

Abstract
Cell death is clearly an important factor in development, homeostasis, pathology and in aging, but medical efforts based on controlling cell death have not become major aspects of medicine. There are several reasons why hopes have been slow to be fulfilled, and they present indications for new directions in research. Most effort has focused on the machinery of cell death, or the proximate effectors of apoptosis and their closely associated and interacting proteins. But cells have many options other than apoptosis. These include autophagy, necrosis, atrophy and stepwise or other alternate means of self-disassembly. The response of a cell to a noxious or otherwise intimidating signal will depend heavily on the history, lineage and current status of the cell. Many metabolic and other processes adjust the sensitivity of cells to signals, and viruses aggressively attempt to regulate the death of their host cells. Another complicating factor is that many deathassociated proteins may have functions totally unrelated to their role in cell death, generating the possibility of undesirable side effects if one interferes with them. In the future, the challenge will be more to understand the challenge to the cell from a more global standpoint, including many more aspects of metabolism, and work toward alleviating or provoking the challenge in a targeted fashion.
Keywords: apoptosis, autophagy, lysosomes, cathepsins, non-apoptotic cell death, autophagic cell death, necrosis
Cell death is important in medicine
There are now over 200,000 publications listed by the United States Library of Medicine for the topics ‘apoptosis OR cell death OR programmed cell death’ and a new publication appears every 24 min. One could therefore reasonably conclude that this group of topics would be a major aspect of clinical medicine. By three criteria, this is true: we are well accustomed to the idea that, with the exception of the targets of natural killer cells, very few cells are murdered. By far the majority commit suicide under specific conditions. Second, many developmental and chronic diseases can be traced to genetic or other problems with genes known to influence cell death, such as the cancers that arise because of mutations or translocations of anti-apoptotic genes such as bcl-2 or damage surveillance apoptosis activators such as p53, and autoimmune diseases associated with mutations of Fas or Fas Ligand. Third, in many diseases even when the origin is not inherently genetic, the pathological state is characterized by substantial apoptosis or other form of cell death. Thus neurodegenerative diseases, HIV-AIDS, inflammatory or autoimmune disease, infarct or ischaemia, several types of viral infection, infertility and many other pathological or undesirable situations are described as deriving from abnormalities in the expected pattern of cell death, or even as being diseases of deranged cell death.These topics are the subject of numerous reviews, including the following excellent and most recent ones [1–9].
The importance of cell death was first recognized in developing embryos. Developmental biologists realized that cell death must be under genetic regulation as deregulated cell death generated many mutant phenotypes including both developmental and adult abnormalities. Complete blockage of cell death is lethal in most embryos (though not Caenorhabditis); and cell death starts at very stages of development - as early as eight cell stage - and is essential for organogenesis [10–13]. Half to two-thirds of developing neurons die before the completion of development and huge numbers of T- and B-lymphocytes likewise undergo developmental selection. Though the numbers are not so massive, cell death can be seen in almost all other developing organs. Some of the best-known examples, such as the cornea [14–16] and interdigital tissues of the limbs of amniotes that do not have webs, are known primarily because their accessibility has allowed experimentation and evaluation [17–23]; but the sculpting of all complex organs, particularly those such as the kidney in which structure is vital for function, involves cell death. Sexual differentiation begins with elimination of either the Müllerian or Wolffian ducts and includes several other instances of controlled cell death throughout life.
Cell death is likewise important in normal cellular and organismic homeostasis. Billions of cells die every day. By far the largest number are red blood cells (about 250 × 109/day), the death of which has been described as a delayed, stepwise death (first, elimination of nucleus and mitochondria; later, though still controlled, phagocytosis of the stiffening, aged erythrocyte) [24–28]. Replacement of skin and digestive tract lining run a close second, with several diseases being related to differences in the kinetics of turnover of epithelial cells. Responsive cells of the immune system must down-regulate after the initial stimulus has been resolved. Even in the liver, the loss of one cell in 1000 every day, in what is described as normal turnover’, adds up to approximately 180,000,000 cells lost per day [29]. Both lack of and inappropriate level or timing of cell death can cause major havoc.
Why do we not have a medicine based on apoptosis?
Nevertheless, several attempts to treat symptoms or disease by manipulating cell death have not succeeded. Among these are efforts to inhibit or activate caspases, to provide supporting trophic factors, to initiate type II or cell surface receptor-induced apoptosis, to correct genetic defects that lead to apoptosis by targeting the products of the altered genes, and to exploit the differences between the availabilities of cell death pathways in malignant and normal cells. By and large, these efforts up to now have looked far more promising in the laboratory than they have proven to be in patients, although there is still much potential [30].
What might be the cause of such a discrepancy? There are several reasons why our understanding of mechanisms of cell death and the vigorous efforts of many laboratories, research institutions and biotech companies, specific applications have not yet reached the clinical horizon. Among the most important of these reasons are the facts that (1) in most instances of pathology, the source of the problem is not the machinery of apoptosis, which remains intact, but rather its regulation; (2) Although the machinery stays intact, the choice of machinery or path can change the measurement or the outcome; (3) our current understanding that a cell will take alternate paths if one is blocked makes it more difficult to do directed drug therapy since attacking and disabling one part of the machinery can be overcome by turning on another path; (4) to specifically target a drug to a specific type of cell is a far more challenging task than to create an inhibitor; (5) many researchers neglect the fact that the death of a cell is a symptom and outcome of a more pervasive problem and (6) when conditions become intolerable for survival-meaning chemical, osmotic, oxidative or other damage to cell or organelle membranes, failure of adenosine triphosphate (ATP)-generating systems, subversion of resources toward virus production or precipitation of proteins because of inappropriate pH, ion composition or temperature-cells will succumb whether or not their preferred method of suicide is available.We will consider each of these issues in turn.
The machinery of cell death
Studies using mutant animals in which lack of cell death could be overcome with the use of drugs [31] or activation or deactivation of specific genes [32, 33] led to the idea that a cell death machinery exists in all cells. The term ‘machinery’ is used in two senses: in the larger sense, it refers to the ability of a cell to generate and interpret signals that tell it to die as well as to the activation of the specific enzymes, primarily caspases, that kill it. In a narrower sense, ‘cell death machinery’ refers only to the caspase cascade and those molecules that directly interact with either the extrinsic or intrinsic pathway. Here, we refer to the machinery in the narrow sense to emphasize that the interaction, engagement and activation of this machinery is very complex and only crudely understood.
The brilliant hypothesis and generalization that a pattern of cell death was common to many organisms, cell types and situations was quickly validated and led both to relatively easy and reliable means of detecting apoptosis and to identification of the mechanisms that created the identifiable morphology and biochemical characteristics of apoptosis. The process was so general and so extensible - ranging from nematode worms to humans - that (following the binary mindset common to most styles of thinking) the perception arose that all cells die either in a controlled apoptotic death or by uncontrolled necrotic death. Today we recognize that the issue is more subtle, as the history, descent, stage of differentiation and environment of a cell can alter its sensitivity to death or the manner in which it dies. However, the paths of apoptosis are so common that they are frequently if incorrectly believed to be an unequivocal or exclusive criterion for cell death. Thus, most of the criteria by which we identify cell death in reality are markers of apoptosis and derive directly or indirectly from the activation of effector caspases. These markers include direct assessment of the activation of (usually) caspase 3; shrinkage and blebbing of cells; margination of chromatin and fragmentation of nuclei and exteriorization of phosphatidylserine while the plasma membrane retains osmotic integrity. The importance of this process is established because there are many mutations, knockouts, transfections, translocations and other genetic alterations either directly to the machinery of cell death (caspase cascade and membrane-associated or mitochondrial components leading to its activation) or to important regulators of the threshold at which the cascade is activated, such as the pro- and anti-apoptotic members of the bcl-2 family of regulators, Most of these, whether spontaneously appearing in humans or engineered in rodents, produce phenotypes or diseases consistent with maladjusted patterns of apoptosis. Similarly, somatic mutation of regulators of apoptosis such as p53 considerably darkens prognosis in many cancers. Thus it is clear that regulation of cell death is an important factor in at least two classes of cancer, and many researchers have associated other severe diseases with imperfect regulation of cell death. For instance, excess cell death characterizes several neurodegenerative diseases and congenital neural anomalies, and autoimmune diseases may arise either from stimulation of T cells by apoptotic fragments resulting from chronic inflammation or from failure of the immune system to down-regulate after stimulation (meaning insufficient death of the reacting cells). However, in many or most situations that have sufficient impact to warrant our attention, either the cell death machinery is intact but the threshold at which it is activated has changed - this is true for the B cell lymphomas and p53-exacerbated digestive tract tumours - or the machinery is appropriately activated because of a proximate cause such as viral infection or oxidative or other injury to the cell. In both instances, direct attack on the machinery, for instance caspase 3, does not alleviate the primary source of the problem. It becomes very difficult to activate caspase 3 in cells possessing a bcl-2 translocation or mutation in p53 without affecting normal cells. Alternatively, while an autoimmune disease may possibly have arisen because of an accumulation of T cells, itself perhaps of genetic origin, excess cell death in inflammation derives from the inflammation rather than from a genetic defect, and the proximate cause of the inflammation must be addressed. In neurodegenerative disease, cells die after prolonged agony because they are subject to chronic metabolic or other stress. Relieving the stress is likely to be more effective than preventing death. In any event, mere survival is not the criterion. Cells may survive in an atrophied state, for instance when they are deprived of growth factors, without being functional. Nerve cells may have minimal resting potential and be incapable of generating or propagating an action potential. Other cells, subjected to lethal toxins such as anti-mitotics, may persist for extended periods of time, existing as ‘zombies’ and incapable of future division [34, 35] yet not undergoing apoptosis.
Thus, apoptosis by itself is neither pathology nor a healthy physiological situation. There is modest evidence that in a few specific, acute situations direct targeting of the cell death machinery may have value. For instance, in an infarct most of the hypoxic cells reversibly accumulate lactic acid and ultimately fail only hours later or, if circulation is re-established, shortly after reperfusion begins. In the latter case, the osmotic disequilibrium triggers either immediate osmotic lysis (necrosis) or the stressed cells undergo apoptosis a bit later. If this apoptosis is prevented, many of the cells ultimately survive and the size of the infarct may be drastically reduced, with substantial clinical benefit. Note however that this is an acute situation in which the stress is ultimately relieved. Delaying apoptosis buys time, and the critical issue is to prevent the suicide until the condition can be corrected, rather like delivering oxygen or water through a tube to an individual trapped in a collapsed building or mine. Where the stress is chronic, unknown or generic, preventing apoptosis may simply postpone the inevitable, or brute-force approaches, such as injecting caspase inhibitors, may seriously compromise healthy cells or have other untoward effects.
The ‘untoward effects’ come in at least three categories: cells have many options other than apoptosis; the response of a cell depends heavily on its history, lineage and current status; and proteins now assumed to be cell death proteins may in fact have ‘day jobs’ or multiple functions not appreciated by the researcher or physician.
Cells have many options other than apoptosis
It stands to reason that one does not need an instruction manual to die. The biological consequence of this platitude is that absence of the phenotype of apoptosis is not necessarily survival. In the laboratory, cells exposed to staurosporin, cycloheximide, ethanol or any of several chemotherapeutic drugs used experimentally may not, under certain circumstances, manifest apoptosis and they may survive a few hours longer than other cells, but they surely will die. Likewise, in an experimental or clinical situation, chemical or genetic blockage of the central machinery of apoptosis may yield decreased apoptosis but not necessarily increased survival of cells. We may have learned something about apoptosis without learning about the pathology. What most commonly happens is that the cell reverts to an alternative path of self-destruction.
There are many alternative paths. Typically, a large, cytoplasm-rich, post-mitotic or poorly mitotic cell such as a mammary epithelial cell has a big problem eliminating its cytoplasm, whereas a small, short-lived, highly mitotic cell subject to many mutagenic situations is more threatening because of its potentially damaged or mutated DNA. Thus apoptosis, with its rapid and complete destruction of DNA, is easily and promptly activated in cells of haematopoietic lineage, while mammary epithelial cells and others undertake massive autophagy. In insects, during metamorphosis the bulk of the larval tissue is destroyed, and cell death is easily timed and is tolerably synchronous. Here, apoptosis is an inconspicuous aspect of the cell death. During metamorphosis, the first 90% of the period during which the cell is dying is occupied by autophagic processes, with no activation of caspases, margination of chromatin, fragmentation of DNA or exteriorization of phosphatidyl serine. At the very end, when almost all cytoplasm has been consumed, one encounters evidence of apoptosis: condensation and margination of chromatin, fragmentation of DNA into nucleosome ladders, exteriorization of phosphatidyl serine and (though insect caspases differ from mammalian caspases and are harder to document) apparent activation of a caspase (Fig. 1). This is what we and others have called autophagic cell death, but it appears today in a different light. Although in the metamorphosing insect nutritional supplies are good (the adult or imaginal tissues are growing rapidly) the larval tissues do not thrive and undergo a type of autophagy that is normally associated with starving cells, cells deprived of trophic hormones or cells that have suffered substantial damage to organelles. It appears that, as in the case in many laboratory experiments, the cell dies only when autophagy fails to sustain it beyond a point of no return. In PC-12 cells deprived of nerve growth factor, the point of no return is the autophagic destruction of all their mitochondria. We do not know the limitation of the larval insect cells. Their tracheae (insect direct-to-cell breathing tubes) collapse and mitochondria are subjected to autophagy, suggesting an energy limitation, but they retain sufficient ATP ultimately to undergo apoptosis. We have no knowledge of the status of hormone receptors or translocators. The suggestion is that the autophagy is a survival mechanism carried beyond its limits.
1.
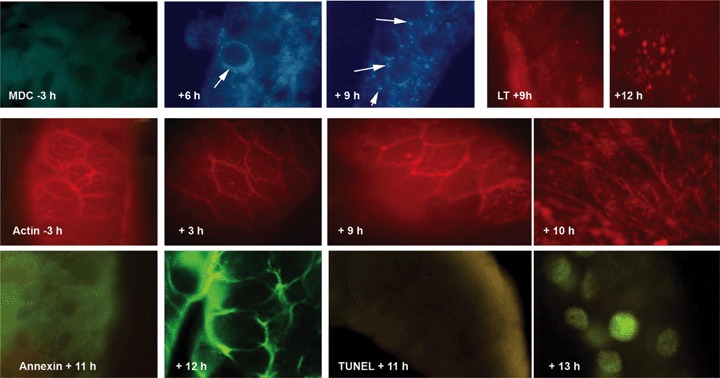
The very late appearance of characteristics of apoptosis in metamorphosing insect cells. Upper row: appearance of lysosomes during the involution of the salivary gland of Drosophila, which in our hands has completely collapsed by 13 hrs after the beginning of puparium formation. Monodansylcadaverine detects no resolvable organelles prior to onset of metamorphosis, but by 6 hrs there are numerous perinuclear autophagic vacuoles (arrows) and by 9 hrs the vacuoles fill the cytoplasm (three figures on left). Lysotracker red ® detects modest activity by 9 hrs and large autophagic vacuoles by 12 hrs. Middle row: By 3 hrs into metamorphosis, the fine filamentous actin network, detected by rhodamine phalloidin, has given way to fine granular clumps of actin, which become more pronounced by 9 hrs. By 10 hrs the actin is in vacuoles that appear to be lysosomes, or clustered near the cell membranes. Lower row: Evidence for apoptosis occurs only very late. Both exteriorization of phosphatidylserine as detected by annexin V (left two panels) and appearance of TUNEL-positive nuclei (right two panels) occur after the 11th hr.We also confirmed the presence of caspase-positive granules at 12 hrs but, failing an interpretation of the significance of the granules, we withhold an interpretation. From doctoral research of Farhan S. Khan.
Typically, autophagy is a slower process than apoptosis. In cell culture, when apoptosis is blocked and cells are subjected to challenge, they manifest substantial autophagy and die somewhat later than they would have died by apoptosis. One gets the impression that autophagy, an apparently evolutionarily older and more general process, is less efficient, slower and less certain than apoptosis as a means of killing cells, but it will kill cells.
This image is relevant to the clinical world because by this interpretation the issue is to explain the failure of the cell to survive in a milieu in which other cells do very well. Death, perhaps conventionally by apoptosis, is the product of the struggle and, though the point of no return may (or may not) represent an important clinical turning point, the bigger clinical effort is likely to be to prevent the cell from approaching the threshold. In other words-and this may be the case for chronic conditions such as neurodegenerative disease-cells may be agonizing for prolonged periods, and relieving the stress may be the means of having the most profound impact.
Many publications ignore the fact cells can live and die without cell death machinery. The caricature version of this argument is that one can pour acid onto a cell or place it in hypotonic medium, and it will expire without the assistance of caspase-3 or other enzymes, in a form of death commonly known as necrosis. However, stepping back from the caricature, it should also be self-evident that certain types of stresses will kill cells even if apoptosis is blocked. Thus, cells poisoned with metabolic inhibitors, cytoskeleton disrupters, inhibitors of DNA synthesis or replication, inhibitors of RNA or protein synthesis, strong oxidants or other materials, will not survive. For most cells examined in culture, apoptosis is the preferred physiological response to stress but the cells can also undergo autophagy and, failing that, invoke other cell-destructive activities, or ultimately lyse. One's interpretation of the result of the experiment depends very much on how one measures viability - exclusion of trypan blue or certain fluorescent molecules, failure to activate caspase 3 or extrude phosphatidyl serine, morphological appearance of the cell or its nucleus, failure to fragment DNA as measured by electrophoresis or TUNEL staining or ability to proliferate. Another operational element is the timing of the experiment: autophagy is often slower than apoptosis, and an injured cell may fail to undergo apoptosis, thus appearing viable for a few hours after it should have died; but it might well die through alternative pathways a few hours later. Quite frequently statements in the literature assume that short-term apparent survival translates into longterm survival. If it does not, the clinical expectation may change considerably.
Note that we are avoiding the term ‘autophagic cell death’. Although this term has been popular and does describe a type of cell death characterized by intense autophagy of the cytoplasm, it is much less certain that a cell can be killed by autophagy. Indeed, cells dependent on trophic hormones can atrophy and become quiescent, to the extent of shrinking to an inactive nucleus surrounded by token cytoplasm [36–40]. This shrinkage may involve autophagy or other proteolytic processes. These atrophic cells are alive, as may be demonstrated by returning their trophic factors. There is as yet no clear experimental evidence that the autophagy or proteolysis manifest by these cells differs in any meaningful way from the autophagy or proteolysis leading to the death of the cell. Rather, a different scenario seems possible. It is perhaps most clearly described for the case of insect larval tissues at metamorphosis described above. These tissues typically undergo a highly autophagic death while the adult tissues are rapidly growing. Thus, one might suggest the following scenario: The hormones that induce the growth of the adult, or imaginal, tissues render the larval tissues - by unknown means, perhaps relating to receptors or transporters of hormones or substrates - insensitive or unresponsive to hormonal activation or uptake of nutrients. In the absence of sufficient effective nourishment (remember that the imaginal cells are thriving in the same hemolymph or extracellular fluid) the cells undertake the normal physiological process of consuming their own resources. They gradually dwindle but, unlike the case of the atrophic tissue, they cannot maintain equilibrium at idle speed (even in atrophic tissue, a substantial number of cells die) and ultimately they reach a point of no return, perhaps when they have consumed even their mitochondria. At this point they die, perhaps by apoptosis. In this optic, autophagy is a survival mechanism that ultimately runs out of steam, and the question is why the cell is unresponsive.
The response of a cell depends heavily on its history, lineage and current status
A typical laboratory experiment to study apoptosis is conducted on a cell line - that is, a malignant strain of cells selected for continual growth in a Petri dish - exposed to an apoptosis-inducing situation in a medium containing approximately 1% serum. If one asks why the cell is not cultured in a medium more closely approximating the physiological extracellular medium, approximately 10% serum, one learns that ‘you don't get very good apoptosis in 10% serum’. In other words, apoptosis is studied most effectively only in severely stressed cells. This anecdote is relevant to the clinical situation in that typically, wellnourished, trophic factor-rich cells are not easily led into apoptosis. The most appropriate interpretation of this observation is that, though the pathway to apoptosis may be rather clean and linear, either the initiation of the pathway or its progress is subject to far more controls and influences than we have seriously considered. For instance, viruses have a vital interest in the survival of their host cell. At first, they need the cell to survive in order to exploit its resources to build more virus. Later, many viruses require rupture of the cell so that they can disperse. Meanwhile, it is in the interest of the organism to detect infection and to destroy the cell in a manner that also destroys the virus - apoptosis - as quickly and efficiently as possible. It is thus not surprising that the bulk of pro- and anti-apoptotic molecules that we know are either viral or cell anti-viral materials. Viruses adapt many strategies, ranging from rapid killing of cells to full inhibition of host-activated cell death [41]. Some viruses induce the death of cells as soon as synthesis and packaging are complete, allowing the virus to escape and continue to spread [42]. Other viruses co-opt host defences, maturing by use of death-associated eukaryotic proteases [43]. Others provoke destruction of B cells or helper cells, thus evading host defences [44]. Many oncogenic viruses inhibit cell death so effectively that mitosis surpasses apoptosis, allowing the tumour to grow and maximizing viral propagation [45].
Many other metabolic and lineage- or historyderived components affect the sensitivity of cells to pro- or anti-apoptotic stimuli. Kroemer, Golstein and others have recently been reflecting on the interactions between autophagic and apoptotic processes and, in a series of eloquent and carefully reasoned papers, argue that the interactions are extremely circumstance- dependent. Thus, for instance, autophagy may eliminate damaged mitochondria, preventing escape to the cytoplasm of cytochrome C, apoptosisinducing factor and other products; or it may terminate the production of reactive oxygen species, which could oxidize membranes; or it may eliminate mitochondria, leading to energy depletion; or lysosomal or other proteases may activate apical or executioner caspases; or these proteases may destroy caspases or other activating components of apoptosis, thus blocking apoptosis [46, 47]. Clearly, we have much to learn about the intermediary metabolism of threatened cells.
Proteins now assumed to be cell death proteins may in fact have ‘day jobs’ or multiple functions not appreciated by the researcher or physician
When a function is identified for a novel protein, we have an unfortunate tendency to assume that we understand the role of the protein and its ‘biological purpose’. But we may be wrong. Larger proteins may have several domains, not all of which are identified. Thus a protein might bind another protein, affecting the function of the latter. But the first protein might also have a hydrophobic domain or a nuclear localization signal. Does this mean that it transports the second protein into the nucleus, or binds it to a cell membrane? Does its binding to a membrane affect the motility of the cell, or its ability to pinocytose, phagocytose or fuse organelles? There are now many examples of proteins with clear physiological functions that have been discovered to perform other very important but distinctly different other physiological functions, often contingent on their transport to a new intracellular or extracellular location.
This issue may be defined by asking, what is the ‘day job’ of caspases? Caspases were proteases before they were drafted into service as killers of cells. Some of the caspases, notably interleukin-converting enzyme, function as immune system effectors rather than cell death enzymes. It seems improbable that the entire panoply of caspases, caspase activators and caspase inhibitors floats around the cell only to function at one final instant in the life of the cell. Such a hypothesis is supported by the observation of the pleiotropic effects of caspases.We now know that caspases appear to be involved in differentiation of erythrocytes and lens fibres, proliferation of T lymphocytes, cell migration and sperm differentiation [48–50]. Most of the functions of caspases have been defined in terms of apoptosis, through which they were first recognized, but the enzymes are typically present, though inactive, in viable cells, and activation leads to the demise of the cell. Is it truly reasonable to assume that they play no role in the physiology of the cell other than to wait for a serious threat to the cell? It seems far more likely that one or more initiator or executioner caspases might be activated in very controlled circumstances to fulfil a very different role, or that a pro-caspase might interact with another protein, sequestering it or otherwise affecting the biological activity of the protein; or caspases or interacting molecules might be trafficked from one location to another, limiting the substrates that they could attack. Such an interaction might lead to phenotypes that have no immediate or obvious connection to cell death, as has been reported for several caspase knockouts [51–57]. In another example, inhibition of caspases leads to a pronounced notochordal deformation in zebrafish embryos (Fig. 2).
2.
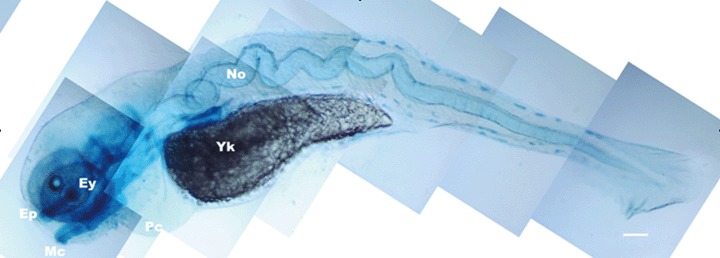
Phenotype resulting from application of the pan-caspase inhibitor, zVAD.fmk, to zebrafish embryos. The notochord (No) is longer than normal and consequently buckles, leading to severe deformity of the embryo.We have not been able to establish that the increased size results from failure of cells to die or directly from the inhibition of caspases. Ey: eye; Yk: yolk. From doctoral research of Nathaniel Abraham.
Some promise for the future?
In spite of the frustrations of the earlier and cruder efforts, there is growing evidence that apoptosisbased therapy will join the medical armamentarium. First, the development of antibodies designed to initiate cell death pathways in specific cells or circumstances [58–62] is likely to lead to specifically targeted means of attacking cancer cells, an approach that promises far lower general toxicity than most chemotherapy. Second, for acute situations short-term general blockage of apoptosis may still prove valuable [63–66]. Third, recognition of alternative forms of cell death has led to approaches to address cell death on a broader scale than narrowly as apoptosis [67–69].
To conclude, the eras of cell culture and molecular biology have been marvellous and have taught us far more than we ever dared to hope to know about the molecular interactions that drive the lives of cells. Yet there is still something missing, something that eludes us even when we know the shapes, sequences, and interactions of molecules. An image of where we might be going, and what we might need, is that of the systems engineer—that we might be going, not smaller, more micro and more nano, but to a larger scale. A systems engineer tries to understand the components of a large complex, for instance a bridge or airplane, but more importantly, he or she attempts to picture how it all works together: how unevenly distributed weight, vibrations, wind, metal fatigue, substratum strength and flexibility, etc, all interact to determine the stability of the structure. Though even the simplest cell is vastly more complex than a bridge, we will have to develop algorithms by which we can envision a cell not as a static or acutely changing mix of components, but as a complex of dynamic and mobile interacting components, some of which are sufficiently stable or long-lived to bear witness to the history and lineage of the cell, others of which reflect the immediate status of the cell, and which interact across distances and pathways that we are not currently considering. This is a vastly larger goal than we can hope to achieve immediately, but even the most modest access will help us to better target, and therefore control, cell death. As the hundreds of thousands of publications attest, cell death is a medically urgent topic, and movement to the next level should cross the clinical horizon.
References
- 1.Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death. Autophagy. 2005;1:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- 2.Bialik S, Kimchi A. The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem. 2006;75:189–210. doi: 10.1146/annurev.biochem.75.103004.142615. [DOI] [PubMed] [Google Scholar]
- 3.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green DR, Chipuk JE. p53 and metabolism: Inside the TIGAR. Cell. 2006;126:30–2. doi: 10.1016/j.cell.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 5.Penaloza C, Lin L, Lockshin RA, Zakeri Z. Cell death in development: shaping the embryo. Histochem Cell Biol. 2006;126:149–58. doi: 10.1007/s00418-006-0214-1. [DOI] [PubMed] [Google Scholar]
- 6.Ferraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem Biophys. 2007;462:210–9. doi: 10.1016/j.abb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Kunchithapautham K, Rohrer B. Autophagy is one of the multiple mechanisms active in photoreceptor degeneration. Autophagy. 2007;3:65–6. doi: 10.4161/auto.3431. [DOI] [PubMed] [Google Scholar]
- 9.Yoshimori T. Autophagy: paying Charon's toll. Cell. 2007;128:833–6. doi: 10.1016/j.cell.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 10.Hardy K. Cell death in the mammalian blastocyst. Mol Hum Reprod. 1997;3:919–25. doi: 10.1093/molehr/3.10.919. [DOI] [PubMed] [Google Scholar]
- 11.Spanos S, Rice S, Karagiannis P, Taylor D, Becker DL, Winston RM, Hardy K. Caspase activity and expression of cell death genes during development of human preimplantation embryos. Reproduction. 2002;124:353–63. doi: 10.1530/rep.0.1240353. [DOI] [PubMed] [Google Scholar]
- 12.Zakeri Z, Lockshin RA, Criado-Rodriguez LM, Martinez-A C. A generalized caspase inhibitor disrupts early mammalian development. Int J Dev Biol. 2005;49:43–7. doi: 10.1387/ijdb.041920zz. [DOI] [PubMed] [Google Scholar]
- 13.Penaloza C, Lin L, Lockshin RA, Zakeri Z. Cell death in development: shaping the embryo. Histochem Cell Biol. 2006;126:149–58. doi: 10.1007/s00418-006-0214-1. [DOI] [PubMed] [Google Scholar]
- 14.Cole LK, Ross LS. Apoptosis in the developing zebrafish embryo. Dev Biol. 2001;240:123–42. doi: 10.1006/dbio.2001.0432. [DOI] [PubMed] [Google Scholar]
- 15.Jeffery WR. Cavefish as a model system in evolutionary developmental biology. Dev Biol. 2001;231:1–12. doi: 10.1006/dbio.2000.0121. [DOI] [PubMed] [Google Scholar]
- 16.Kubo E, Singh DP, Fatma N, Shinohara T, Zelenka P, Reddy VN, Chylack LT., Jr Cellular distribution of lens epithelium-derived growth factor (LEDGF) in the rat eye: loss of LEDGF from nuclei of differentiating cells. Histochem Cell Biol. 2003;119:289–99. doi: 10.1007/s00418-003-0518-3. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Martinez V, Macias D, Gañan Y, Garcia-Lobo JM, Francia MV, Fernandez-Teran MA, Hurle JM. Internucleosomal DNA fragmentation and programmed cell death (apoptosis) in the interdigital tissue of the embryonic chick leg bud. J Cell Sci. 1993;106:201–8. doi: 10.1242/jcs.106.1.201. [DOI] [PubMed] [Google Scholar]
- 18.Zakeri Z, Quaglino D, Ahuja HS. Apoptotic cell death in the mouse limb and its suppression in the hammertoe mutant. Dev Biol. 1994;165:294–7. doi: 10.1006/dbio.1994.1255. [DOI] [PubMed] [Google Scholar]
- 19.Gañan Y, Macias D, Hurle JM. Pattern regulation in the chick autopodium at advanced stages of embryonic development. Dev Dyn. 1994;199:64–72. doi: 10.1002/aja.1001990107. [DOI] [PubMed] [Google Scholar]
- 20.Mori C, Nakamura N, Kimura S, Irie H, Takigawa T, Shiota K. Programmed cell death in the interdigital tissue of the fetal mouse limb is apoptosis with DNA fragmentation. Anat Rec. 1995;242:103–10. doi: 10.1002/ar.1092420114. [DOI] [PubMed] [Google Scholar]
- 21.Hurle JM, Colombatti A. Extracellular matrix modifications in the interdigital spaces of the chick embryo leg bud during the formation of ectopic digits. Anat Embryol. 1996;193:355–64. doi: 10.1007/BF00186692. [DOI] [PubMed] [Google Scholar]
- 22.Merino R, Ganan Y, Macias D, Rodriguez-Leon J, Hurle JM. Bone morphogenetic proteins regulate interdigital cell death in the avian embryo. Ann NY Acad Sci. 1999;887:120–32. doi: 10.1111/j.1749-6632.1999.tb07927.x. [DOI] [PubMed] [Google Scholar]
- 23.Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol. 1999;9:967–70. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- 24.Daugas E, Cande C, Kroemer G. Erythrocytes: death of a mummy. Cell Death Differ. 2001;8:1131–3. doi: 10.1038/sj.cdd.4400953. [DOI] [PubMed] [Google Scholar]
- 25.Bratosin D, Estaquier J, Petit F, Arnoult D, Quatannens B, Tissier JP, Slomianny C, Sartiaux C, Alonso C, Huart JJ, Montreuil J, Ameisen JC. Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ. 2001;8:1143–56. doi: 10.1038/sj.cdd.4400946. [DOI] [PubMed] [Google Scholar]
- 26.Bratosin D, Estaquier J, Ameisen JC, Montreuil J. Molecular and cellular mechanisms of erythrocyte programmed cell death: impact on blood transfusion. Vox Sang. 2002;83:307–10. doi: 10.1111/j.1423-0410.2002.tb05324.x. [DOI] [PubMed] [Google Scholar]
- 27.Lang KS, Duranton C, Poehlmann H, Myssina S, Bauer C, Lang F, Wieder T, Huber SM. Cation channels trigger apoptotic death of erythrocytes. Cell Death Differ. 2003;10:249–56. doi: 10.1038/sj.cdd.4401144. [DOI] [PubMed] [Google Scholar]
- 28.Lockshin RA, Zakeri Z. Caspase-independent cell death. Oncogene. 2004;23:2766–73. doi: 10.1038/sj.onc.1207514. [DOI] [PubMed] [Google Scholar]
- 29.Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36:2405–19. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 30.Reeve JL, Duffy AM, O'Brien T, Samali A. Don't lose heart-therapeutic value of apoptosis prevention in the treatment of cardiovascular disease. J Cell Mol Med. 2005;9:609–22. doi: 10.1111/j.1582-4934.2005.tb00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahuja HS, James W, Zakeri Z. Rescue of the limb deformity in hammertoe mutant mice by retinoic acidinduced cell death. Dev Dyn. 1997;208:466–81. doi: 10.1002/(SICI)1097-0177(199704)208:4<466::AID-AJA3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 32.Horvitz HR, Sternberg PW, Greenwald IS, Fixsen W, Ellis HM. Mutations that affect neural cell lineages and cell fates during the development of the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol. 1983;48:453–63. doi: 10.1101/sqb.1983.048.01.050. [DOI] [PubMed] [Google Scholar]
- 33.Horvitz HR. Nobel lecture. Worms, life and death. Biosci Rep. 2003;23:239–303. doi: 10.1023/b:bire.0000019187.19019.e6. [DOI] [PubMed] [Google Scholar]
- 34.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–54. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deshmukh M, Johnson EM., Jr Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- 36.Wigglesworth VB. The action of growth hormones in insects. Symp Soc Exp Biol. 1957;54:204–27. [PubMed] [Google Scholar]
- 37.Schiaffino S, Hanzlikova V. Studies on the effect of denervation in developing muscle. II. The lysosomal system. J Ultrastruct Res. 1972;39:1–14. doi: 10.1016/s0022-5320(72)80002-9. [DOI] [PubMed] [Google Scholar]
- 38.Hanzlikova V, Schiaffino S. Studies on the effect of denervation in developing muscle. 3. Diversification of myofibrillar structure and origin of the heterogeneity of muscle fiber types. Z Zellforsch Mikrosk Anat. 1973;147:75–85. doi: 10.1007/BF00306601. [DOI] [PubMed] [Google Scholar]
- 39.Gutmann E, Hanzlikova V. Denervation, reinnervation and regeneration of senile muscle. Adv Exp Med Biol. 1975;53:431–40. doi: 10.1007/978-1-4757-0731-1_36. [DOI] [PubMed] [Google Scholar]
- 40.Hanzlikova V, Gutmann E. Effect of castration and testosterone administration on the neuromuscular junction in the levator ani muscle of the rat. Cell Tissue Res. 1978;189:155–66. doi: 10.1007/BF00223126. [DOI] [PubMed] [Google Scholar]
- 41.Hilleman MR. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc Natl Acad Sci USA. 2004;101:14560–6. doi: 10.1073/pnas.0404758101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roulston A, Marcellus RC, Branton PE. Viruses and apoptosis. Annu Rev Microbiol. 1999;53:577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- 43.Wurzer WJ, Planz O, Ehrhardt C, Giner M, Silberzahn T, Pleschka S, Ludwig S. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J. 2003;22:2717–28. doi: 10.1093/emboj/cdg279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casella CR, Rapaport EL, Finkel TH. Vpu increases susceptibility of human immunodeficiency virus type 1-infected cells to fas killing. J Virol. 1999;73:92–100. doi: 10.1128/jvi.73.1.92-100.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar A, Zhao Y, Meng G, Zeng M, Srinivasan S, Delmolino LM, Gao Q, Dimri G, Weber GF, Wazer DE, Band H, Band V. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol Cell Biol. 2002;22:5801–12. doi: 10.1128/MCB.22.16.5801-5812.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Golstein P, Kroemer G. A multiplicity of cell death pathways. Symposium on apoptotic and non-apoptotic cell death pathways. EMBO Rep. 2007;8:829–33. doi: 10.1038/sj.embor.7401042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Selfeating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 48.Fadeel B, Orrenius S, Zhivotovsky B. The most unkindest cut of all: on the multiple roles of mammalian caspases. Leukemia. 2000;14:1514–25. doi: 10.1038/sj.leu.2401871. [DOI] [PubMed] [Google Scholar]
- 49.Kumar S. Migrate, differentiate, proliferate, or die. pleiotropic functions of an apical “apoptotic caspase”. Sci STKE. 2004:49. doi: 10.1126/stke.2542004pe49. [DOI] [PubMed] [Google Scholar]
- 50.Launay S, Hermine O, Fontenay M, Kroemer G, Solary E, Garrido C. Vital functions for lethal caspases. Oncogene. 2005;24:5137–48. doi: 10.1038/sj.onc.1208524. [DOI] [PubMed] [Google Scholar]
- 51.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–76. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–50. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 53.Roth KA, Kuan C, Haydar TF, D'Sa-Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P. Epistatic and independent functions of caspase-3 and Bcl-X(L) in developmental programmed cell death. Proc Natl Acad Sci USA. 2000;97:466–71. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leonard JR, Klocke BJ, D'Sa C, Flavell RA, Roth KA. Strain-dependent neurodevelopmental abnormalities in caspase-3-deficient mice. J Neuropathol Exp Neurol. 2002;61:673–7. doi: 10.1093/jnen/61.8.673. [DOI] [PubMed] [Google Scholar]
- 55.Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science. 2003;302:1560–3. doi: 10.1126/science.1087621. [DOI] [PubMed] [Google Scholar]
- 56.Houde C, Banks KG, Coulombe N, Rasper D, Grimm E, Roy S, Simpson EM, Nicholson DW. Caspase-7 expanded function and intrinsic expression level underlies strain-specific brain phenotype of caspase-3-null mice. J Neurosci. 2004;24:9977–84. doi: 10.1523/JNEUROSCI.3356-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsden VS, Ekert PG, Van Delft M, Vaux DL, Adams JM, Strasser A. Bcl-2-regulated apoptosis and cytochrome c release can occur independently of both caspase-2 and caspase-9. J Cell Biol. 2004;165:775–80. doi: 10.1083/jcb.200312030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonavida B, Ng CP, Jazirehi A, Schiller G, Mizutani Y. Selectivity of TRAIL-mediated apoptosis of cancer cells and synergy with drugs: the trail to non-toxic cancer therapeutics. Int J Oncol. 1999;15:793–802. doi: 10.3892/ijo.15.4.793. [DOI] [PubMed] [Google Scholar]
- 59.Cal C, Garban H, Jazirehi A, Yeh C, Mizutani Y, Bonavida B. Resveratrol and cancer: chemoprevention, apoptosis, and chemo-immunosensitizing activities. Curr Med Chem Anticancer Agents. 2003;3:77–93. doi: 10.2174/1568011033353443. [DOI] [PubMed] [Google Scholar]
- 60.Jazirehi AR, Bonavida B. Cellular and molecular signal transduction pathways modulated by rituximab (rituxan, anti-CD20 mAb) in non-Hodgkin's lymphoma: implications in chemosensitization and therapeutic intervention. Oncogene. 2005;24:2121–43. doi: 10.1038/sj.onc.1208349. [DOI] [PubMed] [Google Scholar]
- 61.Bonavida B. Rituximab-induced inhibition of antiapoptotic cell survival pathways. implications chemo/immunoresistance, rituximab unresponsiveness, prognostic and novel therapeutic interventions. Oncogene. 2007;26:3629–36. doi: 10.1038/sj.onc.1210365. [DOI] [PubMed] [Google Scholar]
- 62.Daniel D, Yang B, Lawrence DA, Totpal K, Balter I, Lee WP, Gogineni A, Cole MJ, Fong Yee S, Ross S, Ashkenazi A. Cooperation of the pro-apoptotic receptor agonist rhApo2L/TRAIL with the CD20 antibody rituximab against non-Hodgkin's lymphoma xenografts. Blood. 2007;110:4037–46. doi: 10.1182/blood-2007-02-076075. [DOI] [PubMed] [Google Scholar]
- 63.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW, 2nd, Armstrong RC, Kitsis RN. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice. Circulation. 2003;108:3036–41. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 64.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–70. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 65.Tarzami ST, Calderon TM, Deguzman A, Lopez L, Kitsis RN, Berman JW. MCP-1/CCL2 protects cardiac myocytes from hypoxia-induced apoptosis by a G(alphai)-independent pathway. Biochem Biophys Res Commun. 2005;335:1008–16. doi: 10.1016/j.bbrc.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 66.Kitsis RN, Jialal I. Limiting myocardial damage during acute myocardial infarction by inhibiting C-reactive protein. N Engl J Med. 2006;355:513–5. doi: 10.1056/NEJMcibr063197. [DOI] [PubMed] [Google Scholar]
- 67.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 68.Teng X, Degterev A, Jagtap P, Xing X, Choi S, Denu R, Yuan J, Cuny GD. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg Med Chem Lett. 2005;15:5039–44. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 69.Hu X, Han W, Li L. Targeting the weak point of cancer by induction of necroptosis. Autophagy. 2007;3:490–2. doi: 10.4161/auto.4592. [DOI] [PubMed] [Google Scholar]