

Abstract
Molecular imprinting is a technique for the synthesis of polymers capable to bind target molecules selectively. The imprinting of large proteins, such as cell adhesion proteins or cell receptors, opens the way to important and innovative biomedical applications. However, such molecules can incur into important conformational changes during the preparation of the imprinted polymer impairing the specificity of the recognition cavities. The “epitope approach” can overcome this limit by adopting, as template, a short peptide sequence representative of an accessible fragment of a larger protein. The resulting imprinted polymer can recognize both the template and the whole molecule thanks to the specific cavities for the epitope. In this work two molecularly imprinted polymer formulations (a macroporous monolith and nanospheres) were obtained using the protected peptide Z-Thr-Ala-Ala-OMe, as template, and Z-Thr-Ile-Leu-OMe, as analogue for the selectivity evaluation, methacrylic acid, as functional monomer, and trimethylolpropane trimethacrylate and pentaerythritol triacrylate (PETRA), as cross-linkers. Polymers were synthesized by precipitation polymerization and characterized by standard techniques. Polymerization and rebinding solutions were analyzed by high performance liquid chromatography. The highly cross-linked polymers retained about 70% of the total template amount, against (20% for the less cross-linked ones). The extracted template amount and the rebinding capacity decreased with the cross-linking degree, while the selectivity showed the opposite behaviour. The PETRA cross-linked polymers showed the best recognition (MIP 2−, α= 1.71) and selectivity (MIP 2+, α′= 5.58) capabilities. The cytotoxicity tests showed normal adhesion and proliferation of fibroblasts cultured in the medium that was put in contact with the imprinted polymers.
Keywords: molecular imprinting, biomaterials, tissue engineering, cell adhesion
Introduction
The construction of biological substitutes containing living functioning cells, capable of restoring, maintaining or improving specific tissue functions is the final goal of tissue engineering [1, 2]. The transplanted cells are incorporated into a supporting matrix, the scaffold, which in turn is implanted into the patient's body. Interest in tissue engineering is currently focused on the development of scaffold structures capable of guiding the growth and the organization of transplanted cells [3]. The knowledge of how cells organize themselves within the body is important in designing the properties of the scaffold.
Cellular processes such as adhesion, migration, growth and gene expression are controlled by interactions with the surrounding environment. The molecular mechanism through which the cells recognize substrates suitable for the adhesion is widely described in literature [4]. Cell adhesion occurs through specific membrane receptors. In particular, the integrins, a family of trans-membrane proteins linked to the cytoskeleton on the cytoplasmatic side of the membrane, recognize only particular peptide sequences present in some proteins (collagen, laminin, fibronectin, etc.), which are the components of the extra-cellular matrix.
The inclusion of these proteins on artificial substrates showed them to be poorly effective, because of the degradation phenomena following conformational changes in the protein structure. On the other hand, it is possible to improve cellular adhesion by including on the artificial surface short chains of oligopeptides or oligosaccarides. An approach for the creation of advanced scaffolds for cellular adhesion and proliferation is represented by molecular imprinting technology. This technology permits the introduction, into a polymeric material, of recognition sites for specific molecular species (templates) through the polymerization of a monomer in the presence of a template [5, 6], or through the dissolving of the preformed polymer in a solution containing the molecule to be recognized [7]. In both cases, the spatial arrangement is maintained by the polymer, after the template extraction, and confers a selective “memory” towards this molecule.
The molecular imprinting technology is a valid alternative to molecular recognition systems present in biological systems, such as those activated by antibodies [8]. The macromolecular matrices prepared with this procedure, in fact, can be stable even in critical chemical and physical conditions [9], can have a life of several years without any apparent reduction in performance, and can be used repeatedly without any alteration to the “memory.” The most extensively studied applications of the molecularly imprinted polymers (MIPs) are mainly in the separation by affinity sector [10–12], and also in determining the level of drugs in human serum in substitution of natural antibodies. Other applications are in the field of the biomimetic sensors [13, 14] and the intelligent polymers [15]. Interesting developments in the biomaterial field in general [16, 17] and in tissue engineering in particular can be envisaged.
The present work represents a first step towards the realization of polymeric materials for applications in tissue engineering, which will operate as “smart” systems for recognizing specific peptide sequences in extra-cellular matrix proteins, in order to guide and control the adhesion process.
The use of entire proteins as template molecules in molecular imprinting, showed to be, often unsuccessful because of the large molecular dimensions and to the flexibility of the chains, which limit the polymer molecular recognition capacity and selectivity. To overcome these limitations, stable short peptide sequences, representative of an accessible fragment of a larger protein of interest (i.e. collagen, fibronectin, laminin, vitronectin, etc.), can be used, these often being located in the receptor domains or in other parts directly involved in the molecular recognition process (epitopes). Therefore, if the material can recognize a peptide that represents the exposed part of a protein structure, it will also be able to bind the entire protein [18]. In this way, it is possible to increase the adhesion and growth characteristics of the cells on the MIP scaffold, thanks to the presence of sites complementary and selective towards specific sequences within the extra-cellular proteins and with which the integrin receptorial domains will interact.
Here we report the study of different MIPs capable of recognizing a tripeptide segment of an exposed part of a fibronectin functional domain [19]. The fibronectin is the main extra-cellular matrix adhesive glycoprotein and plays a fundamental role in the cell adhesion, migration and repair processes.
The tripeptide Z-Thr-Ala-Ala-OMe was used as template molecule maintaining the terminal functional groups protection, in order to avoid undesired inter- and intra-molecular interactions and to enhance the affinity of the peptide hydroxyl group to the polymer functional groups. The polymers were prepared by polymerization of methacrylic acid (MAA) as the functional monomer in the presence of the template molecule and of an appropriate crosslinking agent, trimethylolpropane trimethacrylate (TRIM) or pentaerythritol triacrylate (PETRA), to provide a sufficient rigidity to the recognition sites in the polymer structure.Two different forms of the polymeric materials were obtained by varying the cross-linking agent concentration: a macroporous monolith at the higher cross-linking degree and nanospheres at the lower cross-linking degree [20]. The obtained polymeric materials were characterized for their thermal, spectroscopic and morphological properties. The template molecule was removed from the polymers by solvent extraction, and the performances of the obtained polymers in selectively rebinding the template molecule were verified. Moreover, the selectivity test using the analogue peptide Z-Thr-Ile- Leu-OMe was carried out.
Cytotoxicity tests were also performed by seeding mouse fibroblasts in a culture medium previously put in contact, for a fixed time, with the polymer.
In tissue engineering applications, the loading of nanoparticles on appropriate biodegradable scaffolds, the biocompatibility tests and the functional verification for improving cell adhesion are needed. These important investigations are currently in progress and will be reported separately.
Materials and methods
Materials
MAA (>99%), from Aldrich, was distilled prior to use to remove the inhibitor. TRIM and PETRA, from Aldrich, and azobisisobutyronitrile (>98%), from Fluka, were used as supplied. The protected peptides TAA and TIL (Z-Thr-Ala- Ala-OMe and Z-Thr-Ile-Leu-OMe peptide sequences) were synthesized by the group of Prof. Peggion, University of Padua according to the standard protocols for peptide synthesis in solution using 1-hydroxybenzotriazole/1-ethyl-3(3- dimethylaminopropyl)carbodiimmide as coupling reagents in the presence of diisopropylethylamine and characterized by high performance liquid chromatography (HPLC), nuclear magnetic resonance and electrospray-ionization mass spectroscopy. Acetonitrile (>99.9%), methanol (>99.9%), bidistilled water and acetic acid glacial (>99.9%), from Carlo Erba Reagenti, were of HPLC grade purity. Trifluoroacetic acid (>99%) was from Aldrich. Dulbecco's modification of Eagle's medium was from Cambrex. Formaldehyde and phosphate buffered saline were from Sigma. Coomassie blue solution was from Fluka.
Preparation of MIPs
The MIPs were synthesized via precipitation polymerization at the compositions shown in Table 1. The reactors were borosilicate 20 ml glass tubes, sealed with screw caps. The template molecule was dissolved in acetonitrile, then the functional monomer MAA, the cross-linker and the initiator were added. In the end, acetonitrile was added to the final volume and the tubes were sealed under dry nitrogen. The polymerization was thermally initiated at 60°C and carried out for 20 hrs. A constant agitation of the tubes was provided during all the process. Depending on the polymerization conditions, in particular on the level of porogen coupled with the level of cross-linker, MIPs in the form of nanospheres or a macroporous monolith were obtained. The nanospheres were collected by centrifugation at 14,000 rpm for 15 min (MIKRO 200 centrifuge, Hettich GmbH & Co., Tuttlingen, Germany). The macroporous monolith was crushed and washed with acetonitrile to remove the residual monomer and cross-linker and the not imprinted peptide. The template was extracted by washing the polymers in Soxhlet apparatus with a MeOH/acetic acid mixture 70/70 (v/v) for 10 hrs. The controls were prepared with the same procedure of the imprinted polymers, but in the absence of the template molecule.
1.
Preparation of molecularly imprinted polymers
| Polymer† | TAA (mmol/l) | MAA (mmol/l) | TRIM (mmol/l) | PETRA (mmol/l) | MAA/CL‡ (mol/mol) | (MAA + CL)%§ (v/v) |
|---|---|---|---|---|---|---|
| MIP 1− | 8.4 | 67.4 | 67.4 | – | 1/1 | 2.7 |
| CP 1− | – | 67.4 | 67.4 | – | 1/1 | 2.7 |
| MIP 2− | 8.4 | 67.4 | – | 67.4 | 1/1 | 2.3 |
| CP 2− | – | 67.4 | – | 67.4 | 1/1 | 2.3 |
| MIP 1+ | 8.4 | 67.4 | 296.7 | – | 1/4.4 | 10.1 |
| CP 1+ | – | 67.4 | 296.7 | – | 1/4.4 | 10.1 |
| MIP 2+ | 8.4 | 67.4 | – | 296.7 | 1/4.4 | 8.1 |
| CP 2+ | – | 67.4 | – | 296.7 | 1/4.4 | 8.1 |
MIP:molecularly imprinted polymer;CP:control polymer.
Molar ratio of functional monomer:cross-linker.
Total monomer and cross-linker concentration with respect to porogen.
Physico-chemical characterization
The morphology of the MIPs nanospheres was examined with a JSM 5600 scanning electron microscopy (SEM) (Jeol Ltd., Peabody, MA, USA). The dimensions of the particles were also estimated. Fourier transformed infrared spectroscopy (FT-IR) spectra were recorded on a Spectrum-One instrument, Perkin Elmer Inc., Beaconsfield, England. KBr pellets of the material samples were examined. The thermal characterization was performed with a TGA 6 instrument, Perkin Elmer Inc., Sittard, Netherlands (scan from 30°C to 800°C at 10°C/min).
Chromatographic analysis
The monomer and the cross-linker conversion, and the amount of template entrapped by the polymer, were determined measuring the corresponding residual amount in the polymerization solution by HPLC (200 Series HPLC system, Perkin Elmer Inc., Shelton, CT, USA, with a UV/VIS detector). To determine the monomer and cross-linker concentrations, we used a Alltima C18 5u column (250 mm length × 4.5 mm i.d.) (Alltech Associates Inc., Deerfield, IL, USA). The mobile phase was: A = 0.085% trifluoroacetic acid (w/v) in acetonitrile; B = 0.1% trifluoroacetic acid (w/v) in water. The elution condition was an isocratic elution for 15 min with a mobile phase composition of 54% A and 46% B at a flow rate of 1 ml/min. The injection volume was 100 μl. The detector wavelength was set at λ= 215 nm.
To determine the peptide concentrations, we used a HP Prosphere C4 300A 5u column (250 mm length × 4.5 mm i.d.) (Alltech Associates Inc.). The elution condition was a linear binary gradient at a flow rate of 1ml/min and the gradient was from 30% A and 70% B to 60% A and 40% B in 15 min. The injection volume was 100 μl. The detector wavelength was set at λ= 215 nm.
Recognition experiments
To evaluate the rebinding capacity of MIPs, 10 mg of each polymer sample were put in contact with 1.5 ml of a solution of the template (0.1 mg/ml) in acetonitrile. The tubes were sonicated for 10 min and then maintained under constant agitation for 30 min. The supernatant was separated from the polymer by centrifugation for 15 min at 14,000 rpm. The procedure was repeated twice, adding every time 1 ml of fresh solution. To evaluate the selectivity of MIPs, the same procedure was applied using a solution of the analogue in acetonitrile at the same concentration. For each polymer the amount of template and analogue was determined on five samples and the average values were reported.
Cytotoxicity test
Cytotoxicity tests were carried out on a control polymer. CP 2+ materials were sterilized by UV ray for 30 min followed by immersion in ethanol/water 70/30 mixture for three times. A fixed quantity (20 mg) of CP 2+ was dipped in 1 ml complete culture medium normally used for fibroblasts culture (Dulbecco's modification of Eagle's medium) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 μg/ml streptomycin and 100 IU/ml penicillin. At different times (4 hrs, 24 hrs, 7 days and 15 days) a fixed quantity of this medium was collected and stored at 4°C. At the end of this two weeks NIH-3T3 mouse fibroblasts were seeded in a 24 multiwell plate with a density 5 × 105 cells/ml, cultured in the collected media and placed in an incubator at 37°C under 5% CO2. At several times (24, 48, 72 hrs) the cells were fixed with 4% (v/v) formaldehyde solution in phosphate buffered saline and stained with Coomassie blue solution. Fibroblasts cultured in pure culture medium were used as control. The stained cells were analyzed under an optical microscope (AX70, Olympus Optical, London, UK) and the ratio between the number of cells cultured in the collected media and those cultured in control media was taken as a cytotoxicity index. The cytotoxicity tests were repeated three times and for each experiment three samples of CP 2+ were used.
Results
Physico-chemical characterization
MIPs and CPs were obtained by precipitation polymerization under the conditions indicated in Table 1. After 20 hrs of reaction a high monomer conversion was observed (Table 2). The conversion rate reached almost 100% for the more cross-linked resins and it is slightly lower in the case of MIP 2− and the related control. An incomplete conversion of the monomers to MIP 1− and CP 1− occurred at 20 hrs was also confirmed by calorimetric data.
2.
Monomer and cross-linkers conversion
| Polymer | Conversion† (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| MAA | TRIM | PETRA | ||||||
| MIP 1− | 86.9 | 65.5 | – | |||||
| CP 1− | 81.6 | 64.2 | – | |||||
| MIP 2− | 99.2 | – | 93.2 | |||||
| CP 2− | 98.2 | – | 91.5 | |||||
| MIP 1+ | 98.7 | 100 | – | |||||
| CP 1+ | 97.7 | 100 | – | |||||
| MIP 2+ | 97.9 | – | 100 | |||||
| CP 2+ | 98.1 | – | 100 | |||||
Monomer M conversion = ([M initial mole − M final mole]/M initial mole) × 100.
In the case of loading capacity, a remarkably larger amount of template was retained by the more highly cross-linked polymers (Table 3).
3.
Template fraction retained and extracted
| Polymer | TAA fraction retained†/% | TAA fraction extracted‡ (%) |
|---|---|---|
| MIP 1− | 20 | 40.1 |
| CP 1− | – | – |
| MIP 2− | 26.7 | 40.4 |
| CP 2− | – | – |
| MIP 1+ | 71.1 | 25.0 |
| CP 1+ | – | – |
| MIP 2+ | 70.2 | 33.0 |
| CP 2+ | – | – |
TAA fraction retained = ([TAA initial mole − TAA final mole]/TAA initial mole) × 100.
TAA fraction extracted = ([TAA retained mole − TAA extracted mole]/TAA retained mole) × 100.
SEM micrographs showed large fused aggregates of microgel particles interconnecting to form a labyrinth of macropores in the case of the more cross-linked resins as reported in Figure 1a. The images of MIP 1− showed a powder of nano-sized particles (Fig. 1c). Further investigation on samples obtained by suspension of the MIP 1− in methanol confirmed the spherical shape and the monodisperse size distribution of the nanospheres (Fig. 1e). Also for MIP 2− (Fig. 1f) a mass of particles was observed, but the particles seemed to form small aggregates, these were more clearly revealed by the analysis of the methanol suspension (Fig. 1h).
1.

(a) SEM of MIP 1+ resin showing the fused microgel particles of the macroporous monolith. (b) SEM of CP 1+ resin showing the fused microgel particles of the macroporous monolith. (c) SEM of MIP 1− resin showing aggregates of nanospheres. (d) SEM of CP 1− resin showing aggregates of nanospheres. (e) SEM of MIP 1− resin after suspension in methanol showing discrete and monodispersed nanospheres. (f) SEM of MIP 2− resin showing aggregates of nanospheres. (g) SEM of CP 2− resin showing aggregates of nanospheres. (h) SEM of MIP 2− resin after suspension in methanol showing small aggregates of nanospheres.
FT-IR spectra showed the characteristic absorption peaks of the expected chemical structure (ν O H = 3450 cm−1; ν C-H = 2970−1cm; ν C = O = 1730 cm−1; ν C(= O)−O = 1160 cm−1). When a lower cross-linker amount was used (for example CP 1− with respect to CP 1+) a more intense band at 3450/cm due to the O-H stretching of the MAA carboxyl group was observed (Fig. 2). When PETRA was used instead of TRIM as cross-linker agent (Fig. 3) a stronger ν O-H band, due to the addition of the side chain C-OH group absorption, and a peak at 1065 cm−1, due to the ν C-O, were noted. The comparison between MIPs and CPs did not highlight the presence of the target molecule, because of the small amount (<3%) of TAA entrapped by the polymers. The spectra of CP 1− and MIP 1−, in Figure 4, do not show any difference in the region around (1550 cm−1) where the target molecule presents an intense absorption peak.
2.
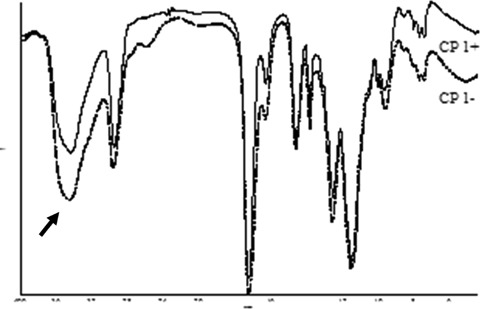
FT-IR spectra of CP 1+ and CP 1− showing an increase in the intensity of the band at 3450 cm−1 for CP 1−, containing a higher concentration of MAA hydroxyl groups.
3.
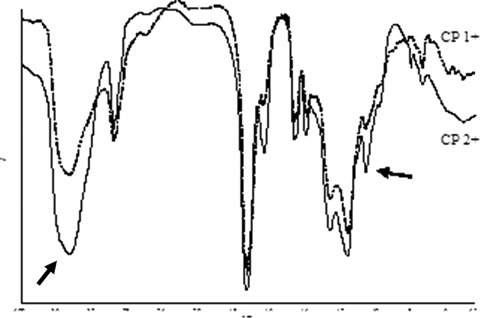
FT-IR spectra of CP 1+ and CP 2+ showing an increase of the band at 3450 cm−1 and the peak at 1065 cm−1 for CP 2+, containing additional OH groups due to the cross-linker PETRA.
4.
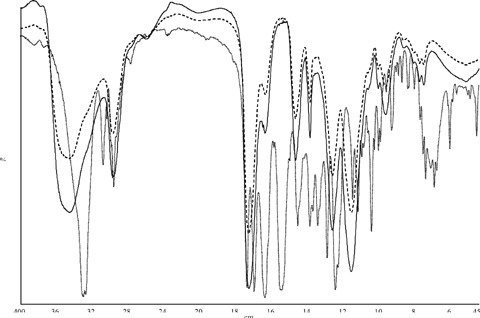
FT-IR superimposed spectra of TAA peptide (dotted line) and a couple of imprinted and related control polymers. CP 1− (continuous line) and MIP 1− (dashed line) spectra are identical in spite of the presence of the peptide in MIP 1−.
The thermal stability of the prepared polymers was investigated by thermogravimetric analysis. The derivative thermogram of PETRA containing resins exhibits a peak centred at about 460°C in the less cross-linked polymers, but it is shifted to higher temperatures by increasing the cross-linking degree.The typical derivative thermograms are presented in Fig. 5. No remarkable differences in the thermal degradation of TRIM containing polymers were noted, but a higher thermal stability (shift of the onset temperature from 450 to 460°C) was obtained enhancing cross-linking rate.
5.
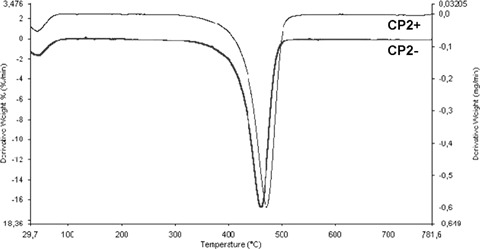
Derivative thermograms: shift of the maximum degradation rate of the more highly cross-linked CP 2+ with respect to CP 2− to higher temperature.
Recognition experiments
The template molecule was removed from the polymers by methanol/acetic acid extraction. The polymer monoliths were crashed and ground in a mortar before this step. As expected the amount of template removed from the nanospheres was higher than from the macroporous resins (Table 3).
The recognition properties of the prepared MIPs were investigated by HPLC analysis. The chromatographic results concerning the binding capacity, that is the amount of template bound per mass of polymer, are shown in Table 4. In all cases the MIPs were more efficient in template recognition than the related controls, indicating the formation of specific binding sites in the polymer network. The binding factor, expressed as the ratio of template bound by the molecular imprinted material compared to the control polymer, indicated that the best performances in terms of template recognition were obtained by the MIP 2− nanospheres. The ability of a prepared MIP to differentiate between the template and a peptide of similar structure was obtained by calculating the selectivity factor, that is the ratio of template compared to analogue bound following the same rebinding procedure. As indicated in Table 4, in all cases higher selectivity for the target molecule than the analogue was measured.
4.
Binding and selectivity tests for TAA imprinted polymers and related controls
| Polymer | Bound TAA | Bound TIL | Binding factor† | Selectivity factor‡ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [μmol/g polymer] | [μmol/g polymer] | α MIP/CP | α′ template/analogue | ||||||||||
| MIP 1− | 4.42 ± 0.09 | 3.58 ± 0.08 | 1.09 | 1.23 | |||||||||
| CP 1− | 4.06 ± 0.11 | 2.51 ± 0.14 | – | – | |||||||||
| MIP 2− | 4.75 ± 0.06 | 2.18 ± 0.09 | 1.71 | 2.18 | |||||||||
| CP 2− | 2.78 ± 0.04 | 3.48 ± 0.07 | – | – | |||||||||
| MIP 1+ | 1.58 ± 0.05 | 0.91 ± 0.08 | 1.07 | 1.73 | |||||||||
| CP 1+ | 1.48 ± 0.09 | 2.28 ± 0.10 | – | – | |||||||||
| MIP 2+ | 1.74 ± 0.04 | 0.31 ± 0.04 | 1.12 | 5.58 | |||||||||
| CP 2+ | 1.55 ± 0.08 | 2.22 ± 0.05 | – | – | |||||||||
†α=μmol TAA bound by MIP/μmol TAA bound by CP.
‡α′=μmol TAA bound by MIP/μmol TIL bound by MIP.
Cytotoxicity test
Figure 6 presents the effects of CP 2+ nanoparticles incubated with media culture at several times (4 hrs, 24 hrs, 7 days and 15 days) time on the cell adhesion at proliferation as a function of incubation time (4, 24, 48 hrs). The ratio between the number of cells cultured in conditioned media and those in pure media was considered as an index of cytotoxicity.
6.
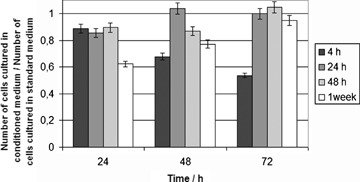
Cytotoxicity index of CP 2+ using NIH-3T3 cultured in conditioned media culture as function of cell culture time after incubation (at 24, 48 and 72 hrs). Number of cells cultured in medium not incubated with nanoparticles was used as control.
Discussion
The polymerization method involved an initially homogeneous solution polymerization which, ultimately produced insoluble resins. The amount of peptide entrapped inside the resins strongly depended on the matrix rigidity. An enhanced cross-linking rate was found to produce materials that blocked the template inside the resin more efficiently. MIPs with different morphology were obtained by changing the reaction conditions (as e.g. amount of porogen amount of cross-linker). For lower porogen levels and higher cross-linker concentration (MIP+ and CP+), the materials obtained were macroporous resins. In a more dilute solution and at lower cross-linker concentration (MIP− and CP−) the microgel particles did not fully coalesce and appeared as a microgel powder dispersion in accordance to what described by Sherrington [21]. The presence of aggregates of particles observed for MIP 2− explained, on the basis of the higher monomer conversion, to a further polymerization in the porogen phase after phase separation of the discrete nanospheres, creating additional polymer which helped fuse the particles together. FTIR-ATR spectra were performed in order to study the interactions of the template molecules with the resins, but the analysis was unsuccessful because of the too low amount of peptide in the mixture to produce detectable signals. No remarkable differences in the thermal degradation of the imprinted polymers were noted, but a higher thermal stability was obtained enhancing the cross-linking rate.
The polymers were washed in a Soxhlet apparatus to remove the template molecule before functional characterization. The template extraction would have been expected to be difficult because of the large dimension of the peptide; it has been indicated [22] that bulky templates cannot easily slip out of a polymer network. The smaller dimension of the particles promote the extraction because the area to volume ratio is increased improving the efficiency of solid-liquid contact, even though the physical configuration of macroporous MIPs allow the solvent to have access to an essential fraction of the polymer mass. Also the enhanced hydrophilicity of the material, obtained with the PETRA cross-linker, promotes polymer wettability with polar solvents favouring the extraction process.
Template and analogue recognition was studied by rebinding experiments. The nanoparticles exhibited better template recognition properties than the macroporous systems. The binding sites on these discrete particles are supposed to be more accessible; on the contrary large templates cannot easily diffuse through the highly cross-linked polymer matrix due to the steric hindrance. Diffusion of a bulky template into the imprinted cavities should be made easier by the low cross-linking rate and the high area to volume ratio, but the comparison between MIP 1− with MIP 2− clearly suggested that the nature of the cross-linker has a more remarkable effect on template affinity. On the other hand, MIP 2− did not exhibit the highest selectivity for the target with respect to the analogue TIL, a peptide with a very similar chemical structure. MIP 2+ exhibited the α′ maximum value of 5.58 and for MIP 2−α′ was equal to 2.18. Even if a higher molar amount of functional monomer was used for MIP 2− with respect to MIP 2+ in order to increase complexation between functional monomer and the template molecule [18], the resulting increased non-specific binding leads to a loss of selectivity. An increased cross-linking content inhibits the diffusion of the template reducing the efficiency of the extraction process and rebinding; on the other hand, it rigidly fixes the arrangement of the functional monomer around the template, favouring the formation of highly selective sites.
In vitro cell culture tests were performed to investigate the cytotoxicity of the particles. CP 2+ particles after 4 hrs of incubation in medium displayed a cytotoxic effect that was a function of time culture. In particular, the cell number decreased of more than 40% as compared to the untreated cells. On the contrary, CP 2+ after 24 hrs of incubation in medium had only a negligible effect on the cell number and an increased trend of cell proliferation was detected for all conditioning times. Actually, the low cell proliferation value at 4 hrs was attributed to the presence of residual solvent used in the preparation phase – that after 72 hrs seemed to be completely removed or metabolized under the experimental conditions. Moreover, the best result of cell viability was obtained after 72 hrs of incubation time for all samples. These results suggested that the developed material did not presented any detectable cytotoxicity.
Conclusions
Two formulations of MIPs, specific to TAA peptide sequence, were synthesized with TRIM and PETRA as cross-linker agents. For each formulation, polymers in the form of a macroporous monolith and nanospheres were prepared, depending on the level of porogen coupled with the level of cross-linker. The analogue used in the selectivity studies was the TIL peptide sequence. It was observed that the template is better retained by the higher cross-linked MIPs and it is better extracted from the lower cross-linked ones. The recognition capacity and factor are superior for the nanosphere formulations, while the selectivity factor is increased in the macroporous monoliths. These results are in accordance with the expected increasing rigidity of the polymer network at higher cross-linking degree. The rigidity of the recognition cavities decreases the rate of diffusion phenomena and increases the selectivity of the binding sites. The MIPs synthesized with PETRA showed the best performance in terms of recognition capacity and factor (MIP 2−, microspheres) and in terms of selectivity (MIP 2+, monolith), compared to TRIM polymers. Our results are in accordance with the molecular imprinting theory and with the conclusions of the reference works. In addition, screening biological tests suggest the absence of toxic effects.
Specific tests on cell adhesion and proliferation on porous scaffold surface modified with imprinted nanoparticles will be also essential for the evaluation of the functional properties of the developed material in view of a perspective application in tissue engineering. The rebinding in aqueous solution, in conditions close to the biological environment will be reported in a separate contribution. The efficient and selective recognition of the TAA epitope is an important step forward in the synthesis of MIPs specific to cell adhesion proteins, cell receptors, extra-cellular matrix molecules, enzymes, growth factors, DNA fragments and complex drugs. Therefore, the technique adopted will lead in the next future to important application in the fields of tissue engineering, synthetic receptors or drug delivery.
Acknowledgments
Co-financing by the Italian Ministry of research through the PRIN project 2005091572_002, year 2005 is acknowledged.
References
- 1.Fournier RL. Basic transport phenomena in biomedical engineering. Philadelphia: Taylor and Francis; 1998. Tissue engineering; pp. 201–36. [Google Scholar]
- 2.Seal BL, Otero TC, Panitch A. Polymeric biomaterials for tissue and organ regeneration. Mater Sci Eng. 2001;34:147–230. [Google Scholar]
- 3.Hutmacher DW. Scaffold design and fabrication technologies for engineering tissues. State of the art and future perspectives. Biomater Sci Polym Edn. 2001;12:107–24. doi: 10.1163/156856201744489. [DOI] [PubMed] [Google Scholar]
- 4.Folk A, Toner M. Microengineering of cellular interaction. Ann Rev Biomed Eng. 2000;2:227–56. doi: 10.1146/annurev.bioeng.2.1.227. [DOI] [PubMed] [Google Scholar]
- 5.Mosbach K. Molecular imprinting. Trends Biochem. Sci. 1994;19:9–14. doi: 10.1016/0968-0004(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 6.Wulff G, Karsten K. Stoichiometric noncovalent interaction in molecular imprinting. Bioseparation. 2002;10:257–76. doi: 10.1023/a:1021585518592. [DOI] [PubMed] [Google Scholar]
- 7.Wang HI, Kobayashi T, Fujii N. Molecular imprint membranes prepared by the phase inversion precipitation technique. Langmuir. 1996;12:4850–6. [Google Scholar]
- 8.Wulff G. Molecular Imprinting in cross-linked materials with the aid of molecular templates: a way towards artificial antibodies. Angew Chem Int Ed Engl. 1995;34:1812–32. [Google Scholar]
- 9.Nicholls IA, Svenson J. On the thermal and chemical stability of molecularly imprinted polymers. Analytica Chimica Acta. 2001;435:19–24. [Google Scholar]
- 10.Sellergren B, Shea KJ. Chiral ion exchange chromatography: correlation between solute retention and a theoretical ion-exchange model using imprinted polymers. J Chromatogr A. 1993;654:17–28. doi: 10.1016/0021-9673(93)83061-V. [DOI] [PubMed] [Google Scholar]
- 11.Mosbach K, Kempe M. Separation of amino acids, peptides and proteins on molecularly imprinted stationary phases. J Chromatogr A. 1995;691:317–23. doi: 10.1016/0021-9673(94)00820-y. [DOI] [PubMed] [Google Scholar]
- 12.Hart BR, Shea KJ. Synthetic peptide receptors: molecularly imprinted polymers for the recognition of peptides using peptide-metal interactions. J Am Chem Soc. 2001;123:2072–3. doi: 10.1021/ja005661a. [DOI] [PubMed] [Google Scholar]
- 13.Mosbach K, Ramstrom O. The emerging technique of molecular imprinting and its future impact on biotechnology. Biotechnology. 1996;14:163–70. [Google Scholar]
- 14.Shea KJ. Molecular imprinting of synthetic network polymers: de novo synthesis of macromolecular binding and catalytic sites. Trends Polym Sci. 1994;2:166–73. [Google Scholar]
- 15.Cormack PAG, Mosbach K. Molecular imprinting: recent developments and the road ahead. React Funct Polym. 1999;41:115–24. [Google Scholar]
- 16.Cristallini C, Ciardelli G, Giusti P, Barbani N. Acrylonitrile-acrylic acid copolymer membrane imprinted with uric acid for clinical uses. Macromolecular Bioscience. 2004;4:31–8. doi: 10.1002/mabi.200300026. [DOI] [PubMed] [Google Scholar]
- 17.Silvestri D, Borrelli C, Giusti P, Cristallini C, Ciardelli G. Polymeric devices containing imprinted nanospheres: a novel approach to improve recognition in water for clinical uses. Analytica Chimica Acta. 2005;542:3–13. [Google Scholar]
- 18.Rachkov A, Minoura N. Towards molecularly imprinted polymers selective to peptides and proteins. the epitope approach. Biochimica et Biophysica Acta. 2001;1544:255–66. doi: 10.1016/s0167-4838(00)00226-0. [DOI] [PubMed] [Google Scholar]
- 19.Dickinson CD, Veerapandian B, Dai XP, Hamlin RC, Xuong NH, Ruoslahti E, Ely KR. Crystal structure of the tenth type III cell adhesion module of human fibronectin. J Mol Biol. 1994;236:1079–92. doi: 10.1016/0022-2836(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 20.Ye L, Weiss R, Mosbach K. Synthesis and characterization of molecularly imprinted microspheres. Macromolecules. 2000;33:8239–45. [Google Scholar]
- 21.Sherrington DC. Preparation, structure and morphology of polymer supports. Chem Commun. 1998:2275–86. [Google Scholar]
- 22.Flam F. Molecular imprints make a mark. Science. 1994;263:1221–2. doi: 10.1126/science.8122101. [DOI] [PubMed] [Google Scholar]