MSCs towards vascular smooth muscle lineages
MSC populations fated to become SMCs with self-expansion abilities have been identified [76]. Several studies have demonstrated MSCs express abundant SM -actin [77, 78], suggesting an enhanced vascular SMC-like differentiation. However, SM α-actin expression alone is not sufficient to determine a SMC-like differentiation event. Vascular SMCs are not terminally differentiated and exhibit a spectrum of phenotypic states, ranging from contractile to synthetic [79]. Within the medial layer of a vascular wall, SMCs display a contractile state, characterized by being quiescent and containing a complex array of organized contractile filaments, with SM myosin heavy chain expression defining the differentiated contractile phenotype. Synthetic SMCs are characterized as being highly proliferative and secrete abundant ECM proteins, but in addition, also express early contractile state markers, SM α-actin and calponin. Our analysis of the SMC-like characteristics of cultured MSCs, revealed the transcript expression profile was similar to synthetic state vascular SMCs [80]. In addition to a wide range of extracellular transcripts, MSCs expressed transcripts and intracellular filaments for several early contractile markers, SM α-actin and calponin, as well as smoothelin-B [80, 81], a novel marker for the late differentiated contractile phenotype [82], demonstrating MSCs possess inherent SMC-like characteristics.
Myocardin is a crucial transcriptional co-factor for serum response factor, which selectively binds to CArG box sequence motifs found in the promoters for several SMC genes [83, 84], including SM α-actin and SM myosin heavy chain, but not smoothelin-B.
MSCs normally contain low levels of myocardin transcript, but when transfected with an adenovirus expressing myocardin, specifically induced SM myosin heavy chain [85]. Activation of the small GTPase RhoA has been shown to promote SM α-actin gene expression, which was mediated by serum response factor [86]. Our studies have demonstrated that MSCs have a high ratio of PDGFRα : PDGFRβ and that PDGFRα signalling activated RhoA, resulting in enhanced SM α-actin transcription and filament polymerization [10]. PDGFRα phosphorylation activates RhoA which, through Rho-associated kinase (ROCK)-dependent cofilin phosphorylation and myosin light chain kinase-dependent pathways, enhances SM α-actin filament polymerization. In contrast, PDGFRβ signalling inhibits SM α-actin filaments by up-regulating RhoE, which inhibits ROCK and by PDGF-BB induced cofilin-mediated filament destabilization [10]. Thus the cell surface PDGFRα : PDGFRβ ratio and ligand concentration are critical determinants for commitment to a vascular SMC fate. A diagram showing PDGF receptor regulation of SM α-actin filament polymerization in MSCs, is shown in Figure 3.
3.
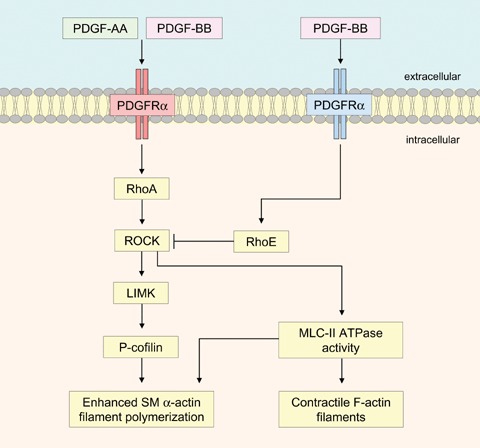
Schematic diagram showing distinct differences between PDGFRα and PDGFRβ signalling, which results in the regulation of contractile SM -actin filaments within MSCs. PDGF-AA induced PDGFRα signalling activates RhoA and increases cofilin phosphorylation via LIM kinase, resulting in enhanced SM α-actin filament polymerization. In addition, ROCK activates myosin light chain (MLC)-II ATPase activity, which is necessary for both SM α-actin and F-actin filament polymerization. In contrast, PDGF-BB induced PDGFRβ signalling increases RhoE, which inhibits ROCK activity, promoting SM α-actin filament depolymerization.
MSCs during vascular injury
Increasing evidence suggests MSCs play an important role in the pathogenesis of vascular disease, such as atherogenesis. It was previously thought that endothelial injury stimulated medial layer contractile SMCs to dedifferentiate and migrate to the damaged intimal layer, where their excessive proliferation and ECM deposition was the principal cause of neointimal hyperplasia [87]. More recently, several studies have demonstrated bone marrow-derived SMC precursor cells make a significant contribution to neointimal SMCs accumulation [88–92]. Using experimental rodent models of allograft vasculopathy, different investigations have demonstrated the majority of neointimal cells (up to 90%) were of bone-marrow origin [90, 91]. However, other studies have reported a lower level of bone-marrow-derived cells (approximately 11%) contributing to neointimal SMCs [89], which may reflect vascular injury heterogeneity, since the contribution of bone-marrow-derived progenitor cells coincides with increased intimal lesion severity [93].
Vascular injury is presumed to generate specific signals to mobilize and recruit MSCs to sites of vessel wall damage, where local stimuli will induce differentiation events. While the mechanisms of recruitment and differentiation are ill-defined, both VEGF and PDGF signalling play predominant roles. Disruption of the intimal endothelial cell layer is known to up-regulate both VEGF and PDGF-BB expression [94], while during cardiovascular disease, PDGF ligands and both receptors are up-regulated [95]. Embryonic vascular progenitor cells can differentiate into endothelial or SMC, in response to VEGF or PDGF-BB stimulation respectively [96]. Similarly, bone marrow-derived endothelial progenitor cells differentiated towards SMC following PDGF-BB exposure [97].
Mechanical stress induced by occluded blood flow during lesion formation, is an important determinant which regulates the pathological phenotype. Flow-induced mechanical stress has been demonstrated to promote bone marrow-derived stromal cells to differentiate towards SMCs [98]. Furthermore, mechanical stress has also been shown to increase PDGFRα and PDGFRβ expression [99, 100], while we observed that mechanical stress-induced MSCs to increase VEGF-A secretion. Thus, blood flow mediated mechanical stress may regulate the differentiation of MSCs during their mobilization, recruitment through the circulation, as well as at sites of vascular injury.
Contribution of MSCs to vasculogenesis
In the adult, new blood vessel growth, known as neovascularization, occurs by two separate mechanisms using cells from different sources. Either differentiated vascular cells derived from pre-existing vessels are utilized, a process known as angiogenesis, or undifferentiated bone marrow-derived cells are recruited and incorporated into a growing vessel wall, a process termed vasculogenesis. While the potent angiogenic growth factor VEGF is crucial in regulating both angiogenesis and vasculogenesis, the mechanisms involved in blood vessel formation are distinct. During angiogenesis, VEGF activates endothelial cells in pre-existing blood vessels, inducing local endothelial cell proliferation and migration to form new blood vessel sprouts, then primitive tubular-like vascular structures. The nascent vascular tubes secrete PDGF-BB which induces proliferation and recruitment of PDGFRβ -positive mural cells (vascular SMCs or pericytes), from vessel walls [41]. The mural cells surround and coat the immature endothelial tubes, which promotes endothelial cell survival, stability and maturation, primarily resulting from mural cell VEGF secretion [101].
In contrast, adult vasculogenesis is similar to mechanisms occurring during inflammation. Local tissue hypoxia was determined as the principal stimulus which induces adult vasculogenesis, which was presumed to be mediated by increased VEGF release [102]. Elevated VEGF levels create a chemoattractive gradient, which has been shown to mobilize bone marrow-derived endothelial progenitor cells into the circulation [103]. Using a novel murine model of ischaemia, bone marrow-derived endothelial progenitor cells were recruited into the circulation, then attracted and arrested to the most severe ischaemic region, where they egressed into the tissue to form cell clusters and proliferated [102]. Gradients of ischaemia induced the formation of cord-like vascular structures towards regions of greatest hypoxia, which formed tubular vessels and connected to the existing vasculature. Functional microvessels were shown to increase perfusion in the injured tissue 2 weeks after injury [102]. Thus, hypoxia has been shown to be a crucial determinant for neovascularization, which is most likely mediated, at least in part, by VEGF concentration gradients. While the proposed mechanism for adult vasculogenesis was established using bone marrow-derived VEGF receptor-positive endothelial progenitor cells, a crucial question is whether VEGF receptor-negative MSCs are regulated by VEGF induced PDGF receptor signalling during vasculogenesis.
MSCs during ischaemic myocardial tissue regeneration
Myocardial infarction or coronary artery occlusion induces tissue ischaemia, resulting in the permanent loss of a proportion of cardiomyocytes. Subsequent ventricular remodelling produces scar tissue, which compromises the contractile function of the remaining cells, which may result in ischaemic cardiomyopathy. Within the infarcted myocardium, ischaemia induces a marked increase in VEGF-A expression [104], which can stimulate the growth of bypass collateral arteries around the injured tissue to improve blood flow perfusion.
Localized administration of human MSCs into the infarcted murine myocardium, resulted in MSC differentiation towards cardiomyocytes, which improved cardiac function [105, 106]. Interestingly, PDGF-AB which stimulates either PDGFRα homodimer or heterodimer formation and activation, was shown to induce the differentiation of adult bone marrow-derived cells to cardiomyocytes in the injured rodent myocardium [44]. Clearly, within an environment rich in VEGF, a PDGF receptor-mediated MSC differentiation towards cardiomyocytes, suggests the involvement of VEGF-A induced PDGF receptor signalling. Other animal studies have also demonstrated the potential for MSCs to differentiate into contractile cardiomyocytes, but in addition, have also shown MSCs were involved in revascularization of the injured myocardium [3]. Using a canine model of myocardial ischaemia, intramyocardial administration of MSCs significantly improved myocardial function with increased vascularity [2]. In this study, the results suggested that after 60 days, the MSCs had differentiated towards both vascular SMCs and endothelial cells, but not cardiomyocytes. Furthermore, the results indicated MSCs were incorporated into the neovasculature. Thus a number of studies indicate MSC incorporate into vessel walls of the growing neovasculature during cardiac tissue regeneration.
Other studies, however, have shown that rather than MSCs being incorporated into new blood vessel walls, they promote neovascularization through paracrine mechanisms. Using a murine model of hindlimb ischaemia, MSCs were shown to release arteriogenic factors, such as VEGF, which stimulated neovascularization from pre-existing arterial vessels [107]. A number of factors may account for the variable level of MSC incorporation into growing blood vessel walls, as observed utilizing animal models of ischaemia. MSC recruitment and differentiation will undoubtedly depend on the severity of the injury, which will modulate the composition of the ECM at the site of tissue damage, as well as the bioavailability of growth factors and cytokines. These local environmental factors will contribute to regulating the fate of MSCs, either to cardiomyocytes, incorporation into vessel walls during vasculogenesis, or as accessory cells for angiogenesis. What is clear is that within the injured myocardium, VEGF and MSCs promote myocardial tissue regeneration, which suggests a potential role for VEGF-A induced PDGF receptor signalling.
Involvement of MSC during tumour vasculogenesis
The formation of tumour blood vessels and associated connective tissue stroma is regulated by VEGF-A, which is expressed by the majority of human tumours [108]. In general, tumours above 1–2 mm in size requires a blood supply to provide nutrients and oxygen for growth. As the tumour enlarges there is a concomitant increase in the level of hypoxia and VEGF-A expression, which stimulates blood vessel formation. While some tumours induces a blood supply by angiogenesis, others also utilize vasculogenesis, by recruiting bone marrow-derived cells [109].
As previously discussed, several studies provide evidence that VEGF-A stimulates the differentiation of VEGF receptor-negative MSCs towards endothe-lial cells [72–74]. Moreover, using a murine tumour model, VEGFR2-negative MSCs were shown to be actively recruited to areas of neovascularization, where they differentiated into CD31-positive endothelial-like cells and increased vascularization [4]. Furthermore, systemically administered MSCs have been shown to home to sites of growing tumours [110, 111]. Murine models of tumour growth have clearly demonstrated that enhanced VEGF levels promote vasculogenesis, by inducing the mobilization and recruitment of bone marrow-derived cells to tumour sites [112]. Thus, the recruitment of MSCs to sites of active tumour neovascularization, together with their differentiation towards endothelial cells, would be predicted to involve VEGF-A induced PDGFR signalling.
Our studies have demonstrated that MSCs possess inherent vascular SMC characteristics [80, 81], which together with their ability to secrete VEGF-A [18], suggests they have the potential to act as mural cells. Characteristically, most tumour vessels lack or have abnormal mural cell coverage, which varies considerably, depending in part on the tumour type [113]. The efficient recruitment of PDGF receptor-positive mural cells by tumour vessel-derived PDGF-BB, is dependent on short-range paracrine signalling and the formation of an extracellular gradient [114]. A gradient of extracellular retained PDGF-BB has been shown to be essential to recruit adequate numbers of mural cells, as well as promoting efficient vessel wall integration, necessary for tumour vessel stability [114]. A relatively low level of mural cell coverage can provide sufficient VEGF-A to promote stability and maturation to tumour vessels. Consequently, the efficiency of anti-VEGF therapies to suppress tumour growth are significantly decreased, thus, mechanisms which regulate mural cell recruitment to tumour vessels are extremely important. The recruitment of MSCs to tumour vessels is likely to be influenced by VEGF-A competitively binding to PDGF receptors. Our studies have shown that VEGF-A165 localized at the cell surface can inhibit PDGF-BB-induced MSC migration [9], while hypoxia stimulates MSCs to increase VEGF-A secretion. Thus, MSC recruitment to tumour vessels may be regulated by hypoxia-induced autocrine VEGF-A165, competing with tumour vessel-derived paracrine PDGF-BB for PDGF receptor occupancy. In addition, extracellular retained VEGF-A165 and PDGF-BB concentration gradients may compete. Thus a role for autocrine VEGF-A in regulating paracrine PDGF signalling is conceptually appealing, as represented in Fig. 1A.
Tumour blood vessels whether derived by angio-genesis or vasculogenesis are structurally highly irregular in shape and size, lacking the defined structural features of arterioles, capillaries or venules [115]. Their vascular patterns are haphazard and tortuous, which often terminate prematurely. Increased VEGF-A expression results in tumour vessels having a high permeability, which allows fibrinogen to leak into the extravascular tissue, where fibrin forms a provisional proangiogenic stroma [108, 116]. Tumour stroma is similar to the provisional matrix deposited during wound healing, however, due to the constant high levels of VEGF-A expression, tumours have been compared to wounds that do not heal [117]. Furthermore, tumour blood vessels produce basement membranes having a distinct composition to normal vessels [118]. Thus tumour stroma is quite different from normal connective tissue, which may well produce tumour-specific regulation of VEGF-A bioavailability and modulate PDGF receptor activation.
The contribution of bone marrow-derived cells to tumour neovascularization in animal models is highly variable, ranging from near to 100% to virtually negligible [119]. Part of this variation is due to differences in the experimental tumour models. Implanted tumours which require an immediate blood supply show a high variability, whereas tumours that form spontaneously from a single cell, which are more representative of human tumours, display a more consistent requirement for utilizing vasculogenesis [119]. Moreover, advanced tumours have been shown to preferentially utilize vasculogenesis to accommodate the increasing requirement for oxygen and nutrients [120], however, multiple factors will regulate the dependency of a particular tumour to utilize vasculogenesis for a blood supply. Each tumour type is likely to have a specific environment, such as a tumour-specific stroma, which will contribute to regulating MSC recruitment and differentiation. It is not known whether a tumour vasculature emanating from bone marrow-derived MSC during vasculogenesis, is appreciably different from a vasculature originating from pre-existing vascular cells during angiogenesis. In this respect, it is tempting to speculate that a MSC-derived vasculature could present a potential therapeutic target.
In addition to MSCs contributing to tumour neovascularization, MSCs may also engraft into tumour lesions and become incorporated into the tumour stroma [110, 121]. MSCs systemically administered into a murine tumour model, were shown to localize and engraft into tumour lesions, where they proliferated, differentiated into vWF- and CD31-positive endothelial-like cells and formed a significant fraction of the tumour stroma [121]. MSC engraftment into tumour microenvironments forms the basis for their use as cellular vehicles for the targeted delivery of anticancer agents to specific tumour sites. Genetically modified MSCs are now being utilized to successfully suppress the growth of different types of tumour in various animal models [122, 123]. Since tumours secrete high levels of VEGF-A, the recruitment of MSCs, their effective engraftment into tumour lesions, together with their subsequent proliferation and differentiation, may well involve VEGF-A induced PDGF receptor signalling.
Summary
While MSC differentiation towards vascular cell lineages and their incorporation into a growing vessel wall is rather ill-defined, their important contribution to promoting postnatal vascularization during ischaemic myocardial tissue regeneration and tumour vasculogenesis is now becoming increasingly recognized. To therapeutically manipulate MSCs in these environments, mechanisms regulating their mobilization and recruitment to sites of neovascularization, as well as their ultimate differentiation to a vascular cell fate need to be identified. As the potent angiogenic stimulant VEGF-A is crucial to directing blood vessel formation, both during physiological and pathological situations, it is a decisive factor in regulating MSC recruitment and fate in the vascular environment. Our finding that VEGF-A can stimulate PDGF receptors, expands the mechanisms by which VEGF can control MSCs during early postnatal vessel formation. Thus, further studies are required to fully understand how PDGF receptors elicit ligand-specific biological responses. It is becoming increasingly apparent that PDGF receptor signalling is not only modified by ligand binding, but is also profoundly influenced by the cellular microenvironment. Identifying mechanisms which regulate VEGF-A induced PDGF receptor signalling, will ultimately facilitate the development of more effective MSC-based vascular repair and regeneration strategies.
References
- 1.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Silva GV, Litovsky S, Assad JA, Sousa AL, Martin BJ, Vela D, Coulter SC, Lin J, Ober J, Vaughn WK, Branco RV, Oliveira EM, He R, Geng Y-J, Willerson JT, Perin EC. Mesenchymal stem cells differentiate into a endothelial phenotype, enhance vascular density, and improve heart function in a canine chronic ischemia model. Circulation. 2005;111:150–6. doi: 10.1161/01.CIR.0000151812.86142.45. [DOI] [PubMed] [Google Scholar]
- 3.Nagaya N, Fujii T, Iwase T, Ohgushi H, Itoh T, Uematsu M, Yamagishi M, Mori H, Kangawa K, Kitamura S. Intravenous administration of mesenchymal stem cells improves cardiac function in rats with acute myocardial infarction through angiogenesis and myogenesis. Am J Physiol Heart Circ Physiol. 2004;287:H2670–6. doi: 10.1152/ajpheart.01071.2003. [DOI] [PubMed] [Google Scholar]
- 4.Annabi B, Naud E, Lee Y-T, Eliopoulos N, Galipeau J. Vascular progenitors derived from murine bone marrow stromal cells are regulated by fibroblast growth factor and are avidly recruited by vascularizing tumors. J Cell Biochem. 2004;91:1146–58. doi: 10.1002/jcb.10763. [DOI] [PubMed] [Google Scholar]
- 5.Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 2005;6:209–19. doi: 10.1186/gb-2005-6-2-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamazaki Y, Morita T. Molecular and functional diversity of vascular endothelial growth factors. Mol Divers. 2006;10:515–27. doi: 10.1007/s11030-006-9027-3. [DOI] [PubMed] [Google Scholar]
- 7.Betsholtz C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev. 2004;15:215–28. doi: 10.1016/j.cytogfr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Hoch RV, Soriano P. Roles of PDGF signaling in animal development. Development. 2003;130:4769–84. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- 9.Ball SG, Shuttleworth CA, Kielty CM. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol. 2007;177:489–500. doi: 10.1083/jcb.200608093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ball SG, Shuttleworth CA, Kielty CM. Platelet-derived growth factor receptor-α is a key determinant of smooth muscle α-actin filaments in bone marrow-derived mesenchymal stem cells. Int J Biochem Cell Biol. 2007;39:379–91. doi: 10.1016/j.biocel.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Vitt UA, Hsu SY, Hsueh AJ. Evolution and classification of cystine knot-containing hormones and related extracellular signaling molecules. Mol Endocrinol. 2001;15:681–94. doi: 10.1210/mend.15.5.0639. [DOI] [PubMed] [Google Scholar]
- 12.Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- 13.Cho NK, Keyes L, Johnson E, Heller J, Ryner L, Karim F, Krasnow MA. Development control of blood cell migration by the Drosophila VEGF pathway. Cell. 2002;108:865–76. doi: 10.1016/s0092-8674(02)00676-1. [DOI] [PubMed] [Google Scholar]
- 14.Bruckner K, Kockel L, Duchek P, Luque CM, Rorth P, Perrimon N. The PDGF/VEGF receptor controls blood cell survival in Drosophila. Developmental Cell. 2004;7:73–84. doi: 10.1016/j.devcel.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Tarsitano M, De Falco S, Colonna V, McGhee JD, Persico MG. The C. elegans pvf-1 gene encodes a PDGF/VEGF-like factor able to bind mammalian VEGF receptors and to induce angiogenesis. FASEB J. 2006;20:227–33. doi: 10.1096/fj.05-4147com. [DOI] [PubMed] [Google Scholar]
- 16.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 17.Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 18.Mayer H, Bertram H, Lindenmaier W, Korff T, Weber H, Weich H. Vascular endothelial growth factor (VEGF-A) expression in human mesenchymal stem cells: autocrine and paracrine role on osteoblastic and endothelial differentiation. J Cellular Biochem. 2005;95:827–39. doi: 10.1002/jcb.20462. [DOI] [PubMed] [Google Scholar]
- 19.Asahara T, Bauters C, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM. Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesis in vivo. Circulation. 1995;92:II-365–71. doi: 10.1161/01.cir.92.9.365. [DOI] [PubMed] [Google Scholar]
- 20.Kano MR, Morishita Y, Iwata C, Iwasaka S, Watabe T, Ouchi Y, Miyazono K, Miyazawa K. VEGF-A and FGF-2 synergistically promote neoan-giogenesis through enhancement of endogenous PDGF-B-PDGFRβ signaling. J Cell Sci. 2005;118:3759–68. doi: 10.1242/jcs.02483. [DOI] [PubMed] [Google Scholar]
- 21.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 22.Liu YX, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells, identification of a 5 enhancer. Circ Res. 1995;77:638–43. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- 23.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, Yang H, Sorokina I, Conaway RC, Conaway JW, Kaelin WG. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA. 2002;99:459–64. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fredriksson L, Li H, Eriksson U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J. 2005;272:5723–41. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- 26.Kondo K, Hiratuska S, Subbalakshmi E, Matsushime H, Shibuya M. Genomic organization of the flt-1 gene encoding for vascular endothelial growth factor (VEGF) receptor-1 suggests an intimate evolutionary relationship between the 7-Ig and the 5-Ig tyrosine kinase receptors. Gene. 1998;208:297–305. doi: 10.1016/s0378-1119(98)00006-7. [DOI] [PubMed] [Google Scholar]
- 27.Neufeld G, Cohen T, Shraga N, Lange T, Kessler O, Herzog Y. The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc Med. 2002;12:13–9. doi: 10.1016/s1050-1738(01)00140-2. [DOI] [PubMed] [Google Scholar]
- 28.Soker S, Miao HQ, Nomi M, Takashima S, Klagsbrun M. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J Cell Biochem. 2002;85:357–68. doi: 10.1002/jcb.10140. [DOI] [PubMed] [Google Scholar]
- 29.Shraga-Heled N, Kessler O, Prahst C, Kroll J, Augustin H, Neufeld G. Neuropilin-1 and neuropilin-2 enhance VEGF121 stimulated signal transduction by the VEGFR-2 receptor. FASEB J. 2007;21:915–26. doi: 10.1096/fj.06-6277com. [DOI] [PubMed] [Google Scholar]
- 30.Gilbertson DG, Duff ME, West JW, Kelly JD, Sheppard PO, Hofstrand PD, Gao Z, Shoemaker K, Bukowski TR, Moore M, Feldhaus AL, Humes JM, Palmer TE, Hart CE. Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF alpha and beta receptor. J Biol Chem. 2001;276:27406–14. doi: 10.1074/jbc.M101056200. [DOI] [PubMed] [Google Scholar]
- 31.Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, Alitalo K, Eriksson U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat Cell Biol. 2001;3:512–6. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- 32.Betsholtz C, Karisson L, Lindahl P. Developmental roles of platelet-derived growth factors. Bioessays. 2001;23:494–507. doi: 10.1002/bies.1069. [DOI] [PubMed] [Google Scholar]
- 33.Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, Hedstrand H, Pekna M, Hellstrom M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Tornell J, Heath JK, Betsholtz C. PDGF-A signaling is a critical event in lung alveolar myofibrob-last development and alveogenesis. Cell. 1996;85:863–73. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- 34.Soriano P. The PDGF alpha receptor is required for neural crest cell survival and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- 35.Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–87. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- 36.Soriano P. Abnormal kidney development and hema-tological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8:1888–96. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- 37.Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Berthold C-H, Heath JK, Betsholtz C, Richardson WD. Defective oligodendrocyte development and severe hypomyeli-nation in PDGF-A knockout mice. Development. 1999;126:457–67. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- 38.Lindahl P, Karlsson L, Hellstrom M, Gebre-Medhin S, Willetts K, Heath JK, Betsholtz C. Alveogenesis failure in PDGF-A deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development. 1997;124:3943–53. doi: 10.1242/dev.124.20.3943. [DOI] [PubMed] [Google Scholar]
- 39.Karlsson L, Bondjers C, Betsholtz C. Roles for PDGF-A and sonic hedgehog in development of mesenchymal components of the hair follicle. Development. 1999;126:2611–21. doi: 10.1242/dev.126.12.2611. [DOI] [PubMed] [Google Scholar]
- 40.Karlsson L, Lindahl P, Heath JK, Betsholtz C. Abnormal gastrointestinal development in PDGF-A and PDGFR-a deficient mice implicates a novel mes-enchymal structure with putative instructive properties in villus morphogenesis. Development. 2000;127:3457–66. doi: 10.1242/dev.127.16.3457. [DOI] [PubMed] [Google Scholar]
- 41.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-β in recruitment of vascular smooth muscle cells and per-icytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–55. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 42.Lindahl P, Hellstrom M, Kalen M, Karlsson L, Pekny M, Pekna M, Soriano P, Betsholtz C. Paracrine PDGF-B/PDGF-Rbeta signaling controls mesangial cell growth in the development of kidney glomeruli. Development. 1998;125:3313–22. doi: 10.1242/dev.125.17.3313. [DOI] [PubMed] [Google Scholar]
- 43.Ekman S, Thuresson ER, Heldin CH, Ronnstrand L. Increased mitogenicity of an alphabeta heterodimeric PDGF receptor complex correlates with lack of RasGAP binding. Oncogene. 1999;18:2481–8. doi: 10.1038/sj.onc.1202606. [DOI] [PubMed] [Google Scholar]
- 44.Xaymardan M, Tang L, Zagreda L, Pallante B, Zheng J, Chazen JL, Chin A, Duignan I, Nahirney P, Rafii S, Mikawa T, Edelberg JM. Platelet-derived growth factor-AB promotes the generation of adult bone marrow-derived cardiac myocytes. Circ Res. 2004;94:e39–45. doi: 10.1161/01.RES.0000122042.51161.B6. [DOI] [PubMed] [Google Scholar]
- 45.Ponte AL, Marais E, Gallay N, Langonne A, Delorme B, Herault P, Charbord P, Domenech J. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25:1737–45. doi: 10.1634/stemcells.2007-0054. [DOI] [PubMed] [Google Scholar]
- 46.Cao R, Brakenhielm E, Li X, Pietras K, Widenfalk J, Ostman A, Eriksson U, Cao Y. Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-α and –β receptors. FASEB J. 2002;16:1575–83. doi: 10.1096/fj.02-0319com. [DOI] [PubMed] [Google Scholar]
- 47.Melaragno MG, Fridell Y-W, Berk BC. The Gas6/Axl system A novel regulator of vascular cell function. Trends Cardiovasc Med. 1999;9:250–53. doi: 10.1016/s1050-1738(00)00027-x. [DOI] [PubMed] [Google Scholar]
- 48.Budagian V, Bulanova E, Orinska Z, Thon L, Mamat U, Bellosta P, Basilico C, Adam D, Paus R, Bulfone-Paus S. A promiscuous liaison between IL-15 receptor and Axl receptor tyrosine kinase in cell death control. EMBO J. 2005;24:4260–70. doi: 10.1038/sj.emboj.7600874. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Gallicchio M, Mitola S, Valdembri D, Fantozzi R, Varnum B, Avanzi GC, Bussolino F. Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood. 2005;105:1970–76. doi: 10.1182/blood-2004-04-1469. [DOI] [PubMed] [Google Scholar]
- 50.Lokker NA, O'Hare JP, Barsoumian A, Tomlinson JE, Ramakrishnan V, Fretto LJ, Giese NA. Functional importance of platelet-derived growth factor (PDGF) receptor extracellular immunoglobulin-like domains. J Biol Chem. 1997;272:33037–44. doi: 10.1074/jbc.272.52.33037. [DOI] [PubMed] [Google Scholar]
- 51.Fuh G, Li B, Crowley C, Cunningham B, Wells JA. Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J Biol Chem. 1998;273:11197–204. doi: 10.1074/jbc.273.18.11197. [DOI] [PubMed] [Google Scholar]
- 52.Ruch C, Skiniotis G, Steinmetz MO, Walz T, Ballmer-Hofer K. Structure of a VEGF-VEGF receptor complex determined by electron microscopy. Nat Struct Mol Biol. 2007;14:249–50. doi: 10.1038/nsmb1202. [DOI] [PubMed] [Google Scholar]
- 53.Seetharam L, Gotoh N, Maru Y, Neufeld G, Yamaguchi S, Shibuya M. A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene. 1995;10:135–47. [PubMed] [Google Scholar]
- 54.McKinnon RD, Waldron S, Kiel ME. PDGF α-receptor signal strength controls an RTK rheostat that integrates phosphoinositol 3-kinase and phospholipase C pathways during oligodendrocyte maturation. J Neurosci. 2005;25:3499–508. doi: 10.1523/JNEUROSCI.5049-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ostman A, Andersson M, Betsholtz C, Westermark B, Heldin CH. Identification of a cell retention signal in the B-chain of PDGF and in the long splice version of the A-chain. Cell Regul. 1991;2:503–12. doi: 10.1091/mbc.2.7.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dijkmans J, Xu J, Masure S, Dhanaraj S, Gosiewska A, Geesin J, Sprengel J, Harris S, Verhasselt P, Gordon R, Yon J. Characterization of platelet-derived growth factor-C (PDGF-C): expression in normal and tumor cells, biological activity and chromosomal localization. Int J Biochem Cell Biol. 2002;34:414–26. doi: 10.1016/s1357-2725(01)00124-8. [DOI] [PubMed] [Google Scholar]
- 57.Park JE, Keller G-A, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–26. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Somasundaram R, Schuppan D. Type I, II, III, IV, V, and VI collagens serve as extracellular ligands for the isoforms of platelet-derived growth factor (AA, BB, and AB) J Biol Chem. 1996;271:26884–91. doi: 10.1074/jbc.271.43.26884. [DOI] [PubMed] [Google Scholar]
- 59.Wijelath ES, Murray J, Rahman S, Patel Y, Ishida A, Strand K, Aziz S, Cardona C, Hammond WP, Savidge GF, Rafii S, Sobel M. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res. 2002;91:25–31. doi: 10.1161/01.res.0000026420.22406.79. [DOI] [PubMed] [Google Scholar]
- 60.Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, Patel Y, Suda Y, Humphries MJ, Sobel M. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ Res. 2006;99:853–60. doi: 10.1161/01.RES.0000246849.17887.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woodard AS, Garcia-Cardena G, Leong M, Madri JA, Sessa WC, Languino LR. The synergistic activity of αvβ3 integrin and PDGF receptor increases cell migration. J Cell Sci. 1998;111:469–78. doi: 10.1242/jcs.111.4.469. [DOI] [PubMed] [Google Scholar]
- 62.Baron W, Shattil SJ, Ffrench-Constant C. The oligodendrocyte precursor mitogen PDGF stimulates proliferation by activation of αvβ3 integrins. EMBO J. 2002;21:1957–66. doi: 10.1093/emboj/21.8.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hollenbeck ST, Itoh H, Louie O, Faries PL, Liu B, Kent KC. Type I collagen synergistically enhances PDGF-induced smooth muscle cell proliferation through pp60src-dependent crosstalk between the α2β1 integrin and PDGFβ receptor. Biochem Biophys Res Comm. 2004;325:328–37. doi: 10.1016/j.bbrc.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 64.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: Role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–32. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 65.Tanimoto T, Lungu AO, Berk BC. Sphingosine 1-phosphate transactivates the platelet-derived growth factor receptor and epidermal growth factor receptor in vascular smooth muscle cells. Circ Res. 2004;94:1050–8. doi: 10.1161/01.RES.0000126404.41421.BE. [DOI] [PubMed] [Google Scholar]
- 66.Lehti K, Allen E, Birkedal-Hansen H, Holmbeck K, Miyake Y, Chun T-H, Weiss SJ. An MT1-MMP-PDGF receptor- axis regulates mural cell investment of the microvasculature. Genes Dev. 2005;19:979–91. doi: 10.1101/gad.1294605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bussolino F, Serini G, Mitola S, Bazzoni G, Dejana E. Dynamic modules and heterogeneity of function: a lesson from tyrosine kinase receptors in endothelial cells. EMBO reports. 2001;2:763–7. doi: 10.1093/embo-reports/kve181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klinghoffer RA, Mueting-Nelsen PF, Faerman A, Shani M, Soriano P. The two PDGF receptors maintain conserved signaling in vivo despite divergent embryological functions. Mol Cell. 2001;7:343–54. doi: 10.1016/s1097-2765(01)00182-4. [DOI] [PubMed] [Google Scholar]
- 69.DeMali KA, Balciunaite E, Kazlauskas A. Integrins enhance platelet-derived growth factor (PDGF)-dependent responses by altering the signal relay enzymes that are recruited to the PDGF β receptor. J Biol Chem. 1999;274:19551–8. doi: 10.1074/jbc.274.28.19551. [DOI] [PubMed] [Google Scholar]
- 70.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–53. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 71.Asahara T, Murohara T, Sullivan A, Silver M, Van Der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 72.Oswald J, Boxberger S, Jorgensen B, Feldmann S, Ehninger G, Bornhauser M, Werner C. Mesenchymal stem cells can differentiate into endothelial cells in vitro. Stem Cells. 2004;22:377–84. doi: 10.1634/stemcells.22-3-377. [DOI] [PubMed] [Google Scholar]
- 73.Wu X, Huang L, Zhou Q, Song Y, Li A, Jin J, Cui B. Mesenchymal stem cells participating in ex vivo endothelial repair and its effect on vascular smooth muscle cells growth. Int J Cardiol. 2005;105:274–82. doi: 10.1016/j.ijcard.2004.12.090. [DOI] [PubMed] [Google Scholar]
- 74.Al-Khaldi A, Eliopoulos N, Martineau D, Lejeune L, Lachapelle K, Galipeau J. Postnatal bone marrow cells elicit a potent VEGF-dependent neoangio-genic response in vivo. Gene Ther. 2003;10:621–9. doi: 10.1038/sj.gt.3301934. [DOI] [PubMed] [Google Scholar]
- 75.Annabi B, Lee Y-T, Turcotte S, Naud E, Desrosiers RR, Champagne M, Eliopoulos N, Galipeau J, Beliveau R. Hypoxia promotes murine bone-marrow-derived stromal cell migration and tube formation. Stem Cells. 2003;21:337–47. doi: 10.1634/stemcells.21-3-337. [DOI] [PubMed] [Google Scholar]
- 76.Kashiwakura Y, Katoh Y, Tamayose K, Konishi H, Takaya N, Yuhara S, Yamada M, Sugimoto K, Daida H. Isolation of bone marrow stromal cell-derived smooth muscle cells by a human SM22 promoter. Circulation. 2003;107:2078–81. doi: 10.1161/01.CIR.0000070082.64414.B5. [DOI] [PubMed] [Google Scholar]
- 77.Galmiche MC, Koteliansky VE, Briere J, Herve P, Charbord P. Stromal cells from human long-term marrow cultures are mesenchymal cells that differentiate following a vascular smooth muscle differentiation pathway. Blood. 2002;82:66–76. [PubMed] [Google Scholar]
- 78.Kinner B, Zaleskas JM, Spector M. Regulation of smooth muscle actin expression and contraction in adult human mesenchymal stem cells. Exp Cell Res. 2002;278:72–83. doi: 10.1006/excr.2002.5561. [DOI] [PubMed] [Google Scholar]
- 79.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 80.Ball SG, Shuttleworth CA, Kielty CM. Direct cell contact influences bone marrow mesenchymal stem cell fate. Int J Biochem Cell Biol. 2004;36:714–27. doi: 10.1016/j.biocel.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 81.Stephan S, Ball SG, Williamson M, Bax DV, Lomas A, Shuttleworth CA, Kielty CM. Cell-matrix biology in vascular tissue engineering. J Anat. 2006;209:495–502. doi: 10.1111/j.1469-7580.2006.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Van Der Loop FT, Gabbiani G, Kohnen G, Ramaekers FC, Van Eys GJ. Differentiation of smooth muscle cells in human blood vessels as defined by smoothelin, a novel marker for the contractile phenotype. Arterioscler Thromb Vasc Biol. 1997;17:665–71. doi: 10.1161/01.atv.17.4.665. [DOI] [PubMed] [Google Scholar]
- 83.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–37. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–64. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 85.Van Tuyn J, Knaan-Shanzer S, Van De Watering MJ, De Graaf M, Van Der Laarse A, Schalij MJ, Van Der Wall EE, De Vries AA, Atsma DE. Activation of cardiac and smooth muscle-specific genes in primary human cells after forced expression of human myocardin. Cardiovasc Res. 2005;67:245–55. doi: 10.1016/j.cardiores.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 86.Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem. 2001;276:341–7. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- 87.Ross R. The pathogenesis of atherosclerosis:a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 88.Han CI, Campbell GR, Campbell JH. Circulating bone marrow cells contribute to neointimal formation. J Vasc Res. 2001;38:113–9. doi: 10.1159/000051038. [DOI] [PubMed] [Google Scholar]
- 89.Hu Y, Mayr M, Metzler B, Erdel M, Davison F, Xu Q. Both donor and recipient origins of smooth muscle cells in vein graft atherosclerotic lesions. Circ Res. 2002;91:E13–20. doi: 10.1161/01.res.0000037090.34760.ee. [DOI] [PubMed] [Google Scholar]
- 90.Saiura A, Sata M, Hirata Y, Nagai R, Makuuchi M. Circulating smooth muscle cells contribute to atherosclerosis. Nat Med. 2001;7:382–3. doi: 10.1038/86394. [DOI] [PubMed] [Google Scholar]
- 91.Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–9. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 92.Shimizu K, Sugiyama S, Aikawa M, Fukumoto Y, Rabkin E, Libby P, Mitchell RN. Host bone-marrow cells are a source of donor intimal smooth muscle-like cells in murine aortic transplant arteriopathy. Nat Med. 2001;7:738–41. doi: 10.1038/89121. [DOI] [PubMed] [Google Scholar]
- 93.Sata M. Circulating vascular progenitor cells contribute to vascular repair, remodelling and lesion formation. Trends Cardiovasc Med. 2003;13:249–53. doi: 10.1016/s1050-1738(03)00106-3. [DOI] [PubMed] [Google Scholar]
- 94.Majesky MW, Reidy MA, Bowen-Pope DF, Hart CE, Wilcox JN, Schwartz SM. PDGF ligand and receptor gene expression during repair of arterial injury. J Cell Biol. 1990;111:2149–58. doi: 10.1083/jcb.111.5.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–54. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 96.Yamashita J, Itoh H, Hirashima M. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000;408:92–6. doi: 10.1038/35040568. [DOI] [PubMed] [Google Scholar]
- 97.Miyata T, Iizasa H, Sai Y, Fujii J, Terasaki T, Nakashima E. Platelet-derived growth factor-BB (PDGF-BB) induces differentiation of bone marrow endothelial progenitor cell-derived cell line TR-BME2 into mural cells, and changes the phenotype. J Cell Physiol. 2005;204:948–55. doi: 10.1002/jcp.20362. [DOI] [PubMed] [Google Scholar]
- 98.Kobayashi N, Yasu T, Ueba H, Sata M, Hashimoto S, Kuroki M, Saito M, Kawakami M. Mechanical stress promotes the expression of smooth muscle-like properties in marrow stromal cells. Exp Hematol. 2004;32:1238–45. doi: 10.1016/j.exphem.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 99.Hu Y, Bock G, Wick G, Xu Q. Activation of PDGF receptor in vascular smooth muscle cells by mechanical stress. FASEB J. 1998;12:1135–42. doi: 10.1096/fasebj.12.12.1135. [DOI] [PubMed] [Google Scholar]
- 100.Ma Y-H, Ling S, Ives HE. Mechanical strain increases PDGF-B and PDGF β receptor expression in vascular smooth muscle cells. Biochem Biophys Res Comm. 1999;265:606–10. doi: 10.1006/bbrc.1999.1718. [DOI] [PubMed] [Google Scholar]
- 101.Darland DC, Massingham LJ, Smith SR, Piek E, Saint-Geniez M, DAmore PA. Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol. 2003;264:275–88. doi: 10.1016/j.ydbio.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 102.Tepper OM, Capla JM, Galiano RD, Ceradini DJ, Callaghan MJ, Kleinman ME. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood. 2005;105:1068–77. doi: 10.1182/blood-2004-03-1051. [DOI] [PubMed] [Google Scholar]
- 103.Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964–72. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kranz A, Rau C, Kochs M, Waltenberger J. Elevation of vascular endothelial growth factor-A serum levels following acute myocardial infarction: evidence for its origin and functional significance. J Mol Cell Cardiol. 2000;32:65–72. doi: 10.1006/jmcc.1999.1062. [DOI] [PubMed] [Google Scholar]
- 105.Liechty KW, MacKenzie TC, Shaaban AF, Radu A, Moseley AB, Deans R, Marshak DR, Flake AW. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000;6:1282–6. doi: 10.1038/81395. [DOI] [PubMed] [Google Scholar]
- 106.Toma C, Pittenger MF, Cahill KS, Byrne BJ, Kessler PD. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation. 2002;105:93–8. doi: 10.1161/hc0102.101442. [DOI] [PubMed] [Google Scholar]
- 107.Kinnaird T, Stabile E, Burnett MS, Shou M, Lee CW, Barr S, Fuchs S, Epstein SE. Local delivery of marrow-derived stromal cells augments collateral perfusion through paracrine mechanisms. Circulation. 2004;109:1543–9. doi: 10.1161/01.CIR.0000124062.31102.57. [DOI] [PubMed] [Google Scholar]
- 108.Dvorak HF. How tumors make bad blood vessels and stroma. Am J Pathol. 2003;162:1747–57. doi: 10.1016/s0002-9440(10)64309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lyden D, Hattori K, Dias S. Impaired recruitment of bone marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 110.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mes-enchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603–8. [PubMed] [Google Scholar]
- 111.Menon LG, Picinich S, Koneru R, Gao H, Yo Lin S, Koneru M, Mayer-Kuckuk P, Glod J, Banerjee D. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem cells. 2007;25:520–8. doi: 10.1634/stemcells.2006-0257. [DOI] [PubMed] [Google Scholar]
- 112.Li B, Sharpe EE, Maupin AB, Teleron AA, Pyle AL, Carmeliet P, Young PP. VEGF and P1GF promote adult vasculogenesis by enhancing EPC recruitment and vessel formation at the site of tumor neovascu-larization. FASEB J. 2006;20:E664–76. doi: 10.1096/fj.05-5137fje. [DOI] [PubMed] [Google Scholar]
- 113.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60:1388–93. [PubMed] [Google Scholar]
- 114.Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. 2003;112:1142–51. doi: 10.1172/JCI18549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ribatti D, Nico B, Crivellato E, Vacca A. The structure of the vascular network of tumors. Cancer Lett. 2007;248:18–23. doi: 10.1016/j.canlet.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 116.Nagy JA, Brown LF, Senger DR, Lanir N, Van de Water L, Dvorak AM, Dvorak HK. Pathogenesis of tumor stroma generation: a critical role for leaky blood vessels and fibrin deposition. Biochim Biophys Acta. 1989;948:305–26. doi: 10.1016/0304-419x(89)90004-8. [DOI] [PubMed] [Google Scholar]
- 117.Dvorak HF. Tumors:wounds that do not heal:similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 118.Baluk P, Morikawa S, Haskell A, Mancuso M, Mc Donald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003;163:1801–15. doi: 10.1016/S0002-9440(10)63540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Aghi M, Chiocca EA. Contribution of bone marrow-derived cells to blood vessels in ischemic tissues and tumors. Mol Ther. 2005;12:994–1005. doi: 10.1016/j.ymthe.2005.07.693. [DOI] [PubMed] [Google Scholar]
- 120.Spring H, Schuler T, Arnold B, Hammerling GJ, Ganss R. Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci USA. 2005;102:18111–6. doi: 10.1073/pnas.0507158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hung S-C, Deng W-P, Yang WK, Liu R-S, Lee C-C, Su T-C, Lin R-J, Yang D-M, Chang C-W, Chen W-H, Wei H-J, Gelovani JG. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. 2005;11:7749–56. doi: 10.1158/1078-0432.CCR-05-0876. [DOI] [PubMed] [Google Scholar]
- 122.Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hasen M, Bekele BN, Champlin RE, Andreeff M. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96:1593–603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 123.Xin H, Kanehira M, Mizuguchi H, Hayakawa T, Kikuchi T, Nukiwa T, Saijo Y. Targeted-delivery of CX3CL1 to multiple lung tumors by mesenchymal stem cells. Stem cells. 2007;25:1618–26. doi: 10.1634/stemcells.2006-0461. [DOI] [PubMed] [Google Scholar]