

Abstract
von Hippel-Lindau disease (VHL) is an autosomal dominant, familial neoplastic disorder with variable interfamilial and intrafamilial expression. VHL is characterized by pre-disposition to development of a combination of benign and malignant tumours affecting multiple organs. We provide molecular evidence of somatic mosaicism in nearly asymptomatic man whose daughter had VHL. The mosaic subject was found to have a cyst of the kidney and an angioma of the glans penis and had had surgery for a mandibular cyst and epididymal cystadenomas. Mosaicism could provide a genetic explanation for the clinical heterogeneity and variable severity of VHL. The real incidence of mosaicism is still unclear and the identification of mosaicism has important consequences in genetic counseling of VHL patients who appear to have de novo VHL mutations and should be considered when evaluating patients with isolated VHL-related tumours. Our results strongly suggest a complete and extensive clinical examination in the parents of each patient affected by an apparently de novo VHL germline mutation.We recommend performing a mutation screening of both parents of a proband with techniques that permit detection of low percentages of mosaicism before concluding that the proband has a de novo VHL mutation.
Keywords: von Hippel-Lindau disease, mosaicism, VHL gene, mutation detection
Introduction
Von Hippel-Lindau disease (VHL; MIM# 193300) is a hereditary multi-systemic tumour syndrome that pre-disposes affected individuals to haemangioblastomas of the central nervous system and retina, pheochromocytomas, clear-cell renal carcinomas, adenomas and carcinomas of the pancreas, paragangliomas, renal and pancreatic cysts, papillary cystadenomas of the epididymis and, rarely, cystadenomas of the endolymphatic sac and broad ligament. VHL affects approximately 1 in 36,000 newborns and is transmitted in an autosomal dominant manner with a penetrance of more than 90% by the age of 65 years [1, 2].
VHL is a tumour suppressor gene located on chromosome 3p25–26 [3, 4]. The gene consists of three exons, is highly conserved across species, and is ubiquitously expressed in both foetal and adult tissues [5, 6]. Expression of the VHL gene is not restricted to the organs affected in VHL [3, 7–9]. The VHL protein pVHL) has been implicated in a variety of functions, including transcriptional regulation, posttranscriptional gene expression, extracellular matrix assembly, protein folding and ubiquitination as reviewed by Kaelin [10].
An increasing number of germline mutations have been reported in VHL-affected individuals [11–14], and genotype-to-phenotype correlations are now emerging [15]. Somatic mutations in VHL have also been detected in several types of sporadic and hereditary tumours [16–18].
Phenotypes vary among families, reflecting genotypic differences [1, 12, 14, 19]. Clinically, VHL is classified in type 1 or type 2 based on the absence or presence of pheochromocytomas. The occurrence of renal cell carcinoma (RCC) allows a further distinction between type 2 A (low risk of RCC) and type 2 B (high risk of RCC). Some type 2 families develop pheochromocytomas only, with no other neoplastic findings of VHL (VHL-type 2 C) [20]. VHL tumours, including pheochromocytomas and paragangliomas, may appear clinically to be sporadic but represent milder cases of VHL, with the attenuated phenotype resulting from either a mild impairment in function of the mutated pVHL or somatic mosaicism. The latter is a condition in which genetically different cells coexist in tissues of the same individual, and the intratumoural mixture of VHL-mutated and VHL-non-mutated cells clearly can modulate the resulting phenotype.
We have analysed the VHL gene in the available members of a VHL family in which the pro-band presented with bilateral pheochromocytomas and multiple paragangliomas. Her father showed what we believe is a very mild and relatively late-onset VHL phenotype. In this study, we describe the somatic mosaicism of the pro-band's father and we reviewed the literature for all the described cases of VHL-mosaicism.
Patients and methods
Clinical evaluation
The clinical features of the female pro-band have been reported in our previously published study [21], briefly, the young girl now 26 years old, underwent surgery at 11 years of age to resect a pheochromocytoma associated with hypertension. At age 18 years, she underwent further surgery to remove a pheochromocytoma in the contralateral adrenal gland and two concurrent paragangliomas of the abdominal aorta and urinary bladder. One year ago, she was also found to have a right-sided, extra-axial, 1.6-cm supratentorial frontal meningioma (Fig. 1). Post-operatively, neuroendocrine serum markers (plasma free metanephrines, chromogranin A, neuron- specific enolase, and gastrointestinal hormones carcinoembryonic antigen [CEA] and calcitonin) have remained negative. A family history obtained from her parents, one brother and one sister was uninformative, except for the father's history. The pro-band's relatives underwent physical examination.
1.
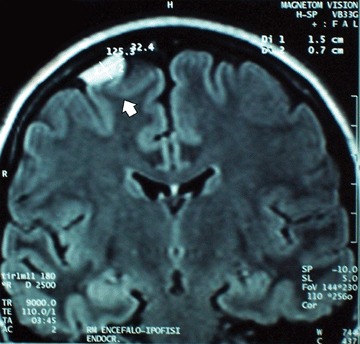
MR image of the pro-band's brain.The arrow indicates a 1.6-cm-diameter right-sided supratentorial frontal meningioma.
The patient's father, now 51 years old, was found to have an angioma of the glans penis and had had surgery for a mandibular cyst and epididymal cystadenomas at age 43 years. Hence, we suspected that the father had an attenuated form of VHL. Indeed, abdominal ultrasonography and total-body magnetic resonance imaging (MRI) revealed a 2.3-cm cyst in his right kidney. His blood pressure and levels of plasma free metanephrines, fractionated urinary metanephrines, chromogranin A were normal. The other three first-degree relatives were normal both clinically and biochemically.
Genetic investigations
Informed consent for genetic testing was obtained from each individual.
Genomic DNA was extracted from peripheral blood lymphocytes (PBLs) of the pro-band and her four first-degree relatives. To assess the possibility that the pro-band's father had an attenuated VHL phenotype caused by mosaicism, somatic DNA was extracted from his oral epithelial cells, hair roots and skin fibroblasts. The DNA was extracted using conventional methods.
The entire coding sequence of the VHL gene was PCR-amplified with 36-cycle reactions using conditions and primers, with minimum variations, described previously [22]. PCR products were analysed by denaturing high-performance liquid chromatography (DHPLC) using a Wave 2100 DNA fragment analysis system (Transgenomic Wave system, Omaha, NE) at column temperatures recommended by the WaveMaker version 4.1.31 software (Transgenomic) and at melt temperatures determined by the DHPLC Melt software (http://insertion.stanford.edu/melt.html). Nucleic acids were separated in the column according to size and degree of denaturation in a gradient of two buffers (A: 0.1 M triethylammonium acetate, [TEAA, ph 7]; B: 0.1 M TEAA, 25% acetonitrile).
DHPLC analysis was performed at a melt temperature of 60°C at a constant flow rate of 0.9 ml/min using a linear gradient of acetonitrile. The gradient started at 8.75%, increased over 2 min. to 13.75%, increased over 5 min. to 16.25%, was kept constant for 1 min., increased over 1 min. to 25%, was kept constant for 1 min. (wash), decreased over 1 min. to 8.75%, and was kept constant for 1 min. (equilibration). At appropriate temperatures, heteroduplex molecules can be detected as an additional peak, or shoulder, in the chromatogram.
Amplimers with abnormal denaturing profiles were purified (Microcon PCR, Millipore, Bedford, MA) and sequenced bidirectionally using an ABI BigDye Terminator cycle sequencing kit v.3.1 and an ABI Prism 310 genetic analyser (both from Applied Biosystems, Foster City, CA, USA). Sequencing results were analysed using the Sequencing Analysis v.3.6.1 and AutoAssembler v.2.1 software packages (both from Applied Biosystems). For semiquantitative analysis, the PCR products were cloned into a vector (TOPO TA cloning, Invitrogen Carlsbad, CA, USA) according to the manufacturer's instructions, directly amplified from bacterial colonies, and sequenced.
Results
The VHL gene sequence was altered in both the proband and her father, though to different extents in each. DHPLC analysis of VHL exon 3 PBLs DNA showed quantitative differences in the DNA elution profiles between daughter and father (Fig. 2A). This finding suggested a heterozygous mutation in both relatives and mosaicism in the father. To test for mosaicism, molecular analysis was performed on tissues of various embryonic origins. DNA exon 3 amplicons extracted from the father's buccal mucosa, hair roots, and skin fibroblasts showed different levels of intensity of the same altered DHPLC peaks, although the intensity of the peaks in these tissues was comparable to that seen in the PBL DNA (Fig. 2B).
2.
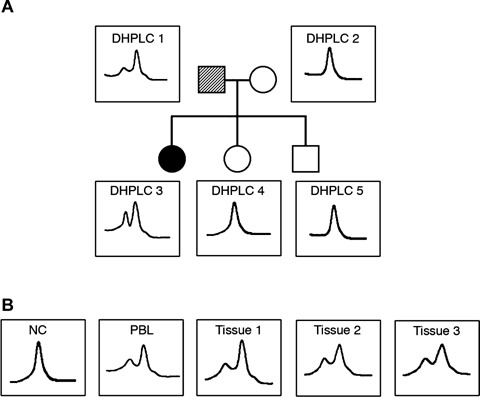
Denaturing high-performance liquid chromatography (DHPLC) analysis of exon 3 of the von Hippel-Lindau disease (VHL) gene in all the family members. (A) DHPLC analysis of the PCR products of exon 3 of the VHL gene in the pro-band (DHPLC 3), her father (DHPLC 1), her mother (DHPLC 2) and her brother and sister (DHPLC 4, 5). In the first-degree pedigree of this family, the pro-band is indicated by number 03, while her father, mother, sister and brother are indicated by the numbers 01, 02, 04 and 05, respectively. The dashed symbol for the pro-band's father indicates genetic mosaicism. DNA was extracted from peripheral blood lymphocytes (PBLs) in all five individuals. DHPLC analysis of the pro-band's DNA shows an extra peak that is barely visible (but reproducibly so) in her father's DNA. (B) DHPLC analysis of DNA extracted from a normal control (NC) sample, the father's PBLs, and the father's oral mucosa (tissue 1), hair roots (tissue 2) and fibroblasts (tissue 3).
Sequence analysis demonstrated that the abnormal elution profile (abnormal peak) seen in the proband and at various levels in the different cells from the pro-band's father was the result of a missense mutation in exon 3. This mutation was a G-to-A substitution at cDNA nucleotide 695, predicting the replacement of wild-type arginine with glutamine codon 161 of pVHL (R161Q). The mutation-related peak in the PBLs DNA of the father was smaller than the same peak in the daughter's PBLs DNA (compare Fig. 3A with Fig. 3B), consistent with the fatherdaughter difference shown by DHPLC. Amplicon cloning revealed that only a few clones contained the G-to-A mutant allele. Of the 40 colonies studied, only six showed sequence mutations in semiquantitative analysis. Therefore, approximately 15% of father's PBLs harboured the mutation. In other words, in the DNA extracted from the father's circulating PBLs, the ratio of wild-type to mutated gene was approximately 85:15, instead of the 50:50 ratio expected in the absence of mosaicism.
3.
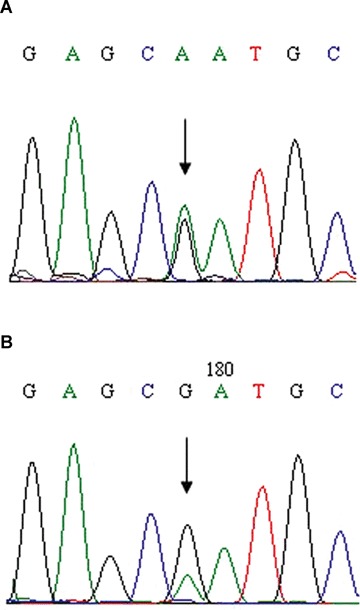
Sequencing analysis of exon 3 of the VHL gene in DNA from PBLs of the pro-band (A) and her father (B).
To summarize, direct sequencing analysis suggested that only a small fraction (about 15%) of PBLs contained the VHL mutation, lower than the typical 50% observed in a heterozygous individual. Furthermore, only in the DNA extracted from buccal cells, hair roots and cultured skin fibroblast cells was this mutation evident. No other mutations were found by DHPLC or sequence analysis.
Discussion
The mosaic individual we described here presented at age 51 years with an angioma of the glans penis and a renal cyst. At age 43 years, he required surgery for epididymal cystadenomas and a mandibular cyst. His daughter, in contrast, had a full-blown germline VHL gene mutation at a much younger age (11 years). In the mosaic subject, the late disease onset and mild VHL phenotype might have been mediated by the presence of two different cell populations, a prevailing population with a normal VHL gene and a smaller one with a mutated VHL gene. Very few abnormal cells are likely to be present in the father's chromaffin cells, thus explaining the absence of pheochromocytomas/paragangliomas, because decreased/lower likelihood to have a so-called ‘second hit’ event with subsequent movement of mutated cells more towards homozygosity, as it has been shown in multiple endocrine neoplasia type 2 associated tumours [23, 24]. The inaccessibility of the chromaffin cells in the father's adrenal medulla and paraganglia precluded the experimental quantification the ratio of normal to mutated VHL. As a consequence of the atypical phenotype of VHL in the father, the presence of familial VHL was not recognized initially.
In recent years, the role of pVHL in the regulation of hypoxia-inducible genes through the targeted ubiquitination and degradation of HIF1α has been elucidated, leading to a model of how disruption of the VHL gene can result in highly vascularized tumours. When pVHL is absent or mutated, HIF1α subunits accumulate, resulting in cell proliferation and neovascularization in VHL tumours [10].
Over 300 germline mutations of the VHL gene have been reported in VHL families [25]. Many of these mutations are missense and occur in any of the three exons. VHL is inherited in an autosomal dominant fashion, with about 80% of cases being familial and about 20% sporadic as a result of a de novo mutation [26]. The family history can sometimes be falsely negative because of failure to recognize the disorder in some family members, reduced penetrance, intrafamilial variability of clinical expression, death of the affected parent before the onset of symptoms or late onset of the disease in the affected parent.
Another reason why VHL may go unrecognized in either parent, so that the disease in the child is erroneously considered to be sporadic, is somatic mosaicism. Sgambati et al. [26] first described VHL mosaicism in 2 (5%) of 42 unrelated families, both of which lacked a history of VHL. The two patients (one man and one woman) had clinical evidence of VHL, but no VHL mutations were detected in the initial genetic test performed on DNA from their PBLs; in contrast, their clinically affected offspring tested positive for VHL mutations in their PBLs. Another case of parental mosaicism was described by Murgia et al. in kindred in whom the pro-band had full-blown VHL [27]. This pro-band presented at age 26 years with cerebellar haemangioblastoma, retinal haemangioma, multiple bilateral renal cysts and bilateral pheochromocytomas. In contrast, his asymptomatic (mosaic) mother, whose PBLs DNA showed a barely visible single-strand conformation polymorphism bandshift identical to that seen in her son, presented with only a small pheochromocytoma and renal microcysts by age 48 years.
In both of the above studies, DNA was also extracted from tissues, such as buccal cells and skin fibroblasts to confirm the somatic mosaicism. In similar families, the mosaic individual may be mildly or minimally affected, generally tends to have less severe disease than his or her offspring, and may be asymptomatic or present with less severe features of the disease. Disease severity varies among mosaic individuals depending on whether mutations occur early or late in embryogenesis [28]. Asymptomatic carriers have been described in a number of heritable tumour syndromes reviewed by Zlotogora [29] and in many other heritable diseases, such as Hutchinson-Gilford progeria [30]. Mosaicism has also been demonstrated to be the cause of many cases of clinical recurrence.
In a mosaic individual, the co-existence of mutated cells with even a small population of normal cells is an important parameter in predicting phenotype and overall prognosis and can increase the difficulty of obtaining a correct diagnosis of VHL. VHL in a mosaic individual may be difficult to recognize merely on clinical grounds, but it should always be considered when evaluating patients with isolated VHL-related tumours or parents of affected individuals. Under these conditions, such individuals should be analysed for low-level mutations by emerging DNA analysis techniques.
It is presumed that the genotype-phenotype correlation in VHL reflects the degree to which the functions of pVHL are quantitatively and qualitatively altered by different mutations. A number of mutation carriers have been described, but not in sufficient numbers to define mutation-based phenotypes. Furthermore, the percentages of symptomatic and asymptomatic VHL mutation carriers and the most important variable affecting disease penetrance and severity (age at diagnosis, sex distribution, genetic co-factors or environmental modifiers) have yet to be evaluated. Actually, studies are focusing on elucidating the genotype -phenotype correlations in VHL [15]. This would allow the development of unified surveillance guidelines for VHL patients or those at risk for this disease.
In summary, we have provided molecular evidence of somatic mosaicism in the father of a patient with full VHL. Because of the incomplete penetrance of the disease the diagnosis of VHL was first not recognized. Because of penetrance age related in VHL, a long-term follow up is warranted. Counselling of patients and closely related family members must take a central place in the management of hereditary multi-organ cancer syndromes, such as VHL. A careful and complete clinical examination in the parents of each patient affected by an apparently de novo VHL germline mutation is warranted.
The evaluation of the parents of a pro-band with an apparent de novo VHL gene mutation should include molecular genetic testing if the VHL disease-causing mutation in the pro-band is known. If the disease-causing VHL mutation in the pro-band is unknown, both parents should be offered a complete and extensive clinical and images examination, such as neurological test including MRI of the craniospinal axis; ophthalmologic evaluation; measurement of plasma free metanephrines, chromogranin A, and fractionated urinary metanephrines; and abdominal ultrasonography, MRI or computed tomography.
The real incidence of mosaicism in VHL remains uncertain, but such a phenomenon has important consequences for molecular testing, clinical diagnosis and genetic counselling, in terms of prediction of phenotype and risk of recurrence after the initial diagnosis. The real incidence of mosaicism will hopefully yield better data on the real incidence of de novo VHL mutations.
Acknowledgments
We thank Gilbert J Cote for critical reading of the manuscript.
References
- 1.Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, Harris R, Sampson J, Williams A, Ferguson-Smith MA, Morton N. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443–7. doi: 10.1136/jmg.28.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel- Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 3.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, Schmidt L, Zhou F, Li H, Wei MH, Chen F, Glenn G, Choyke P, Walther MM, Weng Y, Duan DR, Dean M, Glavǎ D, Richards FM, Crossey PA, Ferguson-Smith MA, Le Paslier D, Chumakov I, Cohen D, Chinault AC, Maher ER, Linehan WM, Zbar B, Lerman MI. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 4.Seizinger BR, Rouleau GA, Ozelius LJ, Lane AH, Farmer GE, Lamiell JM, Haines J, Yuen JW, Collins D, Majoor-Krakauer D, Bonner T, Mathew C, Rubenstein A, Halperin J, McConkie-Rosell A, Green JS, Trofatter JA, Ponder BA, Eierman L, Bowmer MI, Schimke R, Oostra B, Aronin N, Smith DI, Drabkin H, Waziri MH, Hobbs WJ, Martuza RL, Connealy PM, Hsia YE, Gusella JF. von Hippel Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332:268–9. doi: 10.1038/332268a0. [DOI] [PubMed] [Google Scholar]
- 5.Kaelin WG, Iliopoulos O, Lonergan KM, Ohh M. Functions of the von Hippel-Lindau tumour suppressor protein. J Intern Med. 1998;243:535–9. doi: 10.1046/j.1365-2796.1998.00335.x. [DOI] [PubMed] [Google Scholar]
- 6.Ohh M, Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: new perspectives. Mol Med Today. 1999;5:257–63. doi: 10.1016/s1357-4310(99)01481-1. [DOI] [PubMed] [Google Scholar]
- 7.Iliopoulos O, Kibel A, Gray S, Kaelin WG., Jr Tumour suppression by the human von Hippel- Lindau gene product. Nat Med. 1995;1:822–6. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- 8.Renbaum P, Duh FM, Latif F, Zbar B, Lerman MI, Kuzmin I. Isolation and characterization of the fulllength 3' untranslated region of the human von Hippel-Lindau tumor suppressor gene. Hum Genet. 1996;98:666–71. doi: 10.1007/s004390050281. [DOI] [PubMed] [Google Scholar]
- 9.Richards FM, Schofield PN, Fleming S, Maher ER. Expression of the von Hippel-Lindau disease tumour suppressor gene during human embryogenesis. Hum Mol Genet. 1996;5:639–44. doi: 10.1093/hmg/5.5.639. [DOI] [PubMed] [Google Scholar]
- 10.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 11.Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, Lerman MI, Zbar B, Affara NA, Ferguson-Smith MA, Maher ER. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. 1994;3:1303–8. doi: 10.1093/hmg/3.8.1303. [DOI] [PubMed] [Google Scholar]
- 12.Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, Brauch H, Glavac D, Neumann HP, Tisherman S, Mulvihill JJ, Gross DJ, Shuin T, Whaley J, Seizinger B, Kley N, Olschwang S, Boisson C, Richard S, Lips CH, Linhean WM, Lerman M. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8:348–57. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Glavac D, Neuman HP, Wittke C, Jaenig H, Masek O, Streicher T, Pausch F, Engelhardt D, Plate KH, Hofler H, Chen F, Zbar B, Brauch H. Mutations in the VHL tumor suppressor gene and associated lesions in families with von Hippel-Lindau disease from central Europe. Hum Genet. 1996;99:271–80. doi: 10.1007/s004390050206. [DOI] [PubMed] [Google Scholar]
- 14.Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, Pack S, Hurley K, Andrey C, Klausner R, Linehan WM. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417–23. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 15.Ong KR, Woodward ER, Killick P, Lim C, Macdonald F, Maher ER. Genotype-phenotype correlation in von Hippel-Lindau disease. Hum Mutat. 2007;28:143–9. doi: 10.1002/humu.20385. [DOI] [PubMed] [Google Scholar]
- 16.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 17.Koch CA, Brouwers FM, Vortmeyer AO, Tannapfel A, Libutti SK, Zhuang Z, Pacak K, Neumann HP, Paschke R. Somatic VHL gene alterations in MEN2-associated medullary thyroid carcinoma. BMC Cancer. 2006;6:131. doi: 10.1186/1471-2407-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch CA, Huang SC, Zhuang Z, Stolle C, Azumi N, Chrousos GP, Vortmeyer AO, Pacak K. Somatic VHL gene deletion and point mutation in MEN 2A-associated pheochromocytoma. Oncogene. 2002;21:479–82. doi: 10.1038/sj.onc.1205133. [DOI] [PubMed] [Google Scholar]
- 19.Glenn GM, Daniel LN, Choyke P, Linehan WM, Oldfield E, Gorin MB, Hosoe S, Latif F, Weiss G, Walther M, Lerman MI, Zbar B. Von Hippel-Lindau (VHL) disease: distinct phenotypes suggest more than one mutant allele at the VHL locus. Hum Genet. 1991;87:207–10. doi: 10.1007/BF00204184. [DOI] [PubMed] [Google Scholar]
- 20.Koch CA, Mauro D, Walther MM, Linehan WM, Vortmeyer AO, Jaffe R, Pacak K, Chrousos GP, Zhuang Z, Lubensky IA. Pheochromocytoma in von hippel-lindau disease: distinct histopathologic phenotype compared to pheochromocytoma in multiple endocrine neoplasia type 2. Endocr Pathol. 2002;13:17–27. doi: 10.1385/ep:13:1:17. [DOI] [PubMed] [Google Scholar]
- 21.Santarpia L, Lapa D, Benvenga S. Germline mutation of von Hippel-Lindau (VHL) gene 695 G>A (R161Q) in a patient with a peculiar phenotype with type 2C VHL syndrome. Ann N Y Acad Sci. 2006;1073:198–202. doi: 10.1196/annals.1353.021. [DOI] [PubMed] [Google Scholar]
- 22.Ashida S, Okuda H, Chikazawa M, Tanimura M, Sugita O, Yamamoto Y, Nakamura S, Moriyama M, Shuin T. Detection of circulating cancer cells with von Hippel-Lindau gene mutation in peripheral blood of patients with renal cell carcinoma. Clin Cancer Res. 2000;6:3817–22. [PubMed] [Google Scholar]
- 23.Koch CA. Molecular pathogenesis of MEN2-associated tumors. Fam Cancer. 2005;4:3–7. doi: 10.1007/s10689-004-7022-3. [DOI] [PubMed] [Google Scholar]
- 24.Huang SC, Koch CA, Vortmeyer AO, Pack SD, Lichtenauer UD, Mannan P, Lubensky IA, Chrousos GP, Gagel RF, Pacak K, Zhuang Z. Duplication of the mutant RET allele in trisomy 10 or loss of the wild-type allele in multiple endocrine neoplasia type 2-associated pheochromocytomas. Cancer Res. 2000;60:6223–6. [PubMed] [Google Scholar]
- 25.Beroud C, Joly D, Gallou C, Staroz F, Orfanelli MT, Junien C. Software and database for the analysis of mutations in the VHL gene. Nucleic Acids Res. 1998;1:256–8. doi: 10.1093/nar/26.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sgambati MT, Stolle C, Choyke PL, Walther MM, Zbar B, Linehan WM, Glenn GM. Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet. 2000;66:84–91. doi: 10.1086/302726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murgia A, Martella M, Vinanzi C, Polli R, Perilongo G, Opocher G. Somatic mosaicism in von Hippel-Lindau Disease. Hum Mutat. 2000;15:114. doi: 10.1002/(SICI)1098-1004(200001)15:1<114::AID-HUMU20>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 28.Zlotogora J. Penetrance and expressivity in the molecular age. Genet Med. 2003;5:347–52. doi: 10.1097/01.gim.0000086478.87623.69. [DOI] [PubMed] [Google Scholar]
- 29.Zlotogora J. Germ line mosaicism. Hum Genet. 1998;102:381–6. doi: 10.1007/s004390050708. [DOI] [PubMed] [Google Scholar]
- 30.Wuyts W, Biervliet M, Reyniers E, D'Apice MR, Novelli G, Storm K. Somatic and gonadal mosaicism in Hutchinson-Gilford progeria. Am Journal Med Genet A. 2005;135:66–8. doi: 10.1002/ajmg.a.30663. [DOI] [PubMed] [Google Scholar]