

Abstract
Multiple genetic and environmental factors are likely to contribute to the development of Alzheimer's disease (AD). The most important known risk factor for AD is presence of the E4 isoform of apolipoprotein E (apoE). Epidemiological studies demonstrated that apoE4 carriers have a higher risk and develop the disease and an early onset. Moreover, apoE4 is the only molecule that has been associated with all the biochemical disturbances characteristic of the disease: amyloid-beta (Aβ) deposition, tangle formation, oxidative stress, lipid homeostasis deregulation, synaptic plasticity loss and cholinergic dysfunction. This large body of evidence suggest that apoE is a key player in the pathogenesis of AD. This short review examines the current facts and hypotheses of the association between apoE4 and AD, as well as the therapeutic possibilities that apoE might offer for the treatment of this disease.
Keywords: apolipoprotein E, Alzheimer's disease, neurodegeneration
Introduction
Alzheimer's disease (AD) is the leading cause of dementia among people aged 65 and older. The disease is characterized by a slow progressive loss of cognition, behavioural changes and loss of independence that ends in the complete destruction of the personality. AD is characterized by the presence in the brain of extracellular neuritic plaques (mainly constituted of aggregated amyloid β-peptide [Aβ]), and intraneuronal neurofibrillary tangles (NFT) (rich in abnormally phosphorylated tau). The vast majority of AD cases are sporadic and the primary causes of the disease remain elusive. There is a small percentage of familial cases with a clear genetic component due to a mutation in the genes either for the amyloid precursor protein (APP) or pre-senilins [1].
The strongest known risk factor influencing the incidence of sporadic AD is the genotype for apolipoprotein E (ApoE). ApoE is the major carrier of cholesterol in the CNS. In humans, the apoE gene shows polymorphism, with three different alleles (e2, e3 and e4), that give rise to six different phenotypes (E2/2, E2/3, E2/4, E3/3, E3/4 and E4/4). ApoE3 is the most common isoform (77–78%) in the general population, while apoE2 is found in 7–8% and apoE4 in 14–16% of individuals. Individuals with one or two copies of apoE4 have a higher risk of developing AD, compared with carriers of other isoforms. ApoE4 also reduces the median age of AD onset (from 84 in non-carriers to 68 in E4 homozygotes) [2].
Since the discovery of the apoE4/AD association, abundant experimental studies have shown that apoE influences the majority of the AD pathological processes (Aβ generation or deposition, NFT formation, neuronal survival, lipid homeostasis, intracellular signalling, etc.). In all cases, apoE4 was shown to be detrimental (due to either negative or lack of positive effects) compared with other isoforms. However, due to the omnipresence of apoE in all the biochemical disturbances characteristic of AD, it has been difficult to propose a simple and integrative hypothesis that could explain the mechanisms by which apoE4 increases the susceptibility for this disease.
In addition, it has also been shown that the presence of apoE4 has a large impact on longevity, cardiovascular diseases and different other neurological disorders including stroke, cerebral amyloid angiopathy [3], Lewy body disease [4], multiple sclerosis [5], Parkinson's disease [6] and several others. This raises the question whether apoE could participate in some molecular neurodegenerative mechanisms that might be common for several disorders.
Thus, understanding the mechanisms behind the apoE contribution to neurodegenerative disorders would offer new perspectives for the treatment of these diseases. This short review examines the major facts and hypotheses of the contribution of apoE4 to AD and the potential apoE-based therapeutic approaches for this disorder.
ApoE function
The largest production of apoE is found in the liver, followed by the brain. In the brain, apoE is predominantly secreted by astrocytes and microglia [7, 8], although even neurons have been reported to produce it [9, 10].
ApoE is a major determinant of the recognition and uptake of lipoproteins though the low density lipoprotein (LDL) receptor, the LDL receptor related protein (LRP), the apoE receptor 2, the very low density lipoprotein (VLDL) receptor and megalin [11]. ApoE plays an important role in the distribution and metabolism of cholesterol and triglycerides within many organs and cell types in the human body [12]. The apoE polymorphism seen in humans is unique and it has been proposed to appear as a result of evolutionary adaptive changes [13, 14]. The three isoforms E2, E3 and E4 differ from one another at residues 112 and 158. ApoE3 has Cys-112 and Arg-158, whereas apoE4 has arginine at both positions, and apoE2 has cysteine. This substitution affects the three-dimensional structure and the lipid-binding properties between isoforms. In apoE4, the amino acid substitution results in a changed structure with the formation of a salt-bridge between an arginine in position 61 and a glutamic acid in 255 that causes this isoform to bind preferentially to VLDL. ApoE3 and apoE2 bind preferentially high-density lipopro-teins (HDL) [12].
The mechanisms that govern apoE synthesis and secretion in brain are not fully understood. Estrogen has been shown to increase apoE mRNA levels in rat brain in vivo and in astrocytes and mixed glia cultures in vitro[15, 16]. ApoE secretion in rat primary astrocytes can also be modulated by neurotransmitter receptors that regulate intracellular cAMP and protein kinase C (PKC) activity [17]. ApoE secretion from human primary astrocytes can be reduced by the combination of interleukin 1 α and β and interferon γ[18]. Moreover, apoE levels increase dramatically in response to peripheral nerve injury in a rat model [19]. A moderate injury induced enhancement of apoE levels has been also shown in CNS [20]. Such data led to the hypothesis that apoE plays an important role in the maintenance and repair of neu-rons, by distributing lipids necessary for proliferation, synaptogenesis and myelinization of axons.
ApoE role in AD pathology
ApoE and Aβ
The production, oligomerization and deposition of Aβ are known to play a central role in AD. The discovery of autosomal dominant mutations in the APP and presenilin genes (PS) genes, which result in an overall increase in production of Aβ(1–42), emphasized the idea that overproduction of Aβ is a causative agent of AD. Several lines of evidence suggest that at least some of the pathological effects of apoE4 may be mediated by interactions with the Aβ cascade.
Firstly, apoE is present in neuritic plaques and Aβ levels are elevated in brains of AD patients carrying apoE4 [21]. Similar findings have been observed with transgenic mice expressing human apoE isoforms [22, 23].
Secondly, several studies showed differences in the binding of apoE3 and apoE4 to Aβ. However, it remains to be elucidated if apoE4 has an active role in facilitating Aβ aggregation and/or deposition or if, in contrast, apoE3 and E2 have a protective role by inhibiting Aβ aggregation or favouring Aβ clearance. ApoE4, in a lipid-free form has a greater avidity for Aβ than apoE3 [24], but it has also been shown that apoE3 and E2 bind more rapidly to Aβ when associ-ated with lipoproteins [25]. The dosage of apoE was found to be determinant for plaque deposition in a mice overexpressing the mutant human APPV717F[26], which talks in favour of apoE being an active cofactor in plaque formation. However, in APPV717F mice bred onto a mice expressing human apoE in astrocytes, it was found that apoE3 favours Aβ clearance, as apoE3-APPV717F mice showed reduced Aβ deposition compared with apoE4-APPV717F mice [27]. Interestingly, in another mice model overexpressing a mutant variant of human APP, it has been shown that levels of Aβ and apoE in brain increased in parallel with age, at the expense of a decrease of Aβ in plasma [28]. This would suggest that elevations of brain apoE levels during aging would deregulate Aβ clearance and increase Aβ sequestration. Post-mortem studies have shown that Aβ deposition is increased in ApoE4 carriers in both sporadic [21] and genetic AD cases [29]. Recently, it has been shown that the cognitive impairment seen in human APP transgenic mice depends both on apoE and on amyloid formation [30].
Thirdly, neuronal ApoE receptors may also have several roles in APP trafficking and processing [31], in Aβ clearance [32] and apoE4 has been found to enhance the synthesis of Aβ by promoting endocytic recycling of APP [33].
Finally, in vitro data demonstrate that apoE increases the neurotoxicity of Aβ in an isoform-specific manner (E4 > E3) [34, 35]. In addition, a recent study has shown that the impairments in neuroplasticity induced by apoE4 following environmental stimulation in a transgenic mice model are associated with the accumulation of intraneuronal Aβ[36]. These findings suggest the existence of synergistic pathological effects between Aβ and apoE4.
ApoE and neurofibrillary tangles
Abnormal phosphorylation of the tau protein leading to the formation of NFTs is a common feature of AD, and several other neurodegenerative disorders [37]. It is believed that tau hyperphosphorylation compromises the normal functioning of the neuron leading to its death.
The hypothesis that apoE isoforms may differently influence tau pathology derives from in vitro studies, where apoE3 and not apoE4 forms a (SDS)-stable complex with tau. Phosphorylation of tau inhibits its interaction with apoE3 suggesting that apoE3 only binds to non-phosphorylated tau. This further suggests that apoE3 might be able to prevent abnormal tau hyperphosphorylation and destabilization of the neuronal cytoskeleton [38]. Transgenic mice studies have shown increased phosphorylation of tau in mice expressing human apoE4 in neurons, but not in mice expressing apoE4 in astrocytes [39, 40], indicating a neuron-specific effect of apoE4 on tau phosphorylation. In addition, intraneuronal accumulation of hyperphosphorylated tau has been found in apoE KO mice fed with a high cholesterol diet, suggesting a synergic interaction of dietary cholesterol and lack of apoE function [41].
It has been proposed that the amino-terminal domain of apoE3 is responsible for binding to tau [38]. Additionally, studies in transgenic mice have shown that carboxyl-terminal-truncated apoE stimulates tau phosphorylation and intracellular NFT-like inclusion formation in [42]. A direct molecular interaction between the apoE (or apoE fragments) and tau molecules would require that both meet in the same cytosolic compartment. The question of how apoE accesses the neuronal cytoskeleton remains puzzling. An alternative mechanism in which apoE isoforms would differentially contribute to tau hyperphosphorylation is the modulation of tau kinases and phosphatases.
ApoE, cholesterol and synaptic repair
The major function of apoE is to re-distribute lipids and participate in cholesterol homeostasis [12]. In cultured neurons, cholesterol uptake is lower when the lipid is bound to ApoE4 compared to ApoE2 and ApoE3 [43]. ApoE4 is less efficient than other forms in promoting cholesterol efflux from both neurons and astrocytes [44]. The structural differences between different apoE isoforms may account for these alterations. In AD, there is a decrease of cholesterol levels in brain and growing evidence indicates that cholesterol itself is involved in AD pathogenesis [45]. The first indication has come from clinical and epidemiological studies showing that patients with elevated plasma cholesterol levels have increased susceptibility to AD [46–48]. Several studies have shown that the use of statins, drugs that inhibit the cholesterol synthesis, decrease the prevalence [49, 50] and the progression [51] of AD. In addition to apoE, other genes involved in the transport or in the metabolism of cholesterol have been suggested as putative risk factors for AD. Polymorphisms in receptors for the uptake of cholesterol, such as low-density lipoprotein receptor-related protein (LRP) and the very-low-density lipoprotein (VLDL) receptor [52], as well as in enzymes that regulate the choles-terol catabolism, such us Cyp46 [53], have been associated with an increased risk for AD. Furthermore, a number of studies suggest that cholesterol regulates the Aβ production. β-secretase and γ-secretase are localized in cholesterol-rich lipid rafts [54–56], while the non-amyloidogenic α-secretase is associated with the membrane surface, outside raft domains [57]. β-secretase activity is increased by cholesterol [58]. In addition, changes in cholesterol levels or distribution within the membrane have been shown to alter the localization of APP and their availability to be cleaved by these secretases [59, 60]. On the other hand, Aβ modulates the synthesis and the distribution of cholesterol in neurons [61]. Moreover, it has been shown that cholesterol reduces the effects of Aβ on calcium signalling and neurotoxicity in several models [34, 62, 63]. Such data illustrate that the interactions between cholesterol and Aβ are very complex. Regarding the different ability of variant apoE isoforms for carrying lipids, it is likely that the cholesterol/Aβ interactions would be modulated by the apoE genotype.
In the nervous system, interaction between neurons and glial cells is very important in processes of growth, regeneration and synaptic plasticity [64]. There, the apoE-mediated re-distribution of lipids plays a fundamental role. In AD, there is extensive neuronal loss in the limbic system and neocortex, as well as an important synaptic dysfunction that affect the normal being of the surviving neurons [65]. It has been suggested that the variant apoE isoforms participate differently in repair processes and synaptic plasticity. Synaptic plasticity in the CA1 region has been found to be impaired in apoE KO mice [66]. ApoE KO and apoE4−, but not E3−, transgenic mice have an age-dependent disruption of synaptic organization [67, 68]. These apoE isoform differences have also been reported in humans. ApoE4 carriers show a poor compensation of neuronal loss in different brain regions, whereas non-E4 carriers exhibit marked regenerative changes in the same areas [69]. Such data suggest a lack of function of apoE4 in synaptic regeneration, compared with other apoE isoforms. It is likely that this lack of function will also affect synaptic function. Supporting this idea, it has been reported that long-term potentiation (LTP) is reduced in apoE4-transgenic mice compared with wild-type mice and apoE3 mice [70].
ApoE and cholinergic dysfunction
Cholinergic signal transduction is well known to be impaired in AD. ApoE4 carriers with AD show greater deficits than non-carriers in cholinergic activity in the hippocampus and the cortex, as well as a reduction in the total number of cholinergic neurons, and of cholinergic markers, such as choline acetyltransferase activity and nicotinic ACh receptor binding [71–73]. In contrast, there are no significant differences in muscarinic receptor levels between AD patients with different apoE genotypes [73].
Variant apoE isoforms have different effects on ACh muscarinic receptor stimulated signalling in vitro. ApoE4 impaired carbachol-stimulated phosphoinositide hydrolysis, whereas apoE3 alone was without effect. In addition, ApoE3, but not E4, could protect against Aβ(1-42)-mediated disruption [74]. In a double transgenic mice model, it has recently been reported that modulation of AD-like cholinergic deficits depends on the apoE isoform, the overproduction of Aβ, and the age of the animal, but not on plaque deposition [75]. In this study, human APP/apoE4 mice showed synaptic and cholinergic deficits prior to plaque formation. However, old human APP/apoE4 and human APP/apoE3 mice had similar synaptic and cholinergic deficits, despite their differences in plaque load [75].
A direct negative influence of apoE4 on cholinergic signalling may participate in the lower effective-ness of cholinergic replacement treatments reported for apoE4-AD patients [73].
ApoE and signalling
ApoE has been shown to modulate various signalling pathways, some of which are relevant to AD. In several in vitro models, apoE was shown to affect multiple signalling cascades in an isoform-specific manner. ApoE isoforms differentially influence calcium channels causing different increases in free intracellular calcium [76–78]. Also, apoE has isoform-specific effects on the activities of PKC [79], glycogen synthase kinase-3β (GSK-3β) [76], protein kinase B (Akt) [76], extracellular signal-regulated kinase 1/2 (ERK) [80, 81], c-Jun N-terminal kinase 1/2 (JNK) [80] and on the transcriptional activity of the cAMP response element-binding protein (CREB) [81]. Recently, a large microarray study with hippocampal samples from AD patients demonstrated that patterns of gene expression differ substantially between have apoE4 and non-apoE carriers [82]. ApoE4 individuals have more expression of tumour suppressors, and negative regulators of cell growth that may lead to increased apoptosis. In contrast, they showed decreased expression of genes associated with synaptic plasticity, neuronal outgrowth, several neurotransmitter receptors, as well as genes involved in mitochondrial oxidative phosphorylation/-energy metabolism.
Alteration in neurotransmitter receptors and down-stream signalling may contribute to the development of resistance to some pharmacological therapies seen in individuals with apoE4 [73, 83]. Furthermore, the apoE4-mediated disruption of mul-tiple signalling pathways suggests that apoE4 carriers have both a loss of function in cell protection mechanisms and a gain of function in pathways leading to Aβ formation and tau phosphorylation.
ApoE and neurotoxicity
ApoE may also contribute to neurodegenerative processes by being directly toxic to neurons. In this context, lipid-free apoE (E4 > E3) and apoE-derived fragments have been shown to be toxic to neurons in vitro[34, 84, 85]. However, when instead of delipidated apoE, the more physiological apoE-containing lipoproteins were used, this toxicity was avoided [34]. In fact, apoE-containing lipoproteins were shown to promote cell survival and neurite outgrowth also in an isoform-specific manner (E3 > E4) [86, 87]. These data suggest that apoE status or the conformation of apoE-containing lipoproteins might be an important determinant of apoE neurotoxicity.
In vivo studies using transgenic mice that express human apoE3, apoE4 or both, have demonstrated that apoE3/E3 animals were more protected than apoE3/4 against age-induced neurodegeneration and that apoE4/4 showed no protection [88]. This would imply that apoE4 is not only less neuroprotective than apoE3, but also acts as a dominant negative factor interfering with the beneficial function of apoE3.
It has been reported that a N-terminal proteolytic fragment (amino acids 1–191) is responsible for apoE toxicity by increasing intracellular calcium levels [85]. Conversely, several reports from another group have shown that the C-terminal fragment of apoE is neurotoxic in vitro by a mechanism that involves mitochondrial and cytoskeletal alterations [84]. In vivo, apoE4 C-terminal fragments were shown to induce neurodegeneration and behavioural deficits in transgenic mice [42]. Importantly, apoE C-terminal fragments were present at much higher levels in the brains of AD patients (especially associated with NFT) than in controls [40, 89], although the difference between truncated apoE levels in E3 and E4 carriers was not demonstrated.
In view of the different results obtained with lipid-free or lipid-bound apoEs, it is possible that apoE will be more resistant to proteolysis when associated with other lipoproteins or that compositional and/or structural differences of lipoprotein containing apoE particles may be important for neurodegeneration. Since each apoE isoform possesses structurally defined abilities in lipid binding, it is also important to consider that the composition of lipoprotein particles may differ with apoE genotype. Therefore, it is possible that apoE4 is more susceptible to cleavage than apoE3.
Recently, Hatters et al. [90] reported that apoE forms soluble fibrillar aggregates in vitro, and that the rate of aggregation vary substantially between isoforms (E4 > E3 > E2). ApoE fibrils were significantly more toxic to cultured neuronal cells than the tetramers. Previously, the same group demonstrated that lipid-binding protects apolipoproteins from misfolding into amyloid fibrils [91], suggesting that the formation of apoE fibrils may require the presence of lipid-free apoE. However, the presence of apoE fibrils (as well as the isoform differential fibrillation) in AD brain has still to be demonstrated.
Understanding the factors that govern the apoE neurotoxicity (lipid association, proteolytic cleavage, fibrillation, etc) as well as its neuroprotective effects remains crucial for elucidating the role of apoE in neurodegenerative processes.
ApoE as a therapeutic target for AD
A rational strategy for considering the therapeutic potential of apoE would need to take into account whether the apoE4 isoform-specific effects are due to a loss of normal properties (associated with apoE3), to direct deleterious effects, or to both.
If we consider the beneficial effects of apoE, a therapeutic strategy would be to pharmacologically enhance levels of endogenous apoE in brain irrespective of the apoE isotype [92]. This would be useful in restoring lipid homeostasis in AD subjects, in order to promote synaptogenesis and regeneration. However, considering the detrimental effects of apoE4, this strategy it is likely to be useful for apoE3 patients. In addition, most patients possessing the apoE4 are not homozygous, raising the question of how heterozygous E3/E4 patients would respond to such therapy.
An alternative approach would be the creation of an apoE-mimetic compound conserving the functional characteristics of the intact holoprotein, and being able to cross the blood-brain barrier [93]. ApoE-mimetic peptides have been reported to suppress glial inflammatory responses and to protect neurons from excitotoxic injury [94]. The mechanisms behind these positive effects are unknown, but since these peptides do not contain the lipid-binding domain, it is suggested that they are not related to the lipid transport properties of the physiological apoE. A second generation of apoE-mimetic peptides have been recently reported [95]. Those peptides showed improved outcomes in animal models of multiple sclerosis [96] and brain injury [97] without any described side-effect. Thus, the use of apoE-mimetic peptides appears to be an exciting possibility for the treatment of AD and neurodegenerative disorders.
Based on the idea that apoE acts as a chaperon and facilitates Aβ deposition, another therapeutic approach proposes the use of compounds that block the apoE/Aβ interaction [98]. A short synthetic peptide Aβ12–28P, which is non-immunogenic and brain blood barrier-permeable, has been shown to be effective in APPK670L/M671L and APPK670L/M671L/PS1M146L transgenic mice [98]. However, it is still unknown how the increase of soluble circulating Aβ would affect the brain, and how these compounds would influence the potential capacity of apoE for clearing Aβ.
Another therapeutic approach proposed is to change the tri-dimensional structure of apoE4 to an apoE3-like conformation [99]. This requires the use of agents that selectively interact with apoE4 preventing its domain interaction. Several of these molecules have been found, some of which showed positive effects in vitro systems. For example, two compounds, GIND-25, a disulfonate, and GIND-105, a monosulfoalkyl, decreased Aβ production induced by apoE4 to levels very similar to those induced by apoE3 [99]. The use of these molecules would restore the ability of apoE4 for carrying lipids and would reduce the rate of apoE4 cleavage to the levels of apoE3. Theoretically, this strategy would both decrease the negative effects and potentiate the pro-tective effects of apoE4 simultaneously.
Other apoE-based therapeutical strategies have also been proposed. Using an inhibitor of the apoE protease would prevent the generation of the detrimental apoE fragments [84]. Blocking the interaction of apoE4-derived fragments with the mitochondria would prevent the detrimental effects on mitochondrial function [100]. Despite the difficulties, apoE remains as an area of enormous potential for drug development for the treatment of AD and other neurodegenerative disorders.
Conclusions
The presence of apoE4 is the most important known risk factor for AD. ApoE is the only molecule that has been associated with all the biochemical disturbances characteristic of the disease: Aβ deposition, tangle formation, neurodegeneration, lipid dysfunction, loss of synaptic plasticity, cholinergic dysfunction and disruption of signalling (Fig. 1). The presence of apoE4 exacerbates these disturbances, while other apoE isoforms are protective. Several exciting new apoE-based therapeutic strategies for AD are under research (Fig. 2). Drugs are normally designed against a central pathway for a particular disease. Thus, apoE is not a typical target, since it has so many functions and isoform-specific effects. However, this may be an advantage for a multi-etiological disorder such as AD. The omnipresence of apoE in AD pathology may be the key for understanding the mechanisms leading to this complex disorder and for finding new multi-targeted therapeutic approaches.
1.
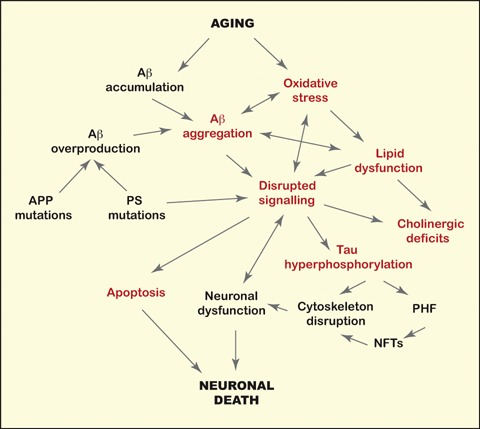
The Alzheimer's disease (AD) puzzle. The molecular mechanisms leading to AD have been shown to include dysfunctions in several biological processes, including oxidative stress, inflammatory responses and altered signal transduction. Aging is the major risk factor AD. Few mutations in the amy-loid precursor protein (APP) or the presenilin genes (PS) cause AD by increasing the production of Aβ. The apoE genotype is the most important genetic factor for AD. The presence of apoE4 has been associated with sever-al aspects (indicated in red) of the AD pathological cascade.
2.
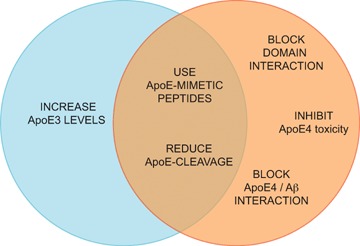
ApoE-based therapeutic approaches for AD. Desired therapeutic strategies would potentiate the ben-eficial effects of apoE3 (blue circle), eliminate the harmful effects of apoE4 (orange circle) or both.
Acknowledgments
I thank Riksbankens jubileumsfond, the Swedish Brain Power project, Loo och Hans Ostermans Stiftelse, Gun och Bertil Stohnes Stiftelse, Karolinska Institutets fund for geriatric research, Åke Wiberg Foundation, Demensförbundet and Stiftelsen för Gamla Tjänarinnor
References
- 1.Blennow K, De Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 2.Ashford JW. APOE genotype effects on Alzheimer's disease onset and epidemiology. J Mol Neurosci. 2004;23:157–65. doi: 10.1385/JMN:23:3:157. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–9. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 4.Josephs KA, Tsuboi Y, Cookson N, Watt H, Dickson DW. Apolipoprotein E epsilon 4 is a determinant for Alzheimer-type pathologic features in tauopathies, synucleinopathies, and frontotemporal degeneration. Arch Neurol. 2004;61:1579–84. doi: 10.1001/archneur.61.10.1579. [DOI] [PubMed] [Google Scholar]
- 5.Masterman T, Hillert J. The telltale scan: APOE epsilon4 in multiple sclerosis. Lancet Neurol. 2004;3:331. doi: 10.1016/S1474-4422(04)00763-X. [DOI] [PubMed] [Google Scholar]
- 6.Martinez M, Brice A, Vaughan JR, Zimprich A, Breteler MM, Meco G, Filla A, Farrer MJ, Betard C, Singleton A, Hardy J, De Michele G, Bonifati V, Oostra BA, Gasser T, Wood NW, Durr A. Apolipoprotein E4 is probably responsible for the chromosome 19 linkage peak for Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. 2005;136:72–4. doi: 10.1002/ajmg.b.30196. [DOI] [PubMed] [Google Scholar]
- 7.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917:148–61. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 8.Uchihara T, Duyckaerts C, He Y, Kobayashi K, Seilhean D, Amouyel P, Hauw JJ. ApoE immunoreactivity and microglial cells in Alzheimer's disease brain. Neurosci Lett. 1995;195:5–8. doi: 10.1016/0304-3940(95)11763-m. [DOI] [PubMed] [Google Scholar]
- 9.Aoki K, Uchihara T, Sanjo N, Nakamura A, Ikeda K, Tsuchiya K, Wakayama Y. Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke. 2003;34:875–80. doi: 10.1161/01.STR.0000064320.73388.C6. [DOI] [PubMed] [Google Scholar]
- 10.Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock- Christian TR, Hulette C, Schmechel DE. Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol. 1999;154:601–11. doi: 10.1016/S0002-9440(10)65305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beffert U, Danik M, Krzywkowski P, Ramassamy C, Berrada F, Poirier J. The neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer's disease. Brain Res Brain Res Rev. 1998;27:119–42. doi: 10.1016/s0165-0173(98)00008-3. [DOI] [PubMed] [Google Scholar]
- 12.Mahley RW, Nathan BP, Pitas RE. Apolipoprotein E. Structure, function, and possible roles in Alzheimer's disease. Ann N Y Acad Sci. 1996;777:139–45. doi: 10.1111/j.1749-6632.1996.tb34412.x. [DOI] [PubMed] [Google Scholar]
- 13.Finch CE, Sapolsky RM. The evolution of Alzheimer disease, the reproductive schedule, and apoE isoforms. Neurobiol Aging. 1999;20:407–28. doi: 10.1016/s0197-4580(99)00053-6. [DOI] [PubMed] [Google Scholar]
- 14.Finch CE, Stanford CB. Meat-adaptive genes and the evolution of slower aging in humans. Q Rev Biol. 2004;79:3–50. doi: 10.1086/381662. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava RA, Srivastava N, Averna M, Lin RC, Korach KS, Lubahn DB, Schonfeld G. Estrogen upregulates apolipoprotein E (ApoE) gene expression by increasing ApoE mRNA in the translating pool via the estrogen receptor alpha-mediated pathway. J Biol Chem. 1997;272:33360–6. doi: 10.1074/jbc.272.52.33360. [DOI] [PubMed] [Google Scholar]
- 16.Stone DJ, Rozovsky I, Morgan TE, Anderson CP, Hajian H, Finch CE. Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp Neurol. 1997;143:313–8. doi: 10.1006/exnr.1996.6360. [DOI] [PubMed] [Google Scholar]
- 17.Cedazo-Minguez A, Hamker U, Meske V, Veh RW, Hellweg R, Jacobi C, Albert F, Cowburn RF, Ohm TG. Regulation of apolipoprotein E secretion in rat primary hippocampal astrocyte cultures. Neuroscience. 2001;105:651–61. doi: 10.1016/s0306-4522(01)00224-x. [DOI] [PubMed] [Google Scholar]
- 18.Baskin F, Smith GM, Fosmire JA, Rosenberg RN. Altered apolipoprotein E secretion in cytokine treated human astrocyte cultures. J Neurol Sci. 1997;148:15–8. doi: 10.1016/s0022-510x(96)05335-x. [DOI] [PubMed] [Google Scholar]
- 19.Ignatius MJ, Gebicke-Harter PJ, Skene JH, Schilling JW, Weisgraber KH, Mahley RW, Shooter EM. Expression of apolipoprotein E during nerve degeneration and regeneration. Proc Natl Acad Sci USA. 1986;83:1125–9. doi: 10.1073/pnas.83.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boschert U, Merlo-Pich E, Higgins G, Roses AD, Catsicas S. Apolipoprotein E expression by neurons surviving excitotoxic stress. Neurobiol Dis. 1999;6:508–14. doi: 10.1006/nbdi.1999.0251. [DOI] [PubMed] [Google Scholar]
- 21.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid betapeptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dolev I, Michaelson DM. A nontransgenic mouse model shows inducible amyloid-beta (Abeta) peptide deposition and elucidates the role of apolipoprotein E in the amyloid cascade. Proc Natl Acad Sci USA. 2004;101:13909–14. doi: 10.1073/pnas.0404458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartman RE, Laurer H, Longhi L, Bales KR, Paul SM, McIntosh TK, Holtzman DM. Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer's disease. J Neurosci. 2002;22:10083–7. doi: 10.1523/JNEUROSCI.22-23-10083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–5. [PMC free article] [PubMed] [Google Scholar]
- 25.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–6. [PubMed] [Google Scholar]
- 26.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 1999;96:15233–8. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999;103:R15–21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuo YM, Crawford F, Mullan M, Kokjohn TA, Emmerling MR, Weller RO, Roher AE. Elevated A beta and apolipoprotein E in A betaPP transgenic mice and its relationship to amyloid accumulation in Alzheimer's disease. Mol Med. 2000;6:430–9. [PMC free article] [PubMed] [Google Scholar]
- 29.Bogdanovic N, Corder E, Lannfelt L, Winblad B. APOE polymorphism and clinical duration determine regional neuropathology in Swedish APP(670, 671) mutation carriers: implications for late-onset Alzheimer's disease. J Cell Mol Med. 2002;6:199–214. doi: 10.1111/j.1582-4934.2002.tb00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nilsson LN, Arendash GW, Leighty RE, Costa DA, Low MA, Garcia MF, Cracciolo JR, Rojiani A, Wu X, Bales KR, Paul SM, Potter H. Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol Aging. 2004;25:1153–67. doi: 10.1016/j.neurobiolaging.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 31.Cam JA, Bu G. Modulation of beta-amyloid precursor protein trafficking and processing by the low density lipoprotein receptor family. Mol Neurodegener. 2006;1:8. doi: 10.1186/1750-1326-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13:1029–31. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci USA. 2005;102:18700–5. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cedazo-Minguez A, Huttinger M, Cowburn RF. Beta-VLDL protects against A beta(1-42) and apoE toxicity in human SH-SY5Y neuroblastoma cells. Neuroreport. 2001;12:201–6. doi: 10.1097/00001756-200102120-00006. [DOI] [PubMed] [Google Scholar]
- 35.Manelli AM, Bulfinch LC, Sullivan PM, LaDu MJ. Abeta42 neurotoxicity in primary co-cultures: effect of apoE isoform and Abeta conformation. Neurobiol Aging. 2007;28:1139–47. doi: 10.1016/j.neurobiolaging.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levi O, Dolev I, Belinson H, Michaelson DM. Intraneuronal amyloid-beta plays a role in mediating the synergistic pathological effects of apoE4 and environmental stimulation. J Neurochem. 2007;103:1031–40. doi: 10.1111/j.1471-4159.2007.04810.x. [DOI] [PubMed] [Google Scholar]
- 37.Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739:179–97. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 38.Strittmatter WJ, Weisgraber KH, Goedert M, Saunders AM, Huang D, Corder EH, Dong LM, Jakes R, Alberts MJ, Gilbert JR, et al. Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp Neurol. 1994;125:163–71. doi: 10.1006/exnr.1994.1019. [DOI] [PubMed] [Google Scholar]
- 39.Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol. 2000;156:951–64. doi: 10.1016/S0002-9440(10)64963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Dee Fish J, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–34. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rahman A, Akterin S, Flores-Morales A, Crisby M, Kivipelto M, Schultzberg M, Cedazo-Minguez A. High cholesterol diet induces tau hyperphosphorylation in apolipoprotein E deficient mice. FEBS Lett. 2005;579:6411–6. doi: 10.1016/j.febslet.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 42.Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci USA. 2003;100:10966–71. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapp A, Gmeiner B, Huttinger M. Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie. 2006;88:473–83. doi: 10.1016/j.biochi.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 44.Michikawa M, Fan QW, Isobe I, Yanagisawa K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem. 2000;74:1008–16. doi: 10.1046/j.1471-4159.2000.0741008.x. [DOI] [PubMed] [Google Scholar]
- 45.Reid PC, Urano Y, Kodama T, Hamakubo T. Alzheimer's Disease: cholesterol, membrane rafts, isoprenoids and statins. J Cell Mol Med. 2007;11:383–92. doi: 10.1111/j.1582-4934.2007.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer's disease: a case-control study. Neurology. 1995;45:1092–6. doi: 10.1212/wnl.45.6.1092. [DOI] [PubMed] [Google Scholar]
- 47.Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, Tuomilehto J, Nissinen A. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology. 1998;17:14–20. doi: 10.1159/000026149. [DOI] [PubMed] [Google Scholar]
- 48.Roher AE, Kuo YM, Kokjohn KM, Emmerling MR, Gracon S. Amyloid and lipids in the pathology of Alzheimer disease. Amyloid. 1999;6:136–45. doi: 10.3109/13506129909007315. [DOI] [PubMed] [Google Scholar]
- 49.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–31. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 50.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–43. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 51.Simons M, Schwarzler F, Lutjohann D, Von Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T, Schulz JB. Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol. 2002;52:346–50. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- 52.Zerbinatti CV, Bu G. LRP and Alzheimer's disease. Rev Neurosci. 2005;16:123–35. doi: 10.1515/revneuro.2005.16.2.123. [DOI] [PubMed] [Google Scholar]
- 53.Wolozin B. Cyp46 (24S-cholesterol hydroxylase): a genetic risk factor for Alzheimer disease. Arch Neurol. 2003;60:16–8. doi: 10.1001/archneur.60.1.16. [DOI] [PubMed] [Google Scholar]
- 54.Chen S, Bawa D, Besshoh S, Gurd JW, Brown IR. Association of heat shock proteins and neuronal membrane components with lipid rafts from the rat brain. J Neurosci Res. 2005;81:522–9. doi: 10.1002/jnr.20575. [DOI] [PubMed] [Google Scholar]
- 55.Riddell DR, Christie G, Hussain I, Dingwall C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol. 2001;11:1288–93. doi: 10.1016/s0960-9822(01)00394-3. [DOI] [PubMed] [Google Scholar]
- 56.Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- 57.Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM. Proc Natl Acad Sci USA. 2001;98:5815–20. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280:36815–23. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- 59.Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004;167:953–60. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer betaamyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–23. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:2070–5. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawahara M, Kuroda Y. Intracellular calcium changes in neuronal cells induced by Alzheimer's beta-amyloid protein are blocked by estradiol and cholesterol. Cell Mol Neurobiol. 2001;21:1–13. doi: 10.1023/A:1007168910582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yip CM, Elton EA, Darabie AA, Morrison MR, McLaurin J. Cholesterol, a modulator of membraneassociated Abeta-fibrillogenesis and neurotoxicity. J Mol Biol. 2001;311:723–34. doi: 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- 64.Verkhratsky A, Toescu EC. Neuronal-glial networks as substrate for CNS integration. J Cell Mol Med. 2006;10:826–36. doi: 10.1111/j.1582-4934.2006.tb00527.x. [DOI] [PubMed] [Google Scholar]
- 65.Forero DA, Casadesus G, Perry G, Arboleda H. Synaptic dysfunction and oxidative stress in Alzheimer's disease: emerging mechanisms. J Cell Mol Med. 2006;10:796–805. doi: 10.1111/j.1582-4934.2006.tb00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Valastro B, Ghribi O, Poirier J, Krzywkowski P, Massicotte G. AMPA receptor regulation and LTP in the hippocampus of young and aged apolipoprotein E-deficient mice. Neurobiol Aging. 2001;22:9–15. doi: 10.1016/s0197-4580(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 67.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/-mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–80. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Veinbergs I, Masliah E. Synaptic alterations in apolipoprotein E knockout mice. Neuroscience. 1999;91:401–3. doi: 10.1016/s0306-4522(98)00602-2. [DOI] [PubMed] [Google Scholar]
- 69.Arendt T. Disturbance of neuronal plasticity is a critical pathogenetic event in Alzheimer's disease. Int J Dev Neurosci. 2001;19:231–45. doi: 10.1016/s0736-5748(01)00007-7. [DOI] [PubMed] [Google Scholar]
- 70.Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Ye GL, Sotak M, Sullivan PM, Pasternak JF, LaDu MJ. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport. 2004;15:2655–8. doi: 10.1097/00001756-200412030-00020. [DOI] [PubMed] [Google Scholar]
- 71.Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, Hui S, Bertrand P, Nalbantoglu J, Gilfix BM, Gauthier S. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:12260–4. doi: 10.1073/pnas.92.26.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reid RT, Sabbagh MN, Thal LJ. Does apolipoprotein E (Apo-E) genotype influence nicotinic receptor binding in Alzheimer's disease. J Neural Transm. 2001;108:1043–50. doi: 10.1007/s007020170023. [DOI] [PubMed] [Google Scholar]
- 73.Soininen H, Kosunen O, Helisalmi S, Mannermaa A, Paljarvi L, Talasniemi S, Ryynanen M, Riekkinen P., Sr A severe loss of choline acetyltransferase in the frontal cortex of Alzheimer patients carrying apolipoprotein epsilon 4 allele. Neurosci Lett. 1995;187:79–82. doi: 10.1016/0304-3940(95)11343-6. [DOI] [PubMed] [Google Scholar]
- 74.Cedazo-Minguez A, Cowburn RF. Apolipoprotein E isoform-specific disruption of phosphoinositide hydrolysis: protection by estrogen and glutathione. FEBS Lett. 2001;504:45–9. doi: 10.1016/s0014-5793(01)02761-2. [DOI] [PubMed] [Google Scholar]
- 75.Buttini M, Yu GQ, Shockley K, Huang Y, Jones B, Masliah E, Mallory M, Yeo T, Longo FM, Mucke L. Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid beta peptides but not on plaque formation. J Neurosci. 2002;22:10539–48. doi: 10.1523/JNEUROSCI.22-24-10539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cedazo-Minguez A, Popescu BO, Blanco-Millan JM, Akterin S, Pei JJ, Winblad B, Cowburn RF. Apolipoprotein E and beta-amyloid (1-42) regulation of glycogen synthase kinase-3beta. J Neurochem. 2003;87:1152–64. doi: 10.1046/j.1471-4159.2003.02088.x. [DOI] [PubMed] [Google Scholar]
- 77.Hartmann H, Eckert A, Muller WE. Apolipoprotein E and cholesterol affect neuronal calcium signalling: the possible relationship to beta-amyloid neurotoxicity. Biochem Biophys Res Commun. 1994;200:1185–92. doi: 10.1006/bbrc.1994.1576. [DOI] [PubMed] [Google Scholar]
- 78.Muller W, Meske V, Berlin K, Scharnagl H, Marz W, Ohm TG. Apolipoprotein E isoforms increase intracellular Ca2+ differentially through a omega-agatoxin IVa-sensitive Ca2+-channel. Brain Pathol. 1998;8:641–53. doi: 10.1111/j.1750-3639.1998.tb00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cedazo-Minguez A, Wiehager B, Winblad B, Huttinger M, Cowburn RF. Effects of apolipoprotein E (apoE) isoforms, beta-amyloid (Abeta) and apoE/Abeta complexes on protein kinase C-alpha (PKC-alpha) translocation and amyloid precursor protein (APP) processing in human SH-SY5Y neuroblastoma cells and fibroblasts. Neurochem Int. 2001;38:615–25. doi: 10.1016/s0197-0186(00)00128-5. [DOI] [PubMed] [Google Scholar]
- 80.Hoe HS, Harris DC, Rebeck GW. Multiple pathways of apolipoprotein E signaling in primary neurons. J Neurochem. 2005;93:145–55. doi: 10.1111/j.1471-4159.2004.03007.x. [DOI] [PubMed] [Google Scholar]
- 81.Ohkubo N, Mitsuda N, Tamatani M, Yamaguchi A, Lee YD, Ogihara T, Vitek MP, Tohyama M. Apolipoprotein E4 stimulates cAMP response element-binding protein transcriptional activity through the extracellular signal-regulated kinase pathway. J Biol Chem. 2001;276:3046–53. doi: 10.1074/jbc.M005070200. [DOI] [PubMed] [Google Scholar]
- 82.Xu PT, Li YJ, Qin XJ, Kroner C, Green-Odlum A, Xu H, Wang TY, Schmechel DE, Hulette CM, Ervin J, Hauser M, Haines J, Pericak-Vance MA, Gilbert JR. A SAGE study of apolipoprotein E3/3, E3/4 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Mol Cell Neurosci. 2007;36:313–31. doi: 10.1016/j.mcn.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sporis D, Sertic J, Henigsberg N, Mahovic D, Bogdanovic N, Babic T. Association of refractory complex partial seizures with a polymorphism of ApoE genotype. J Cell Mol Med. 2005;9:698–703. doi: 10.1111/j.1582-4934.2005.tb00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang Y. Molecular and cellular mechanisms of apolipoprotein E4 neurotoxicity and potential therapeutic strategies. Curr Opin Drug Discov Devel. 2006;9:627–41. [PubMed] [Google Scholar]
- 85.Tolar M, Keller JN, Chan S, Mattson MP, Marques MA, Crutcher KA. Truncated apolipoprotein E (ApoE) causes increased intracellular calcium and may mediate ApoE neurotoxicity. J Neurosci. 1999;19:7100–10. doi: 10.1523/JNEUROSCI.19-16-07100.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DeMattos RB, Curtiss LK, Williams DL. A minimally lipidated form of cell-derived apolipoprotein E exhibits isoform-specific stimulation of neurite outgrowth in the absence of exogenous lipids or lipoproteins. J Biol Chem. 1998;273:4206–12. doi: 10.1074/jbc.273.7.4206. [DOI] [PubMed] [Google Scholar]
- 87.Hayashi H, Campenot RB, Vance DE, Vance JE. Apolipoprotein E-containing lipoproteins protect neurons from apoptosis viaa signaling pathway involving low-density lipoprotein receptor-related protein-1. J Neurosci. 2007;27:1933–41. doi: 10.1523/JNEUROSCI.5471-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Buttini M, Akeefe H, Lin C, Mahley RW, Pitas RE, Wyss-Coray T, Mucke L. Dominant negative effects of apolipoprotein E4 revealed in transgenic models of neurodegenerative disease. Neuroscience. 2000;97:207–10. doi: 10.1016/s0306-4522(00)00069-5. [DOI] [PubMed] [Google Scholar]
- 89.Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci USA. 2001;98:8838–43. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol. 2006;361:932–44. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 91.Hatters DM, Howlett GJ. The structural basis for amyloid formation by plasma apolipoproteins: a review. Eur Biophys J. 2002;31:2–8. doi: 10.1007/s002490100172. [DOI] [PubMed] [Google Scholar]
- 92.Poirier J, Apolipoprotein E. a pharmacogenetic target for the treatment of Alzheimer's disease. Mol Diagn. 1999;4:335–41. doi: 10.1016/s1084-8592(99)80010-1. [DOI] [PubMed] [Google Scholar]
- 93.Laskowitz DT, Vitek MP. Apolipoprotein E and neurological disease: therapeutic potential and pharmacogenomic interactions. Pharmacogenomics. 2007;8:959–69. doi: 10.2217/14622416.8.8.959. [DOI] [PubMed] [Google Scholar]
- 94.Aono M, Bennett ER, Kim KS, Lynch JR, Myers J, Pearlstein RD, Warner DS, Laskowitz DT. Protective effect of apolipoprotein E-mimetic peptides on N-methyl-D-aspartate excitotoxicity in primary rat neuronal-glial cell cultures. Neuroscience. 2003;116:437–45. doi: 10.1016/s0306-4522(02)00709-1. [DOI] [PubMed] [Google Scholar]
- 95.Laskowitz DT, Fillit H, Yeung N, Toku K, Vitek MP. Apolipoprotein E-derived peptides reduce CNS inflammation: implications for therapy of neurological disease. Acta Neurol Scand Suppl. 2006;185:15–20. doi: 10.1111/j.1600-0404.2006.00680.x. [DOI] [PubMed] [Google Scholar]
- 96.Li FQ, Sempowski GD, McKenna SE, Laskowitz DT, Colton CA, Vitek MP. Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. J Pharmacol Exp Ther. 2006;318:956–65. doi: 10.1124/jpet.106.103671. [DOI] [PubMed] [Google Scholar]
- 97.Laskowitz DT, McKenna SE, Song P, Wang H, Durham L, Yeung N, Christensen D, Vitek MP. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24:1093–1107. doi: 10.1089/neu.2006.0192. [DOI] [PubMed] [Google Scholar]
- 98.Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci USA. 2006;103:18787–92. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci USA. 2006;103:5644–51. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roses AD, Saunders AM, Huang Y, Strum J, Weisgraber KH, Mahley RW. Complex disease-associated pharmacogenetics: drug efficacy, drug safety, and confirmation of a pathogenetic hypothesis (Alzheimer's disease) Pharmacogenomics J. 2007;7:10–28. doi: 10.1038/sj.tpj.6500397. [DOI] [PubMed] [Google Scholar]