

Abstract
Oxidative stress has been implicated as a mechanism underlying hyperglycaemia-associated cellular damage and could play a role in the development of diabetes-related complications. This study aimed to evaluate the significance of changes in oxidant–antioxidant status in 176 child and adolescent diabetic patients at clinical onset, during disease progression and when early microvascular complications appeared. Indicative lipid and protein oxidation markers and antioxidant defence activity were measured in plasma and correlated with clinical data, diabetes duration, long-term glycometabolic control and serum lipids. Compared with their respective age-matched controls, diabetic patients had greater oxidative damage to lipids and proteins, demonstrated through the analysis of hydroperoxides, lipoperoxides and oxidation protein products, all of which were significantly raised at onset, decreased during the first 1.5 years of evolution and rose progressively thereafter. Plasma levels of oxidizable lipids were significantly associated with lipid and protein oxidation products. Overall, plasma antioxidant capacity was significantly and consistently lower from clinical onset onwards. These results suggest that insulin therapy in the first year improved metabolic and oxidant homeostasis and consequently hyperglycaemia-derived biomolecular oxidative damage. Diabetes-associated hyperlipidaemia is related to lipid and protein oxidation processes, which supports the concept of glucose toxicity and lipotoxicity being interrelated. The greatest increase in lipid and protein oxidative damage biomarkers in young diabetic patients with premature microangiopathy points to oxidative stress as a possible contributing mechanism of microvascular dysfunction. Consequently, tight lipid and glycometabolic control may have therapeutic potential by diminishing oxidative tissue-damaging effects of hyperglycaemia.
Keywords: antioxidant potential, hyperlipidaemia, young diabetic patients, lipid peroxidation, oxidative stress, protein oxidation
Introduction
In diabetes, increased flux of glucose and free fatty acids is associated with mitochondrial reactive oxygen species (ROS) overproduction and, as a consequence, increased oxidative stress, which results from an imbalance in redox state in which pro-oxidants overwhelm antioxidant capacity, may therefore contribute significantly to disease mechanisms [1]. Several biochemical interacting pathways strictly associated with hyperglycaemia (glucose auto-oxidation, non-enzymatic protein glycation, mitochondrial ROS overproduction and activation of protein kinase C, nitric oxide synthase, xanthine oxidase, aldose reductase and the polyol pathway) boost ROS generation which provokes oxidative stress in diabetes [2–7]. Type 1 diabetes mellitus (T1DM) is one of the most common chronic disorders in children and adolescents in whom an early onset of microvascular complications is a cause for concern. Although hyperglycaemia constitutes the main cause of complications in diabetes and good glycaemic control reduces their rate of development and progression, as established in the conclusions of the Diabetes Control and Complications Trial Research Group [8], other genetic or metabolic factors also appear to play a role in the pathogenesis of vascular diabetes complications. Oxidative stress is believed to be associated with hyperglycaemia-mediated vascular dysfunction and, therefore, implicated in the early development of complications [4, 9, 10]. In T1DM, insulin deficiency not only provokes high blood glucose levels but also lipid metabolism alterations, resulting in perturbations in metabolic homeostasis, with ensuing rises in substrate concentrations in plasma or other tissues. This excess of carbohydrate and lipids in the bloodstream could be a problem per se, since an overload of peroxidizable substrates in the circulation may be an underlying pathogenic mechanism related to deposition and oxidation of lipoproteins in the vascular wall and contribute to the development of diabetesrelated complications. Indeed, patients with diabetes have a two- to five-fold increased risk of coronary atherosclerosis that is associated with diabetesinduced abnormalities in plasma lipids and lipoprotein metabolism [11, 12].
Whether oxidative stress plays any role in the premature development of angiopathy in young diabetic patients remains unclear. In an attempt to elucidate this question and delve further into the pathophysiological processes, we aimed to assess whether increased blood levels of oxidative stress biomarkers at onset in diabetic children fluctuate in the course of the disease and when complications appear, whether dyslipidaemia implies greater susceptibility to oxidation, and if so to what extent, and whether it is related to weakened antioxidant activity or to the glycometabolic control of the disease. With these aims in mind, indicative markers of lipoperoxidation, protein oxidation and changes in antioxidant defence activity were studied in plasma of young diabetic patients at different time points and correlated with diabetes-associated metabolic abnormalities, long-term metabolic control parameters, diabetes duration and the premature appearance of complications. We focused on analysing molecular oxidative damage, which has been increasingly postulated as a major contributor to the pathogenesis of the vascular injury occurring in diabetes; nonetheless, evidence supporting this hypothesis would be further strengthened by the contribution of new findings stemming from studies on oxidative process evolution in children.
Patients and methods
Patients
This study was conducted in 176 children, adolescents and young adults with T1DM and 140 healthy control individuals matched for sex, age and body mass index age [CR]. For this study in which diabetic patients were studied at onset and in different periods of disease progression, they were subgrouped according to diabetes duration into six subgroups: diabetes onset [DO], disease duration less than 1 and a half years [<1.5D], disease duration from 1.5 to 5 years [1–5D], disease duration from 5 to 10 years [5–10D], disease duration more than 10 years [>10D], and patients in the latter group recently diagnosed (<6 months) with clinical symptoms of microvascular complications [+DC]; all these 34 patients had both persistent microalbuminuria (defined as an albumin excretion rate >20 mg/min in two of three overnight urine collections) and background retinopathy. Characteristics of the subgroup of 34 diabetic patients with clinical symptoms of microvascular complications were as follows: 30 had clinical symptoms of retinopathy and nephropathy and four had retinopathy, nephropathy and neuropathy. At 5 years of diabetes clinical onset onwards, albuminuria analyses are determined every 6 months, ophthalmologic examination to detect retinopathy once a year and neurological examination also once a year. Population characteristics of the study groups are shown in Table 1. The number of participants followed during the different time points of disease evolution was: seven diabetic children studied in four different time points; twelve three times; sixty-seven two times and 90 once. The 75 DO patients were evaluated between 7 and 10 days after clinical onset of diabetes when hydroelectrolytic disorders and acidosis had returned to normal with therapy; this group of patients presented the highest levels of glycosylated haemoglobin (HbA1c) and fructosamine (Table 1). The remaining five diabetic groups presented similar plasma values of HbA1c and fructosamine. At the time of sample extraction, all diabetic patients had plasma bicarbonate levels within the normal range of 20–23 mEq/l; serum acidbase electrolytes were also normal, and no traces of ketones were found in urine in any of the 5 days prior to the time blood samples were taken. Diabetic blood samples were drawn during periodic routine control analyses. Diabetic patients were not pre-selected at any time and the HbA1c values obtained at the time of blood extraction were correlated with biomarkers of oxidative stress. Blood samples of healthy controls were obtained from blood analyses prior to minor surgery (hernia, fimosis, wisdom tooth extractions and minor plastic surgery). Informed consent was obtained from all the individuals after the purpose and nature of the study had been explained. All patients were diagnosed and followed up at the Paediatric Diabetes Unit of the Vall d’Hebron Children's Hospital and were invited to participate in the study, approved by the Ethics Committee for Clinical Research of the Vall d’Hebron Hospital.
1.
Clinical and biochemical characteristics of study subjects
| [spanname =“3to8”]Diabetic patients | |||||||
|---|---|---|---|---|---|---|---|
| Controls | At onset [DO] | [<1.5D] | [1.5–5D] | [5–10D] | [10–20D] | [+DC] | |
| n | 140 | 75 | 30 | 48 | 61 | 40 | 34 |
| Age (years, mean ± SD) | 13.7±9.3 | 9.1±3.7 | 9.7±4.3 | 12.2±3.8 | 16.1±4.3 | 20.1±4.5 | 25.9±4.3 |
| (Range) | (1–30) | (1–17) | (3–18) | (5–22) | (7–24) | (13-27) | (20-36) |
| Diabetes duration (years) | – | – | 0.88±0.06 | 3.28±0.16 | 7.44±0.175 | 14.7±0.5 | 18.96±1.1 |
| (Range) | (13-32) | ||||||
| HbA1c (%) | (4.6–6.6)* | 11.6±0.3 | 8.2±0.2 | 9.03±0.22 | 8.7±0.24 | 8.39±0.29 | 8.4±0.3 |
| Fructosamine (μM) | (180–285)* | 488.6±18.7 | 360.4±18.7 | 371.4±15.3 | 399.4±15.3 | 388.5±12.3 | 384.4±15.1 |
| Cholesterol (mM) | 4.04±0.05 | 4.8±0.11c | 4.2±0.11 | 4.6±0.11c | 4.6±0.11c | 4.8±0.14c | 4.85±0.17c |
| Triglycerides (mM) | 0.6±0.02 | 0.81±0.05b | 0.6±0.04 | 0.57±0.03 | 0.69±0.05 | 0.97±0.09c | 1.17±0.14c,d |
| Uric acid μM) | 239.5±7.1 | 154±6.2c | 190±10.7b | 184.6±9.5c | 229.6±10.3 | 204.07±14a | 221.4±22.9 |
| -Tocopherol/TL (μM/mM) | 5.18±0.09 | 4.75±0.14a | 4.63±0.13a | 4.69±0.12a | 4.9±0.1 | 5.15±0.3 | 4.4±0.27c,d |
Data are means ± SEM. *Data in parentheses are normal ranges. a P<0.05, b P<0.005 and c P<0.0001 versus control subjects. d P<0.05 versus [10–20D]. Subgroups of diabetic patients: diabetes onset [DO], disease duration <1.5 years [<1.5D], disease duration from 1–5 years [1.5–5D], disease duration from 5–10 years [5–10D], disease duration from 10–20 years [10–20D], patients with microvascular complications [+DC]. TL = Total lipids.
Chemicals
All chemicals were analytical grade. Butylated hydroxytoluene (BHT), 2-thiobarbituric acid (TBA), chloramine-T, potassium iodide (KI), hydrogen peroxide (H202), 1,1,3,3-tetraethoxypropane (TEP), phosphotungstic acid, α-Tocopherol, β-phycoerythrin from Porphirium cruentum(β-PE), and 5,5’-dithio-bis- (2-nitro-benzoic acid) (DTNB) were purchased from Sigma Chemical Company (St. Louis, MO, USA). High Performance Liquid Chromatography (HPLC) grade acetonitrile from Romil (Cambridge, UK) and n-Butanol, methanol, ethanol and n-Hexane from Merck (Darmstadt, Germany). Highly-purified water (resistivity = 18 MΩ.cm) obtained through a Milli-Q water purification system (Millipore, Bedford, MA, USA) was used for solution preparation and for the mobile phase. Separation of α-tocopherol was carried out using a Nova-Pak C18 (5 μm) stainlesssteel column (3.9 x 150 mm i.d.) (Waters). The guard column was packed with the same material. 6-Hydroxy- 2,5,7,8-tetramethylchroman 2-carboxylic acid (TROLOX) was obtained from Aldrich Chemical Co. (Milwaukee, WI, USA). 2,2’-azobis-(2-amidinopropane) hydrochloride (AAPH) was obtained from Polysciences (Warrington, PA, USA). Triphenylphosphine 99% (TPP) was purchased from Aldrich (Gillingham, Dorset, UK). Ammonium ferrous sulphate in aqueous sulphuric acid and xylenol orange in methanol were obtained as the commercial preparation (PeroXOquant quantitative peroxide assay, lipid-compatible formulation) from Pierce (Rockford, IL, USA). The commercially available synthetic hydroperoxides 5-HpETE and 13- HPODE were purchased from ICN Biomedicals Inc. (Aurora, OH, USA). All other general chemicals used were of the highest purity available.
Blood sample collection, processing and analytical methods
Venous blood samples were drawn in fasting state into tubes containing EDTA or heparin and processed immediately, as previously reported [13–16]. Aliquots of plasma and washed erythrocytes were stored at –85°C until prompt analysis. Analyses of plasma total radical antioxidant potential (TRAP), oxidizability, total thiols (SH groups) and plasma lipid hydroperoxides (HPx) were analysed within 1 week. All the above analytical procedures followed those established by our laboratory and previously described in detail [13–16]. Plasma lipid peroxide levels were measured according to an optimized method with modifications previously introduced by us for acid pH control of the reaction and extractions of lipids, proteins, glucose and water-soluble aldehydes in plasma [15] and concentrations expressed as malondialdehyde (MDA) using 1,1,3,3- tetraethoxypropane as standard (μmol/l). Total lipid hydroperoxide plasma levels were quantified by PeroXOquantTM Quantitative Peroxide Assay FOX (ferrous oxidation/xylenol orange technique), according to the manufacturer's protocols. Plasma HPx concentration in the samples was determined as a function of the mean absorbance difference of samples with and without HPx elimination by TPP at 560 nm using an H2O2 standard curve in the concentration range of 0–20 μmol/l. Sample assays were made in triplicate and the acceptable conduction velocity (CV) had to be under 5%.
Advanced oxidation protein products (AOPP) were measured by the method described by Witko-Sarsat et al.[17] with slight modifications. AOPP were determined spectrophotometrically using a plate reader (Model Anthos HTII) and calibrated with chloramine-T solutions, which in the presence of KI absorb at 340 nm. AOPP concentrations were expressed as μmol/l chloramine-T equivalents. The within-run and between-run CVs were 2.7% and 4.1%, respectively.
Plasma α-tocopherol was determined by reversedphase HPLC with ultraviolet detection at 280 nm (Waters Model 486 tunable absorbance detector) and peak automatic integration as detailed previously [13–16]. Injection volume was 50 μl, and flow-rate 2.0 ml/min at room temperature. Plasma α-tocopherol was extracted with hexane. The mobile phase used was methanol. Tocopherol acetate used as internal standard and serum calibration standard for vitamin E analysis were obtained from Chromsystems (Munich, Germany). The within-run and between-run CVs were 3.2% and 5.7%, respectively; detection limit was 1.3 μM. Given the clear metabolic relationship between plasma α-tocopherol and plasma lipids, we also express α-tocopherol levels as a ratio of α-tocopherol/total lipids (cholesterol + triglycerides). Plasma lipid profile (total cholesterol, triglycerides, low-density lipoprotein [LDL], HDL) and uric acid were measured by quantitative enzymatic assays as previously reported [18].
Whole plasma oxidation was assayed spectrophotometrically according to the method of Kontush and Beisiegel [19]. Plasma oxidizability was measured as an increase in absorbance at 234 nm known to reflect the level of conjugated dienes in samples. Briefly, heparin plasma (20 μl) was diluted 150-fold with phosphate-buffered saline (PBS) and its oxidation induced by the radical initiator AAPH (330 μM) at 37°C over a period of 20 hrs. Total plasma thiol levels were measured spectrophotometrically, using DTNB as described by Hu [20].
Total radical-trapping antioxidant parameter (TRAP) analyses. Plasma antioxidant activity was measured using a modification of Glazer's fluorometric assay [21]. The water-soluble vitamin E analogue Trolox was used as a standard and AAPH as generator of peroxyl radicals. Fluorescence was measured at the emission of 565 nm and excitation of 540 nm. The results were calculated in the following way:% TRAP = 1– (Δabs. sample or Trolox/_ abs. blank) x100. TRAP levels were expressed as arbitrary units equivalent to Trolox antioxidant activity and calculated from a calibration curve determined daily between the TRAP percentage and the concentration of the three standards. The linear regression coefficient of the calibration curve was never below 0.999. Intra- and inter-day coefficients of variation were 2.9% and 6.6%, respectively.
Statistical analysis
Statistical analyses were carried out using the STATVIEW 4.5 statistical program (ABACUS Concepts, Berkeley, CA, USA). Data (expressed as means ± SEMs) were analysed by one-way ANOVA test. Each group of diabetic patients was compared with their corresponding age-matched controls. Linear regression analysis was used to study associations between variables. P values ≤ 0.05 were considered statistically significant.
Results
Metabolic control parameters
Description of the clinical characteristics, biochemical and metabolic control parameters of this wide age range group of diabetic patients between disease onset and up to 20 years of disease evolution and of the control group are summarized in Table 1. Glycated haemoglobin (HbA1c) and fructosamine levels were raised in all diabetic patient subgroups, with the highest values being at [DO]; insulin therapy produced a clear improvement in metabolic control as reflected by marked reductions in HbA1c and fructosamine.
Plasma cholesterol and triglyceride levels were significantly increased at [DO]; however, while triglycerides remained in normal range during the first 10 years, rising significantly thereafter, a slight decrease in cholesterol was observed in the first 1.5 years followed by a gradual, significant increase over the study period. Plasma uric acid levels were statistically lower in diabetic patients than in controls during the first 5 years, with uricaemia yielding the lowest values at [DO].
Oxidative stress indices
Plasma HPx, the major initial reaction products of lipid peroxidation, were significantly raised at [DO] and decreased during the first 1.5 years of evolution to normal values, only to rise progressively thereafter (Fig. 1A). Similarly, concentrations of secondary lipid peroxidation end products were significantly higher in plasma of diabetic patients, except for subgroup [<1.5D], than in age-matched controls (Fig. 1B). From 1.5 years, a significant trend towards greater and progressive increases in MDA formation was clearly observed. Single linear regression analyses revealed significant correlations between plasma levels of oxidizable lipids and lipid peroxide levels (P<0.0001).
1.
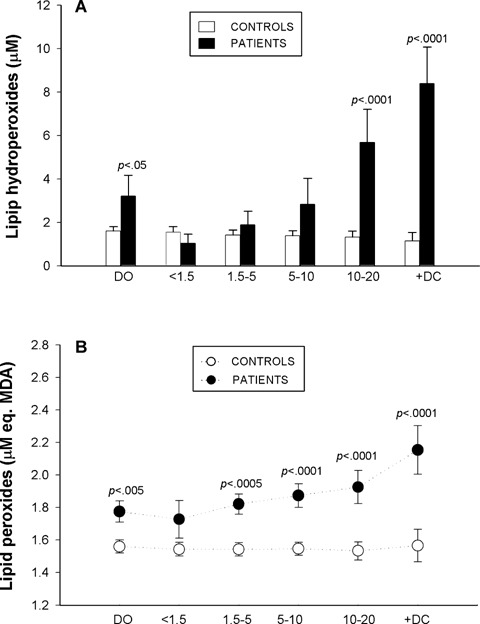
Plasma levels of Lipid hydroperoxides (A) and Lipid peroxides (B) in young diabetic patients at disease onset [DO], during the first 20 years of disease progression [four subgroups: <1.5, 1.5–5, 5–10, 10–20], in diabetic patients with complications [+DC] and in their respective controls. (A) Filled columns:diabetic patients data; open columns:control data. (A) Filled circles:diabetic patients data; open circles: control data. Values are expressed as mean ± SEM and were analysed for statistically-significant differences by analysis of variance (ANOVA); Pvalues <0.05 (versus respective control participants) were considered significant and are shown above each subgroup in the graph.
Whole plasma oxidation-induced assay was used to measure changes in the oxidizability of plasma lipids during evolution of the disease (Fig. 2A). Plasma oxidizability was significantly higher in diabetic patients than in their respective age-matched controls, except in the (<1.5 D) group, with the most significant difference being found in samples from diabetic children at [DO] (P<0.0001). The increase in conjugated diene formation was proportional to disease duration. This measurement correlated significantly and positively with the sum of concentrations of the major oxidizable lipids, cholesterol and triglycerides, (Fig. 2B) and with HbA1c (Fig. 2C).
2.
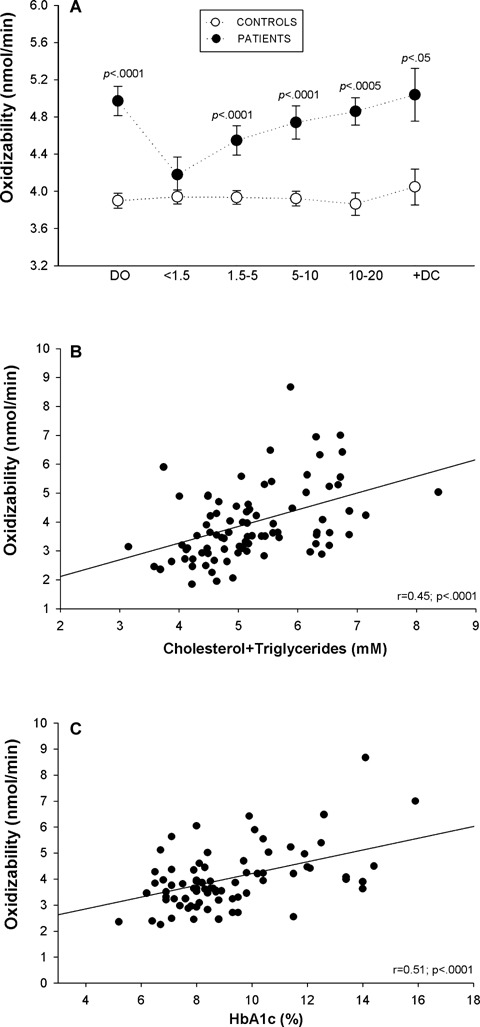
Plasma oxidizability (A) in young diabetic patients at disease onset [DO], during the first 20 years of disease evolution [four subgroups: <1.5, 1.5–5, 5–10, 10–20], in diabetic patients with complications [+DC] and in their respective controls; filled circles: diabetic patients data; open circles: control data. Correlations between plasma oxidizability values and total lipid concentrations in plasma (B) and HbA1c (C). Values represented are mean ± SEM and were analysed for statistically- significant differences by analysis of variance (ANOVA); P values <0.05 (versus respective control subjects) were considered significant and are shown above each subgroup in the graph.
Plasma AOPP, markers of protein oxidation, were significantly higher (P<0.0005) in samples from [DO], [5–10D] and [10–20D] groups and higher still in the group of diabetic patients with complications compared with their respective age-matched controls (Fig. 3A). Linear regression analysis of plasma AOPP showed positive correlations with both triglycerides (r = 0.42; P<0.0005) and total lipids (Fig. 3B).
3.
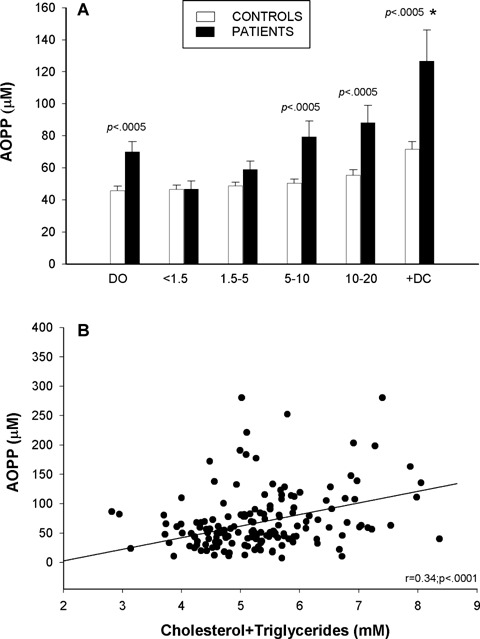
Plasma advanced oxidation protein products (AOPP) (A) in diabetic patients at disease onset [DO], during the first 20 years of disease evolution [four subgroups: <1.5, 1.5–5, 5–10, 10–20], in diabetic patients with complications [+DC] and in their respective controls; filled columns: diabetic patients data; open columns: control data. Correlation between plasma AOPP and total lipid concentrations in plasma (B). Values are expressed as mean ± SEM and were analysed for statistically-significant differences by analysis of variance (ANOVA); P values <0.05 (versusrespective control participants) were considered significant and are shown above each subgroup in the graph. * P<0.05 diabetic patients with complications [+DC] versus[10–20] subgroup of patients.
Antioxidant capacity
Mean plasma TRAP values were significantly lower in diabetic patients throughout disease evolution (Fig. 4A) with the greatest statistical differences being at [DO], after which there appeared to be a progressive 10-year partial recovery period with a sharp decline thereafter. Plasma TRAP values were significantly lower in samples of diabetic patients with complications compared with the values of the [10–20D] subgroup of patients with the same diabetes evolution time. Plasma thiol, a parameter of non-oxidation of protein-SH groups, was significantly lower in all diabetic patients, with the exception of subgroup [<1.5D], than in age-matched controls; the decrease was constant in all groups, although the more marked loss of SH groups occurred in patients at [DO] (Fig. 4B). A significant inverse association was observed between thiols and HbA1c (Fig. 4C). Concentrations of the liposoluble antioxidant (-tocopherol were similar in diabetic patients and controls. However, (-tocopherol levels corrected by total lipids were significantly decreased during the first 5 years of disease evolution and in +DC group (Table 1). Values of oxidative stress parameters expressed by diabetic patient subgroups and those of their respective controls are shown in Table 2. Biochemical and molecular oxidative damage biomarkers and antioxidant activities in plasma were not affected by age or pubertal stage in either controls or diabetic patients (Tables 3A, 3B and 4). Table 4 specifically presents the data of diabetic patients according to age and sex and shows there were no differences between males and females at the ages studied.
4.
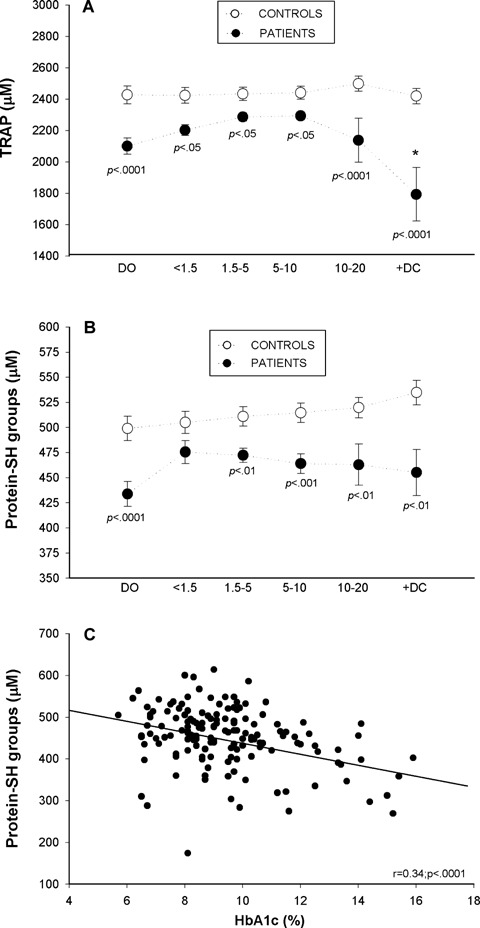
Plasma Total Radicaltrapping Antioxidant Parameter (TRAP) values (A) and protein sulphydryl groups (B) in diabetic patients at disease onset [DO], during the first 20 years of disease evolution [four subgroups: <1.5, 1.5–5, 5–10, 10–20], in diabetic patients with complications [+DC] and in their respective controls; Filled circles: diabetic patients data; open circles: control data. Correlation between protein sulphydryl groups and HbA1c (C). Data are means ± SEM and were analysed for statistically-significant differences by analysis of variance (ANOVA); P values <0.05 (versus respective control participants) were considered significant and are shown above each subgroup in the graph. * P<0.05 diabetic patients with complications versus[10–20] subgroup of patients.
2.
Plasma oxidative stress values of diabetic patients and their respective controls
| Controls | At onset [DO] | Controls | [<1.5D] | Controls | [1.5–5D] | Controls | [5–10D] | Controls | [10–20D] | Controls | [+DC] | |
| Lipid hydroperoxides (μM) | 1.6±0.2 | 3.2±0.9* | 1.55±0.2 | 1.03±0.4 | 1.4±0.2 | 1.88±0.6 | 1.39±0.2 | 2.8±1.2 | 1.32±0.3 | 5.7±1.5• | 1.15±0.4 | 8.4±1.6• |
| Lipid peroxides (μM eq.MDA) | 1.56±0.04 | 1.7±0.06† | 1.54±0.04 | 1.73±0.1 | 1.54±0.04 | 1.8±0.06† | 1.54±0.04 | 1.87±0.1• | 1.53±0.05 | 1.9±0.1• | 1.56±0.1 | 2.15±0.14• |
| Oxidizability (nmol/min) | 3.9±0.08 | 4.97±0.1• | 3.9±0.07 | 4.17±0.2 | 3.9±0.07 | 4.5±0.16• | 3.92±0.08 | 4.7±0.18• | 3.8±0.1 | 4.86±0.1§ | 4.05±0.2 | 5.04±0.2* |
| TRAP (μM) | 2426±57 | 2100±51• | 2423±51 | 2202±33* | 2433±42 | 2286±26* | 2440±42 | 2293±25* | 2498±48 | 2137±98• | 2418±140 | 1792±170• |
| Protein-SH groups (μM) | 499±12 | 433±0.1• | 505±11 | 475±11 | 511±9 | 472±7** | 515±9.7 | 464±9.8‡ | 520±10 | 463±20** | 534±12 | 455±23** |
| AOPP (μM) | 45.8±2.7 | 70.1±6.4§ | 46.7±2.6 | 46.7±4.9 | 48.6±2.4 | 59±5 | 50.4±2.5 | 79.5±9.7§ | 55.4±3.3 | 88.2±11§ | 71.6±4.8 | 126.7±19§ |
Data are means ± SEM and were analysed for statistically-significant differences by analysis of variance (ANOVA): *p<0.05, ** p<.01, †P<0.005, ‡ P<0.001, §P<0.0005, •P<0.0001 versus respective control participants.Subgroups of diabetic patients:diabetes onset [DO], disease duration <1.5 years [<1.5D], disease duration from 1–5 years [1.5–5D], disease duration from 5–10 years [5–10D], disease duration from 10–20 years [10–20D], patients with microvascular complications [+DC].AOPP = Advanced Oxidation Protein Products.TRAP = Total Radical-trapping Antioxidant Parameter.
3A.
Plasma levels of cholesterol, triglycerides, malondialdehyde (MDA), lipid hydroperoxides (HPx) and advanced oxidation protein products (AOPP) in control subjects
| n | Cholesterol (mM) | Triglycerides (mM) | MDA (μM) | HPx (μM) | AOPP (μM) | |
| Age (years) | ||||||
| 1–12 | 73 | 3.9 ± 0.08 | 0.59 ± 0.02 | 1.55 ± 0.05 | 1.6 ± 0.36 | 51.74 ± 17 |
| 13–30 | 67 | 4.1 ± 0.08 | 0.63 ± 0.03 | 1.51 ± 0.06 | 1.3 ± 0.32 | 53.98 ± 16.3 |
| Sex | ||||||
| Male | 77 | 4.03 ± 0.07 | 0.61 ± 0.02 | 1.46 ± 0.05 | 1.39 ± 0.3 | 54.3 ± 15 |
| Female | 63 | 4.06 ± 0.08 | 0.6 ± 0.03 | 1.61 ± 0.05 | 1.51 ± 0.37 | 51.3 ± 16.4 |
Data are means ± SEM
3B.
Plasma oxidizability and plasma levels of total sulphydryl (SH) groups, uric acid and Plasma Total Radical-trapping Antioxidant Parameter (TRAP) values in control subjects
| n | Oxidizability (nmol/min) | -SH groups (μM) | -tocopherol/ total lipids | Uric acid (μM) | TRAP (mM) | |
| Age (years) | ||||||
| 1–12 | 73 | 3.98 ± 0.11 | 490.2 ± 18.3 | 5.3 ± 0.09 | 233.5 ± 9.6 | 2.4 ± 0.8 |
| 13–30 | 67 | 3.91 ± 0.13 | 519.3 ± 11.1 | 5.03 ± 0.18 | 243.3 ± 10.7 | 2.5 ± 0.6 |
| Sex | ||||||
| Male | 77 | 3.95 ± 0.12 | 507.2 ± 14 | 5.1 ± 0.13 | 262.1 ± 9.9 | 2.4 ± 0.5 |
| Female | 63 | 3.93 ± 0.12 | 510.7 ± 13.7 | 5.3 ± 0.11 | 211.8 ± 8.9 | 2.5 ± 0.7 |
Data are means ± SEM
4.
Biochemical characteristics of diabetic patients according to sex
| At onset [DO] | [<1.5D] | [1.5–5D] | [5–10D] | [10–20D] | [+DC] | |||||||
| Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | Males | Females | |
| n | 39 | 36 | 17 | 13 | 23 | 25 | 34 | 27 | 15 | 25 | 9 | 25 |
| HbA1c (%) | 11.5±0.4 | 11.6±0.4 | 7.9±0.3 | 8.7±0.2 | 9.1±0.3 | 8.8±0.3 | 8.5±0.3 | 8.9±0.3 | 8±0.3 | 8.8±0.4 | 8.4±0.4 | 8.4±0.3 |
| Fructosamine (μM) | 467±37 | 502±19 | 391±33 | 333±16 | 352±38 | 384±16 | 401±18 | 395±32 | 391±20 | 387±15 | 397±37 | 380±17 |
| Cholesterol (mM) | 4.9±0.1 | 4.7±0.1 | 4.2±0.16 | 4.2±0.17 | 4.7±0.2 | 4.6±0.16 | 4.5±0.13 | 4.7±0.2 | 4.4±0.3 | 5±0.2 | 4.96±0.2 | 4.8±0.2 |
| Triglycerides (mM) | 0.8±0.06 | 0.8±0.07 | 0.6±0.03 | 0.6±0.08 | 0.5±0.05 | 0.6±0.04 | 0.6±0.04 | 0.8±0.1 | 0.9±0.2 | 1±0.09 | 1.2±0.2 | 1.15±0.2 |
| Uric acid (μM) | 155±7.9 | 152±9.9 | 188±15 | 192±16 | 191±11 | 178±15 | 244±14 | 213±14 | 222±14 | 191±22 | 240±62 | 220±17 |
| α-Tocopherol/TL (μM/mM) | 4.7±0.19 | 4.8±0.2 | 4.7±0.18 | 4.4±0.2 | 4.7±0.2 | 4.6±0.2 | 4.8±0.13 | 4.9±0.15 | 5.4±1.1 | 4.9±0.2 | 4±0.6 | 4.5±0.2 |
| Lipid hydroper oxides (μM) | 2.8±0.6 | 3.7±1.9 | 0.9±0.4 | 1.2±0.6 | 1.2±0.4 | 2.6±1.1 | 2.5±0.1 | 3.6±0.12 | 4.3±1.5 | 6.9±2.4 | 9.5±3.9 | 8.06±1.8 |
| Lipid peroxides (μM eq.MDA) | 1.7±0.07 | 1.8±0.11 | 1.6±0.06 | 1.8±0.12 | 1.7±0.1 | 1.9±0.09 | 1.8±0.09 | 1.9±0.12 | 1.96±0.2 | 1.92±0.1 | 2.1±0.09 | 2.16±0.2 |
| Oxidizability (nmol/min) | 4.9±0.1 | 5.04±0.2 | 4.2±0.3 | 4.1±0.2 | 4.6±0.3 | 4.4±0.2 | 4.6±0.2 | 4.9±0.2 | 4.8±0.1 | 5±0.2 | 4.7±0.4 | 5.1±0.3 |
| TRAP (μM) | 2023±68 | 2203±71 | 2196±42 | 2210±60 | 2280±32 | 2291±43 | 2280±33 | 2312±40 | 2255±32 | 2012±11 | 1844±31 | 1778±21 |
| Protein-SH groups (μM) | 446±16 | 417±19 | 481±13 | 471±13 | 481±11 | 460±10 | 470±15 | 457±13 | 499±11 | 427±35 | 520±17 | 435±26 |
| AOPP (μM) | 63.8±7.3 | 78.2±11 | 48.4±6 | 47±4.8 | 53.3±8.7 | 69±8.2 | 75±8 | 95±21.5 | 75±12 | 100±17 | 136±37 | 123±24 |
Data are means ±SEM.All differences P>0.05.Subgroups of diabetic patients:diabetes onset [DO], disease duration <1.5 years [<1.5D], disease duration from 1–5 years [1.5–5D], disease duration from 5–10 years [5–10D], disease duration from 10–20 years [10–20D], patients with microvascular complications [+DC].AOPP = Advanced Oxidation Protein Products.TRAP = Total Radical-trapping Antioxidant Parameter.
Discussion
Oxidative stress and associated oxidative damage are currently acknowledged as components of molecular and cellular tissue damage mechanisms underlying vascular dysfunction in diabetes [22]. Several biochemical pathways, strictly associated with hyperglycaemia, increase ROS production that causes oxidative stress in diabetes [2–7] and, furthermore, diabetes-associated dyslipidaemia could contribute to cell oxidative damage. The results of this comprehensive study provide evidence that hyperglycaemia-derived oxidative stress was already present at DO and almost disappeared during the first year and a half of insulin treatment. Thus, the metabolic efficacy of insulin is not limited to its direct action on glucose homeostasis maintenance; indeed, insulin exerts other beneficial effects, probably by reducing levels of free fatty acids and triglycerides, substrates that are susceptible to lipoperoxidation. As a consequence of these beneficial effects of insulin, an improvement in levels of cytotoxic products of molecular oxidative damage was observed in this ‘honeymoon’ period. Changes in plasma lipid levels were in line with those observed in the formation of lipoperoxidation products (HPx, MDA) with similar graphic patterns and, again, the nadir was in the <1.5-year ‘honeymoon’ period. Linear regression analyses revealed significant correlations between plasma levels of oxidizable lipids and total peroxide levels in diabetic patients alone, which demonstrates greater oxidizability of plasma lipids in diabetes.
ROS can react with all biological macromolecules, among the more susceptible of which are PUFA, and their peroxidation, mostly in membrane phospholipids, poses a constant threat to cell integrity and function. Our results showed that both the major initial and end products of lipoperoxidation were significantly high in diabetic patients at onset, significantly improved during the first 1.5 years of insulin treatment and raised progressively thereafter with the highest values being found in patients with early diabetic angiopathy. Hence, oxidative damage to lipids appears to be present at onset and throughout the course of the disease and to be associated with the presence of oxidizable lipid substrates in diabetic patients. Dyslipidaemia in diabetes is considered to be a major modifiable coronary risk factor [11, 12, 23] and an association between lipoperoxidation parameters and diabetic angiopathy has been reported in young adults with early nephropathy [24]. Moreover, a direct effect of triacylglycerols has been described as a further underlying mechanism of oxidative stress-mediated lipotoxicity [25].
Lipid peroxidation, a chain reaction that can continue until substrate is completely consumed or termination occurs due to antioxidants, is accompanied by a selective uptake or net consumption of α-tocopherol which appears to have served to limit, though clearly not terminate, lipoperoxidation in young T1DM patients. Greater susceptibility of plasma to in vitro oxidation found in these patients correlated positively with the sum of concentrations of the major oxidizable lipids and with glycaemic control, which concurs with reports that LDL from patients with wellcontrolled T1DM does not differ in composition or in susceptibility to in vitrooxidation compared with LDL from non-diabetic participants [26, 27]. Thus, increased lipid susceptibility to oxidation appears to be secondary to poorly controlled diabetes and its associated hyperlipidaemia, and may account for the accelerated vascular disease seen in diabetes. Should an excess of lipids, the substrate under greatest attack by free radicals, be a major source of increased oxidative stress in diabetes, this pathogenic pathway could be a therapeutic target for attenuating molecular oxidative damage, thereby preventing the acceleration of atherosclerosis from the early stages of diabetes. Therefore, it is worthwhile considering the use of statins, the beneficial effects of which are mainly attributed to their cholesterol-lowering properties, but which also include other possible pleiotropic effects related to their anti-inflammatory and antioxidant activities [28, 29] and attenuate high glucose-induced and diabetes-induced oxidative stress in vitro and in vivo[30], thereby providing new insights into oxidative therapy in diabetes.
Oxidative damage to proteins was also clearly shown to be continuous from onset and throughout the course of the first 20 years of diabetes evolution covered by this study. Elevated oxidative protein damage has been observed in types 1 and 2 diabetes [13, 31, 32]. Plasma concentrations of carbonylated proteins (by the method based on binding of 2, 4-dinitrophenyl-hydrozine (DNPH) to protein carbonyls) have been the most frequently determined marker of oxidative protein damage and constitute a well-validated method [33]. In the present study, evidence of protein oxidative stress was demonstrated by assessment of AOPPs, considered to be an oxidized albumin index [17], which were markedly higher (two-fold) in patients with early diabetic angiopathy; interestingly, blood levels of these abnormal products correlated positively with those of total lipids and with lipid peroxidation by-products in the whole study cohort. In view of the results of our present and previous studies, we consider circulating AOPP levels to be a useful marker for estimating the degree of protein oxidative damage since they provide valid information on the early development of microvascular complications in diabetes and might be used to monitor the severity and progress of the disease; these data concur with those found in type 2 DM and recently reported [34]. This noteworthy finding would point to a role of oxidative injury to proteins in the early development of diabetic microvascular complications, thereby linking protein oxidation to disease severity, which is plausible since oxidative damage to proteins is one mechanism by which oxidants promote inflammatory damage and increase the tissue-destructive effects of proteolytic enzymes, all of which may lead to severe failure of cell biological functions [32]. ROS overload is known to primarily modify proteins and generate lipid peroxidation-derived reactive aldehydes that, in turn, can also produce secondary modifications of proteins [35]. Our findings confirm that both processes took place in newly-diagnosed young diabetic patients and those with longer standing disease and support the hypothesis of carbonyl stress by which diabetes induces a generalized increase in oxidative modification of proteins together with glycoxidation and lipoxidation processes which overgenerate products with cytotoxic effects that disrupt cell metabolism [10, 36].
The degree of oxidative stress and the ensuing endothelial dysfunction might depend on the imbalance between excessive ROS generation and individual antioxidant defence activity. This study showed plasma SH groups to be susceptible to oxidative damage, since levels in children with recent-onset T1DM were the lowest observed in the course of the disease. Insulin administration boosted recovery of plasma thiols and it is noteworthy that a significant inverse relationship was observed between thiol concentration and glycometabolic control which, in poorly controlled T1DM patients, may play a role in the pathogenesis of diabetes complications [37]. The present work also found overall plasma antioxidant capacity to be decreased throughout the study period, particularly in diabetic patients with complications, indicating a possible complicity between the circulating antioxidant activity and subsequent development of microvascular complications. Human plasma protection against free radical injury is offered by a wide spectrum of antioxidants with synergic action. The directly-measured TRAP value, used to assess chain-breaking antioxidants as a whole, was decreased in sera of diabetic children from clinical onset, improving slightly with insulin treatment up to >10 years and declining thereafter. Since the ability of serum to delay peroxidation is largely determined by levels of individual antioxidants and their relative contributions to antioxidant activity, low levels of antioxidants, such as sulfhydryl compounds and uric acid, together with an insufficient α-tocopherol content for the hyperlipidaemia of these diabetic children, appear to be responsible for the inadequate capacity of plasma to protect its environment from free radical aggression and might account for the lack of maintenance of the overall redox network in plasma of diabetic patients [38, 39]. Since uric acid has been proposed as a powerful antioxidant molecule, the hypouricaemia found in diabetic children at levels 23–32% lower than in controls in the first 5 years of childhood diabetes may contribute to loss of physiologic protection against free radical activity. In addition, glucose and free radicals have been shown to provoke structural changes in albumin, resulting in impairment of its antioxidant properties [40].
The results of this novel and comprehensive study, which closely followed oxidative stress progression in children and adolescents with T1DM, provide further solid evidence-linking oxidative processes to diabetes and their potential contribution to the prematue development of complications. The overall increase in lipid and protein oxidative damage biomarkers that precedes or accompanies the early stages of diabetes- related development of complications points to oxidative stress processes as potential pathogenic mechanisms, precursors of microvascular alterations in diabetes. Moreover, we showed that hyperlipidaemia caused by diabetes-associated loss of optimal regulation of lipid metabolism implies greater susceptibility to oxidation that would lead and significantly contribute to the extent of lipid and protein oxidation, thereby supporting the concept of glucose toxicity and lipotoxicity being interrelated. Therefore, concerted efforts should be made to reduce oxidative lipaemia and oxidant stress in an attempt to potentially delay the development and/or progression of early microvascular complications. Consequently, tight lipid and glycometabolic control may have therapeutic potential by diminishing oxidative tissuedamaging effects of hyperglycaemia.
Acknowledgments
This work was supported by grants from FIS (99/1103, 02/0852), Centre Network RCMN (C03/08) and Group Network RGDM (G03/212) financed by FIS of the Carlos III Institute of Health, Spain. We are grateful to Miss C. O’Hara for her help in the English version of this manuscript.
References
- 1.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3. Oxford: Clarendon Press; 1999. [DOI] [PubMed] [Google Scholar]
- 2.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 3.Wolff SP, Dean RT. Glucose autooxidation and protein modification. The potential role of autoxidative glycosylation in diabetes. Biochem J. 1987;245:243–50. doi: 10.1042/bj2450243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. New Engl J Med. 1988;318:1315–21. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 5.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–45. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 6.Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation. 1997;96:25–8. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- 7.Desco MC, Asensi M, Márquez R, Martínez-Valls J, Vento M, Pallardó F, Sastre J, Viña J. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51:1118–24. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 8.Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. New Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 9.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–67. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 10.Baynes JW, Thorpe S. Role of oxidative stress in diabetic complications. A new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Steinberg D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as a partners of crime. Nat Med. 2002;8:1211–17. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]
- 12.Syvanne M, Taskinen MR. Lipids and lipoproteins as coronary risk factors in non-insulin-dependent diabetes mellitus. Lancet. 1997;350:SI20–3. doi: 10.1016/s0140-6736(97)90024-6. [DOI] [PubMed] [Google Scholar]
- 13.Domínguez C, Ruiz E, Gussinyé M, Carrascosa A. Oxidative stress at onset and in early stages of type 1 diabetes mellitus in children and adolescents. Diabetes Care. 1998;21:1736–42. doi: 10.2337/diacare.21.10.1736. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Gallán P, Carrascosa A, Gussinyé M, Domínguez C. Estimation of lipoperoxidative damage and antioxidant status in diabetic children: relationship with individual antioxidants. Free Radic Res. 2005;39:933–42. doi: 10.1080/10715760500156751. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Gallán P, Gussinyé M, Carrascosa A, Domínguez C. Biomarkers of diabetes-associated oxidative stress and antioxidant status in young diabetic patients with or without subclinical complications. Free Radic Biol Med. 2003;34:1563–74. doi: 10.1016/s0891-5849(03)00185-0. [DOI] [PubMed] [Google Scholar]
- 16.Llurba E, Gratacós E, Martín-Gallán P, Cabero L, Domínguez C. A comprehensive study of oxidative stress and antioxidant status in pre-eclampsia and normal pregnancy. Free Radic Biol Med. 2004;37:557–70. doi: 10.1016/j.freeradbiomed.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 17.Witko-Sarsat V, Friedlander M, Capéillere-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, Jungers P, Descamps-Latscha B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996;49:1304–13. doi: 10.1038/ki.1996.186. [DOI] [PubMed] [Google Scholar]
- 18.Llurba E, Casals E, Domínguez C, Delgado J, Mercade I, Crispi F, Martin-Gallán P, Cabero L, Gratacós E. Atherogenic lipoprotein subfraction profile in preeclamptic women with and without high triglycerides: different pathophysiologic subsets in preeclampsia. Metabolism. 2005;54:1504–9. doi: 10.1016/j.metabol.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Kontush A, Beisiegel U. Measurement of oxidizability of blood plasma. Methods Enzymol. 1999;299:35–49. doi: 10.1016/s0076-6879(99)99007-9. [DOI] [PubMed] [Google Scholar]
- 20.Hu ML. Measurement of protein thiol groups and glutathione in plasma. Methods Enzymol. 1994;233:380–85. doi: 10.1016/s0076-6879(94)33044-1. [DOI] [PubMed] [Google Scholar]
- 21.Glazer AN. Phycoerythrin fluorescence-based assay for reactive oxygen species. Methods Enzymol. 1990;186:161–68. doi: 10.1016/0076-6879(90)86106-6. [DOI] [PubMed] [Google Scholar]
- 22.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 23.Weis U, Turner B, Gibney J, Watts GF, Burke V, Shaw KM, Cummings MH. Long-term predictors of coronary artery disease and mortality in type 1 diabetes. QJM. 2001;11:623–30. doi: 10.1093/qjmed/94.11.623. [DOI] [PubMed] [Google Scholar]
- 24.Chiarelli F, Cipollone F, Mohn A, Marini M, Iezzi A, Fazia M, Tumini S, De Cesare D, Pomilio M, Pierdomenico SD, Di Gioacchino M, Cuccurullo F, Mezzetti A. Circulating monocyte chemoattractant protein-1 and early development of nephropathy in type 1 diabetes. Diabetes Care. 2002;25:1829–34. doi: 10.2337/diacare.25.10.1829. [DOI] [PubMed] [Google Scholar]
- 25.Aronis A, Madar Z, Tirosh O. Mechanism underlying oxidative stress-mediated lipotoxicity: exposure of J774. 2 macrophages to triacylglycerols facilitates mitochondrial reactive oxygen species production and cellular necrosis. Free Radic Biol Med. 2005;38:1221–30. doi: 10.1016/j.freeradbiomed.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins AJ, Klein RL, Chassereau CN, Hermayer KL, Lopes-Virella MF. LDL from patients with wellcontrolled IDDM is not more susceptible to in vitro oxidation. Diabetes. 1996;45:762–7. doi: 10.2337/diab.45.6.762. [DOI] [PubMed] [Google Scholar]
- 27.Willems D, Dorchy H, Dufrasne D. Serum antioxidant status and oxidized LDL in well-controlled young type 1 diabetic patients with and without subclinical complications. Atherosclerosis. 1998;137:S61–4. doi: 10.1016/s0021-9150(97)00320-1. [DOI] [PubMed] [Google Scholar]
- 28.Munford RS. Statins and the acute-phase response. N Engl J Med. 2001;344:2016–8. doi: 10.1056/NEJM200106283442609. [DOI] [PubMed] [Google Scholar]
- 29.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy- 3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–9. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 30.Tsubouchi H, Inoguchi T, Sonta T, Sato N, Sekiguchi N, Kobayashi K, Sumimoto H, Utsumi H, Nawata H. Statin attenuates high glucoseinduced and diabetes-induced oxidative stress in vitro and in vivo evaluated by electron spin resonance measurement. Free Radic Biol Med. 2005;39:444–52. doi: 10.1016/j.freeradbiomed.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 31.Cakatay U. Protein oxidation parameters in type 2 diabetic patients with good and poor glycaemic control. Diabetes Metab. 2005;31:551–7. doi: 10.1016/s1262-3636(07)70230-6. [DOI] [PubMed] [Google Scholar]
- 32.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med. 2003;9:169–76. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 33.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 34.Piwowar A, Knapik-Kordecka M, Warwas M. AOPP and its relations with selected markers of oxidative/antioxidative system in type 2 diabetes mellitus. Diabetes Res Clin Pract. 2007;77:188–92. doi: 10.1016/j.diabres.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 35.Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aldini G, Dalle-Donne I, Colombo R, Maffei Facino R, Milzani A, Carini M. Lipoxidation-derived reactive carbonyl species as potential drug targets in preventing protein carbonylation and related cellular dysfunction. Chem Med Chem. 2006;1:1045–58. doi: 10.1002/cmdc.200600075. [DOI] [PubMed] [Google Scholar]
- 37.Darmaun D, Smith SD, Sweeten S, Sager BK, Welch S, Mauras N. Evidence for accelerated rates of glutathione utilization and glutathione depletion in adolescents with poorly controlled type 1 diabetes. Diabetes. 2005;54:190–6. doi: 10.2337/diabetes.54.1.190. [DOI] [PubMed] [Google Scholar]
- 38.Marra G, Cotroneo P, Pitocco D, Manto A, Di Leo MA, Ruotolo V, Caputo S, Giardina B, Ghirlanda G, Santini SA. Early increase of oxidative stress and reduced antioxidant defenses in patients with uncomplicated type 1 diabetes: a case for gender difference. Diabetes Care. 2002;25:370–5. doi: 10.2337/diacare.25.2.370. [DOI] [PubMed] [Google Scholar]
- 39.Niculescu L, Stancu C, Sima A, Toporan D, Simionescu M. The total peroxyl radical trapping potential in serum - an assay to define the stage of atherosclerosis. J Cell Mol Med. 2001;5:285–94. doi: 10.1111/j.1582-4934.2001.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourdon E, Loreau N, Blache D. Glucose and free radicals impair the antioxidant properties of serum albumin. FASEB J. 1999;13:233–44. doi: 10.1096/fasebj.13.2.233. [DOI] [PubMed] [Google Scholar]