

Abstract
The trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine scaffold is a known pharmacophore for mu opioid (MOP), kappa opioid (KOP), and delta opioid (DOP) receptor antagonists; however, it has not been explored in nociceptin opioid (NOP/ORL-1) receptor ligands. We recently found that the selective KOP antagonist JDTic, (3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide, containing this opioid antagonist pharmacophore, has significant binding affinity at the NOP receptor (Ki 16.67 ± 0.76 nM), with no intrinsic activity in the [35S]GTPγS functional assay. Since this is the first ligand containing the trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonist pharmacophore to have affinity for the NOP receptor, we explored the structural determinants of its NOP binding affinity. When rational chemical modifications of JDTic were carried out, based on our previously established NOP pharmacophoric structure–activity relationship (SAR) model, most modifications led to a significant decrease in NOP and opioid binding affinity compared to JDTic. Interestingly, however, removal of the 3,4-dimethyl groups of the trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine antagonist scaffold of JDTic increased the binding affinity at NOP by 10-fold (Ki 1.75 ± 0.74 nM) while maintaining comparable affinity for KOP, MOP, and DOP receptors (Ki 1.14 ± 0.63, 1.67 ± 0.6, and 19.6 ± 1.3 nM, respectively). In vitro functional efficacy studies using the [35S]GTPγS assay showed that this compound AT-076 functions as an antagonist at all four opioid receptors. Detailed characterization of the antagonist activity of AT-076 shows that it has a noncompetitive antagonist profile at the NOP and KOP receptors (insurmountable antagonism), but is a potent competitive antagonist at the MOP and DOP receptors, with Ke values 3–6-fold more potent than those of JDTic. AT-076 is the first opioid pan antagonist with high affinity at all four opioid receptor subtypes. Our SAR studies show that the 3,4-dimethyl groups of the well-known trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonist scaffold may be removed without significant loss in binding affinity or antagonist potency to obtain an opioid pan antagonist such as AT-076.
Keywords: NOP, opioid antagonist, pan antagonist, nociceptin antagonist, nociceptin ligand, JDTic
The trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine scaffold is a known opioid antagonist pharmacophore for the mu, delta, and kappa opioid receptors (MOP, KOP, and DOP, respectively),1 and is present in several opioid antagonists such as the nonselective opioid antagonist LY255582,2 the peripherally restricted mu opioid antagonist LY246736 (Alvimopan; ENTEREG),3 and the kappa opioid antagonist JDTic4 (Figure 1). However, this universal opioid antagonist pharmacophore has not been explored for antagonists at the fourth opioid subtype, nociceptin opioid receptor (NOP). Although opioid peptides and classical morphinan-based opioid ligands do not have appreciable affinity for NOP, phenylpiperidine-type scaffolds are found among both opioid (e.g., fentanyl, lofentanil) as well as NOP ligands (e.g., SB-612111) (Figure 1). As part of our continuing investigation into novel NOP ligands, we examined the affinity of the kappa opioid antagonist JDTic at the NOP receptor, and found it to have surprisingly high affinity at NOP, with a Ki of 16.7 ± 0.76 nM and no intrinsic activity in the [35S]GTPγS functional assay, in agreement with a recent report by Munro and colleagues5 (Table 1). Since this was the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine-containing ligand to show good binding affinity at the NOP receptor, we explored the structural determinants of JDTic’s NOP binding affinity and functional activity, with rational chemical modifications designed to inform the structure–activity relationship (SAR) of the various functionalities of this new ‘antagonist’ molecule at the NOP receptor. The chemical modifications explored are shown in Figure 2.
Figure 1.

Structures of trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine-containing opioid antagonists and phenylpiperidine-containing opioid and nociceptin receptor ligands. (a) From ref (6). (b) From ref (7).
Table 1. Binding Affinities of JDTic Analogues at the Four Opioid Receptors, Determined in Competition Radioligand Displacement Assays in Cloned Human Opioid Receptor-Transfected Cellsa.
| receptor
binding, Ki (nM) |
||||
|---|---|---|---|---|
| compd | NOP | KOP | MOP | DOP |
| JDTic | 16.7 ± 0.76 | 0.43 ± 0.03 | 1.53 ± 0.19 | 10.6 ± 0.17 |
| 1 | >10K | 51.07 ± 8.23 | 40.35 ± 0.89 | 586.16 ± 7.90 |
| 2 | >10K | 51.48 ± 13.32 | 6.01 ± 1.16 | 104.62 ± 14.68 |
| 3 | >10K | 68.12 ± 2.21 | 516.37 ± 51.4 | 2914.4 ± 833.8 |
| 4 | >10K | 410.34 ± 130 | 30.02 ± 8.16 | 205 ± 0.0 |
| 5 | 37.7 ± 8.6 | 19.5 ± 6.6 | 0.98 ± 0.35 | 38.77 ± 9.9 |
| 6 | 1.75 ± 0.74 | 1.14 ± 0.63 | 1.67 ± 0.6 | 19.6 ± 1.3 |
| 7 | 48.38 ± 3.93 | 100.0 ± 0.9 | 164.3 ± 18.7 | 1403 ± 110.9 |
| 8 | 10.93 ± 2.14 | 2.15 ± 0.82 | 15.01 ± 2.44 | >10K |
| SB-612111 | 1.06 ± 0.52 | 541.26 ± 36.3 | 623.11 ± 156.3 | 2894.1 ± 533 |
Ki values were determined by competitive displacement of the respective radioligands: [3H]N/OFQ–NOP, [3H]U69,593–KOP, [3H]DAMGO–MOP, and [3H]Cl-DPDPE–DOP receptor. The Ki was calculated from the IC50 values determined from the binding curves, using the Cheng-Prusoff equation. Values are the mean ± SEM of three independent experiments run in triplicate.
Figure 2.

Structural exploration of JDTic for SAR of the binding affinity and functional activity at the NOP receptor. Target compound numbers are indicated in parentheses in the figure.
Most high affinity NOP ligands possess a central piperidine scaffold, and generally contain two other pharmacophoric groups, namely, substituents on the piperidine nitrogen and at the 4-piperidine position, as represented in our previously proposed NOP ligand 2D pharmacophore model.8 Typically, the N-substituents on the piperidine nitrogen tend to be aromatic or lipophilic alicyclic groups such as those found in the NOP antagonist J-1133979 or SB-612111.10 We therefore examined the importance of the 2-methylpropyl-7-hydroxy-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide N-substituent of JDTic, for its NOP activity by replacing it with lipophilic substituents such as those found in other NOP ligands, namely, the 4-isopropylcyclohexyl and the cyclooctyl groups as in compounds 1–3 (Figure 2, Table 1). The valine-amide moiety at the piperidine nitrogen of JDTic was also removed, retaining the 7-hydroxy-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide as in compounds 4 and 5 (Figure 2, Table 1).
The importance of the 3,4-dimethyl substituents of the ‘opioid antagonist’ pharmacophore in JDTic to its NOP and opioid binding affinity was also investigated with compound 6 (AT-076) (Figure 2, Table 1), which lacks both the 3- and 4-methyl groups. Although the trans-3,4-dimethyl groups have long been considered important for the opioid ‘antagonist’ profile of compounds containing this pharmacophore,1a,1b Kormos et al.11 recently showed that removing the 3- or 4-methyl groups or both, retained opioid antagonist activity in a series of N-methyl and N-phenylpropyl piperidine opioids, albeit with reduced potency and no selectivity.
The phenolic 7-hydroxy group on the tetrahydroisoquinoline moiety was shown to be an important address group for kappa affinity and antagonist activity of JDTic.12 We investigated the importance of both phenolic hydroxy groups on JDTic, namely, the 7-hydroxyl and the 3-hydroxyl groups, to the binding affinity and opioid selectivity, with compounds 7 and compound 8, respectively.
Chemistry
Compounds 1 and 2 (Scheme 1) were prepared from (3R,4R)-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine I-1 and 4-isopropylcyclohexanone, using standard reductive amination conditions. Alternatively, a stepwise sequence using Ti(OiPr)4 followed by NaBH(OAc)3 was employed for cyclooctyl analogue 3 (Scheme 1),13 as standard reductive amination conditions did not afford the desired material in sufficient yield.
Scheme 1. Synthesis of 1–3.

Reagents and conditions: (a) for 1 and 2: 4-isopropylcyclohexanone, HOAc, NaBH(OAc)3, DCE, rt, 24 h, 20–50%; for 3: (i) cyclooctanone, Ti(OiPr)4, THF, 40 °C, 21 h; (ii) NaBH(OAc)3, 40 °C, 1 h, 71%.
Compounds 4 and 5 lacking the valine-amide moiety were synthesized as shown in Scheme 2. HATU-mediated coupling of piperidine I-1 and (3R)-2-(tert-butoxycarbonyl)-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid II-1 or (3R)-2-(tert-butoxycarbonyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid II-2 afforded amides II-3 and II-4, respectively. Removal of the Boc group with trifluoroacetic acid followed by reduction of the amide with borane dimethyl sulfide complex gave the compounds 4 and 5 in moderate yields.
Scheme 2. Synthesis of 4 and 5.

Reagents and conditions: (a) for II-3: HATU, Et3N, MeCN, rt, 21 h, 49%; for II-4: (i) HATU, Et3N, MeCN, rt, 22 h; (ii) LiOH, MeOH, rt, 5 h, 94%; (b) TFA, CH2Cl2, rt, 1.5–2 h, 82–89%; (c) BH3·SMe2, THF(ah), reflux, 5 h, 36–56%.
Des-3R,4R-dimethyl analogue 6 (AT-076) was prepared using a similar synthesis strategy as that used for JDTic4,14 (Scheme 3). BOP-assisted amidation of 4-(3-hydroxyphenyl)piperidine III-1 with Boc-l-valine yielded amide III-2. Boc deprotection and borane reduction gave amine III-3. Subsequent BOP-mediated coupling of amine III-3 with carboxylic acid II-1 gave the corresponding amide III-4, and Boc-deprotection with trifluoroacetic acid afforded 6 in good yields.
Scheme 3. Synthesis of 6 and 7.

Reagents and conditions: (a) Boc-l-valine, BOP, Et3N, THF, rt, 2 h, 97%; (b) TFA, CH2Cl2, rt, 1 h, 91%; (c) BH3·SMe2, THF(ah), reflux, 4 h, 71%; (d) II-1 (for III-4) or II-2 (for III-5), BOP, Et3N, THF, rt, 3–4 h, 67–93%; (e) TFA, CH2Cl2, rt, 1.5–2 h, 56–57%.
Des-hydroxyl analogues 7 and 8 (Schemes 3 and 4) were synthesized using the same strategy as that used for compound 6 (AT-076). Compound 7, lacking the 7-hydroxyl group of the Tic moiety (Scheme 3), was prepared from common intermediate III-3 by BOP-coupling with carboxylic acid II-2, followed by trifluoroacetic acid deprotection of the Boc group. Compound 8, lacking the 3-hydroxyl group on the phenylpiperidine (Scheme 4), was prepared by BOP-coupling of 4-phenylpiperidine IV-1 and Boc-l-valine to give amide IV-2, followed by trifluoroacetic acid deprotection and subsequent borane reduction to give amine IV-3. BOP-assisted coupling of amine IV-3 with carboxylic acid II-1 afforded amide IV-4, followed by deprotection with hydrochloric acid to give 8.
Scheme 4. Synthesis of 8.

Reagents and conditions: (a) Boc-l-valine, BOP, Et3N, THF, rt, 20 h, 85%; (b) TFA, CH2Cl2, rt, 1.5 h, quantitative; (c) BH3·SMe2, THF(ah), reflux, 3.5 h, 73%; (d) II-1, BOP, Et3N, THF, rt, 4 h, quantitative; (e) HCl/ether, ether/MeOH, rt, 5 h, 87%.
In Vitro Pharmacology
The binding affinities of 1–8 for the opioid receptors were determined by radioligand competition experiments, using [3H]N/OFQ for the NOP receptor, [3H]U69,593 for KOP, [3H]DAMGO for MOP, and [3H]Cl-DPDPE for the DOP receptor, as radioligands (Table 1) in receptor-transfected CHO cells, as we have reported previously15 and described in detail.16
The intrinsic activity of the compounds alone was determined by their ability to stimulate [35S]GTPγS binding to cell membranes and compared to the standard agonists N/OFQ (NOP), DAMGO (MOP), U69,593 (KOP), and DPDPE (DOP) (Table 1), as reported previously.15,16b,17
The functional antagonism of the compounds was determined by the ability of the compounds to inhibit agonist-stimulated [35S]GTPγS binding in a dose-dependent fashion. Agonist concentration–response curves were run in the presence of a series of single concentrations of the test compound. The nature of the antagonism at each of the opioid receptors was determined by Schild regression analysis of the data. In the case where the antagonist induced parallel shifts of the agonist concentration–response curves without suppressing the maximum agonist response, the antagonist potency was calculated as the Ke (nM), using the formula
where [L] is the antagonist (test compound) concentration and DR is the dose ratio of the agonist EC50 in the presence and absence of the antagonist.18
In cases where the test compound caused nonparallel shifts of the agonist concentration–response curves and decreased the maximum agonist response, the experiments were carried out with at least five to eight different test compound concentrations to determine the nature of antagonism, according to the principles of insurmountable antagonism described by Kenakin et al.19 The antagonist potency was calculated as the Ke, based on the nature of the concentration–response curves.
Results and Discussion
The compounds were first tested for their binding affinity at the four opioid receptors. JDTic and the NOP antagonist SB-61211110 were also tested for comparison. Consistent with previous reports, JDTic showed a potent nanomolar binding affinity Ki of 0.43 and 1.53 nM at KOP and MOP, respectively, and slightly lower, but still potent binding affinity of 10.6 and 16.7 nM at DOP and NOP, respectively (Table 1).4b,5 Compounds that showed a binding affinity Ki <100 nM were then tested for their intrinsic activity alone in the [35S]GTPγS binding assay at a range of concentrations up to the highest concentration of 10 μM. As expected, JDTic had no intrinsic activity at any of the opioid receptors. The relatively potent binding affinity of JDTic at the NOP receptor was quite surprising, given that JDTic is an atypical ligand for NOP, compared to the usual piperidine-based NOP ligands (Figure 1).20 Exploration of the molecular features of JDTic revealed some interesting SAR.
Replacing the tetrahydroisoquinoline (Tic–OH) and valine-amide N-substituent of JDTic with lipophilic alicyclic groups typically found in NOP ligands, such as the 4-isopropylcyclohexyl (1, 2) and cyclooctyl (3), completely abolished NOP binding affinity and significantly suppressed affinities at MOP, DOP, and KOP. It is interesting to note that the trans-4-isopropylcyclohexyl-containing compound 2 (but not the cis compound 1) has reasonably potent affinity at MOP (Ki = 6 nM), but not at any other opioid receptor. The complete lack of binding affinity of 1–3 at NOP suggests that the trans-3R,4R-dimethylpiperidine-containing ligands are not able to bind to the NOP binding pocket in a manner similar to other piperidine-based NOP ligands not containing the 3R,4R-dimethyl groups (compare with SB-612111 and SR16430, Figure 1). Even with the 7-hydroxy-1,2,3,4-tetrahydroisoquinoline (Tic–OH) reintroduced at the N-substituent, albeit without the 2-isopropylethylamide linkage (as in 4), the compound showed no affinity at NOP and a significant loss of affinity at the other opioid receptors (Table 1). Interestingly, however, the compound without the 7-hydroxy group on the Tic moiety (5) showed a significant increase in binding affinity at all four opioid receptors, particularly at MOP, where it has a potent 1 nM binding affinity. The dramatic increase in NOP affinity with this simple chemical change compared with that of compound 4 suggests that the trans-3R,4R-dimethylpiperidine scaffold (present in 1–5 and in JDTic) must have a specific fit and interactions in the NOP binding pocket, making compounds containing this scaffold very sensitive to structure modifications affecting their NOP binding affinity. Compound 5, however, has no intrinsic activity at any of the opioid receptors (Table 2), suggesting that 5 may be a reasonably selective MOP antagonist.
Table 2. Inhibition of [35S]GTPγS Binding by JDTic Analogues at the Four Opioid Receptors, Determined in Cloned Human Receptor-Transfected Cells.
| functional
activity |
||||||||
|---|---|---|---|---|---|---|---|---|
| [35S]GTPγS NOP |
[35S]GTPγS KOP |
[35S]GTPγS MOP |
[35S]GTPγS DOP |
|||||
| EC50 (nM) | % stim | EC50 (nM) | % stim | EC50 (nM) | % stim | EC50 (nM) | % stim | |
| JDTic | >10K | 0 | >10K | 0 | >10K | 0 | >10K | 0 |
| 1 | NDa | >10K | 0 | >10K | 0 | ND | ||
| 2 | ND | >10K | 0 | >10K | 0 | ND | ||
| 3 | ND | >10K | 0 | ND | ND | |||
| 4 | ND | ND | >10K | 0 | ND | |||
| 5 | >10K | 0 | >10K | 0 | >10K | 0 | >10K | 0 |
| 6 | >10K | 0 | >10K | 0 | >10K | 0 | >10K | 0 |
| 7 | >10K | 7.0 ± 3.2 | >10K | 0 | ND | ND | ||
| 8 | 53.3 ± 12.12 | 18.3 ± 1.3 | 408 ± 41 | 72.3 ± 4 | >10K | 0 | ND | |
| SB-612111 | >10K | 0 | ND | ND | ND | |||
ND = Compounds with binding affinity Ki >100 nM were not tested in functional assays.
The effect of the trans-3R,4R-dimethyl groups on the piperidine was even more evident from the SAR of compound 6 (AT-076). Removal of the trans-3R,4R-dimethyl groups, leaving the rest of the JDTic molecule intact, resulted in compound 6 (AT-076) which showed a significant (∼10-fold) increase in NOP binding affinity (Ki 1.75 nM) compared with JDTic itself, equipotent affinity at MOP and KOP compared with JDTic, and only a slight decrease in affinity at DOP (Table 1). Overall, compound 6 (AT-076) has potent nanomolar binding affinity at all four opioid receptors, comparable to JDTic at KOP, MOP, and DOP, and 10-fold higher affinity at NOP. Compound 6 (AT-076) shows no intrinsic (agonist) activity at any of the opioid receptors in the [35S]GTPγS functional assay (Table 2). The potent binding affinity and lack of agonist activity of 6 (AT-076) compared to JDTic indicates that the trans-3R,4R-dimethyl groups on the piperidine in JDTic are not indispensable for its antagonist activity, as previously thought. Although the trans-3R,4R-dimethylpiperidine has been long considered an opioid “antagonist” pharmacophore,1a,21 removal of the 3- or 4-methyl groups, or both, has been shown to afford opioid antagonists (Kormos et al.11 and this work). For the NOP receptor, the SAR of compounds 1–6 compared to JDTic suggests that the trans-3R,4R-dimethyl groups likely lead to a specific but not optimum binding fit at the NOP receptor, and removal of these groups results in a 10-fold improvement in binding affinity at NOP, without negatively affecting the affinity at the other opioid receptors, making compound 6 (AT-076) a potent, nanomolar affinity ligand at all four opioid receptors.
Interestingly, removal of the 7-hydroxyl group of the Tic moiety of compound 6 (AT-076), as in 7, resulted in a significant drop (20–70-fold) in binding affinity at all opioid receptors, unlike the effect of this same structural change in compound 4 (to compound 5), which increased its binding affinity significantly, particularly at NOP (compare 4 and 5, Table 1). Removal of the 3-hydroxyl group on the phenylpiperidine of compound 6 (AT-076), on the other hand, showed only a small drop in binding affinity (compound 8, Table 1), but resulted in an interesting increase in intrinsic (agonist) activity at the NOP and KOP receptors (Table 2). This appears to suggest that the phenolic 3-OH group on the phenylpiperidine in 6 (AT-076) and in JDTic may participate in binding interactions in NOP and KOP that stabilize the receptors in an inactive conformation. JDTic, bound in the KOP crystal structure 4DJH, does not show direct hydrogen-bond interactions of the 3-phenolic hydroxy group with the receptor, but does show a structured water molecule-mediated polar interaction with C210 and W124.22
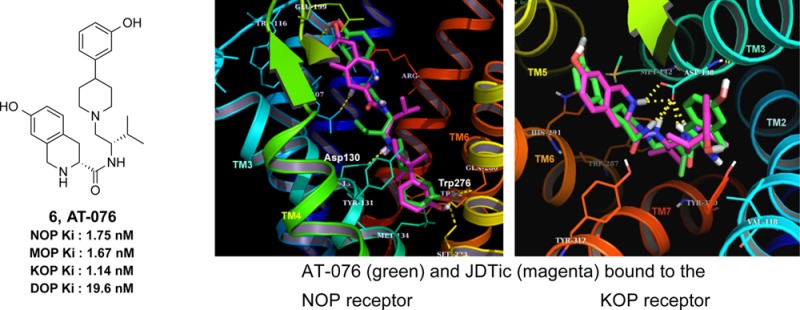
We carried out molecular docking of JDTic and AT-076 into the NOP crystal structure 4EA3 (bound to the NOP antagonist ligand C-2423) using Surflex Dock (Tripos SYBYL X1.2) and compared it to the docked pose of JDTic in the KOP crystal structure 4DJH (Figure 3). Unlike its V-shaped bound orientation in the KOP crystal structure, JDTic binds to NOP in an extended conformation, similar to the binding orientation of the NOP antagonist C-24 (Figure 3A). The 3-OH-containing phenylpiperidine moiety is buried deep inside the binding pocket of NOP, where the piperidine nitrogen makes the classic ionic interaction with the conserved Asp130 of TM3. Another distinctive difference between JDTic’s binding to NOP versus its interaction with KOP is that, in the NOP receptor, only the piperidine nitrogen of JDTic (but not the isoquinoline nitrogen) makes an ionic interaction with the conserved Asp1303.32 (superscript denotes Ballesteros-Weinstein numbering) (Figure 3A), whereas in KOP both the piperidine nitrogen as well as the isoquinoline Tic-nitrogen interact in a bidentate ionic interaction with the conserved Asp1383.32 (Figure 3C). This lack of an additional ionic interaction in NOP may contribute to JDTic’s 10-fold lower binding affinity compared to that at KOP. The 3,4-desmethyl JDTic analogue 6 (AT-076) was also docked into NOP (Figure 3B) and found to bind in an extended orientation similar to and superimposable with JDTic. On the other hand, at the KOP receptor, the top-scoring AT-076 docking pose showed that while it binds in a V-shaped orientation similar to JDTic’s bound conformation in KOP, the phenylpiperidine and isoquinoline rings of AT-076 are oriented opposite to those of JDTic (Figure 3C). The protonated nitrogens of the phenylpiperidine and isoquinoline groups of AT-076 still appear to interact with the Asp138 in KOP, as seen with JDTic.
Figure 3.

(A) Binding model of JDTic in the NOP receptor crystal structure 4EA3. The TM helices are color-coded and annotated. The NOP antagonist C-24 bound in the NOP crystal structure is shown in cyan, whereas JDTic is in magenta. Key interacting amino acids are as indicated. (B) Binding orientation of opioid pan antagonist AT-076 in the NOP crystal structure. AT-076 (green) is superimposed on JDTic (magenta) in the NOP binding pocket. Both ligands bind in an extended conformation in the NOP receptor. (C) Binding model of AT-076 in the KOP receptor crystal structure 4DJH. AT-076 (green) is superimposed on the bound orientation of JDTic (magenta) in the KOP crystal structure. Note that AT-076 binds KOP in a similar V-shaped orientation as JDTic, but with the phenylpiperidine and isoquinoline moieties oriented exactly opposite to those of JDTic.
Since 6 (AT-076) had potent binding affinity at all four opioid receptors and no intrinsic activity, its antagonist potency and nature of antagonism were determined from the inhibition of agonist-stimulated [35S]GTPγS binding curves (Figure 4) in the presence of different concentrations of AT-076, using Schild analysis. At MOP and DOP, AT-076 caused a significant rightward shift of the concentration–response curve of agonist DAMGO and DPDPE, respectively, without reducing Emax, indicating competitive antagonism (Figures 4A and B). The antagonist potency, calculated as the Ke value (similar to what is reported for JDTic and analogues) was 0.58 ± 0.28 and 24.95 ± 13 nM for MOP and DOP, respectively. Note that the Ke values for potency at MOP and DOP are comparable to their binding affinity Ki (1.67 nM for MOP and 19.6 nM for DOP).
Figure 4.

Concentration–response curves of agonist-stimulated [35S]GTPγS binding in the presence of indicated concentrations of AT-076 at the mu (A), delta (B), kappa (C), and nociceptin (D) opioid receptors and JDTic at the NOP receptor (E). The respective agonists used were as follows: nociceptin, GGFTGARKSARKLANQ for NOP; U69,593, (5a,7a,8b)-(−)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]benzeneacetamide for KOP; DAMGO, [d-Ala2, NMe-Phe4, Gly-ol5]-enkephalin for MOP; DPDPE, [d-Pen2,d-Pen5]enkephalin for DOP. For calculation of the Ke values, the experimentally determined EC50 values of the subtype-specific agonists are as follows: nociceptin, 2.76 nM; U69,593, 26.87 nM; DAMGO, 19.78 nM; DPDPE, 4.65 nM.
At KOP and NOP, however, AT-076 at increasing concentrations not only caused a decrease in agonist EC50, but also a reduction in Emax of the agonist–response curve, producing an insurmountable antagonism of the agonist response (Figure 4C and D). Similarly, JDTic at the NOP receptor also caused a reduction in the Emax of the agonist (nociceptin) concentration–response curve and a rightward shift of the agonist EC50 at increasing concentrations. The reduction in maximum agonist response at increasing antagonist concentrations is suggestive of a noncompetitive antagonist mechanism. As put forth by Kenakin et al.,19 such insurmountable antagonism is suggested to result either from a slow offset orthosteric antagonist or due to allosteric modulation of the receptor.19 Given that AT-076 has potent nanomolar binding affinity at both NOP and KOP in radioligand displacement assays using orthosteric agonists, it appears that the former is likely the case, rather than allosteric modulation. As described by Kenakin et al,19 in such cases, the antagonist potency can be determined from these experiments as an apparent Ke value. Using these principles, the antagonist potency for AT-076 at KOP was calculated as Ke = 4.30 nM, and at NOP as Ke = 30 nM. The antagonist Ke value for JDTic at the NOP receptor was calculated to be 680 nM. This weaker antagonist potency of JDTic at NOP, compared with the high binding affinity Ki = 16.7 nM, suggests noncompetitive antagonism and is similar to the results observed by Munro et al.5 for JDTic in the cAMP functional assay at NOP.
Although the antagonist potency of AT-076 at NOP and KOP appeared to be lower than that suggested by its potent binding affinity Ki of 1.75 and 1.14 nM, respectively, AT-076 suppresses maximum agonist response even at the lowest concentration tested (2 nM, Figure 4C and D), indicating that it is a potent antagonist at these receptors. Regardless of the precise molecular mechanism of antagonism, which would require further detailed experimental analysis, AT-076 is a high-affinity pan antagonist at all four opioid receptors. The SAR revealed in this study show that the 3,4-dimethyl groups on the classic opioid antagonist pharmacophore in JDTic are dispensable, while still maintaining the ‘antagonist’ activity at the opioid receptors, but likely contribute to the unusual antagonist profile and pseudoirreversible nature of JDTic’s antagonist activity at the KOP receptor.4,24
In conclusion, simple SAR studies on JDTic’s affinity for the NOP receptor resulted in compound 6 (AT-076), which has higher binding affinity than JDTic at NOP and equally high affinity at the other three opioid receptors, resulting in an opioid ligand which binds well to all the four opioid receptors. It is well-known that even though the NOP receptor belongs in the opioid receptor family and shares a high degree of homology with the other opioid receptors, it has low or no affinity for known opioid ligands.25 Site-directed mutagenesis studies26 and the recent opioid receptor crystal structures22,23,27 suggest residues in the NOP binding pocket that exclude the binding of opioid ligands to the NOP receptor. A pan opioid ligand like AT-076, which binds with high nanomolar affinity to all four opioid receptors, would therefore be a useful tool to investigate a common opioid-binding pharmacophore, through future crystal structure determinations with all four opioid receptors. Our own molecular docking analysis with AT-076 showed that it bound to the kappa opioid receptor in an orientation exactly opposite that of the kappa-selective ligand JDTic, while still binding to the same kappa binding pocket (Figure 3C). Structure-based studies of such pan opioid ligands at the four opioid receptors may likely lead to useful insights for the rational design of selective or designed multifunctional opioid ligands based on this common opioid pharmacophore.
Table 3. Inhibition of Agonist-Stimulated [35S]GTPγS Binding by AT-076 in Cloned Human NOP, KOP, MOP, and DOP Receptorsa.
|
Ke (nM) |
||||
|---|---|---|---|---|
| NOPb | KOPc | MOPd | DOPe | |
| JDTic | 680.15 ± 2.35 | 0.01 ± 0.00f | 3.41 ± 0.36f | 79.3 ± 9.3f |
| 6 (AT-076) | 30.05 ± 21.85 | 4.3 ± 1.86 | 0.58 ± 0.28 | 24.95 ± 13.03 |
For calculation of the Ke values, the experimentally determined EC50 values of the subtype-specific agonists are as follows: nociceptin, 2.76 nM; U69,593, 26.87 nM; DAMGO, 19.78 nM; DPDPE, 4.65 nM.
Agonist used is nociceptin for NOP.
Agonist used is U69,593, (5a,7a,8b)-(−)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4,5]dec-8-yl]benzeneacetamide, for KOP.
Agonist used is DAMGO, [d-Ala2, NMe-Phe4, Gly-ol5]-enkephalin, for MOP.
Agonist used is DPDPE, [d-Pen2,d-Pen5]enkephalin, for DOP.
Data taken from ref (4b).
Methods
Thin layer chromatography was performed on Analtech silica gel GF 250 μm TLC plates. The plates were visualized with a 254 nM UV light and staining with iodine. Flash chromatography was carried out on F60 silica gel, 230–400 mesh, 60 Å (Silicycle SiliaFlash). NMR was recorded on a Varian Mercury Plus NMR (300 MHz) using CDCl3 or MeOD-d6. Mass spectra were obtained on a LCQ Fleet Ion Trap LC/MSn, a micromass ZMD 1000 or PE Sciex API 150EX LC/MS using electrospray ionization (ESI) mode. Elemental analyses were performed by Atlantic Microlabs, Norcross, GA. HRMS analyses were performed by the Mass Spectrometry Service Laboratory, University of Minnesota Department of Chemistry, Minneapolis, MN on a Bruker BioTOF II HRMS using ESI mode. HPLC analysis was performed on a reverse phase Agilent Zorbax SB-Phenyl column (5 μM, 2.1 × 150 mm), using a binary gradient of 95/5 solvent A (95/5 H2O/ACN + 0.1% formic acid)/solvent B (5/95 H2O/ACN + 0.1% formic acid) → 0/100 for 10 min, at a flow rate of 0.4 mL/min. Eluted peaks were monitored at 254 nm with a Shimadzu SPD-10AVP UV–vis detector. All final compounds tested were confirmed to be of >95% purity by the HPLC method described above.
3-((3R,4R)-1-(4-Isopropylcyclohexyl)-3,4-dimethylpiperidin-4-yl)phenol (1, cis diastereomer) and (2, trans diastereomer)
To a suspension of (3R,4R)-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine I-1 (125 mg, 0.609 mmol, 1.00 equiv) in 1,2-dichloroethane (DCE) (6.00 mL) was added 4-isopropylcyclohexanone (0.150 mL, 0.974 mmol, 1.60 equiv), MgSO4 (125 mg), and HOAc (0.066 mL, 1.15 mmol, 1.89 equiv), and the mixture was stirred at room temperature for 1 h. NaBH(OAc)3 (244 mg, 1.15 mmol, 1.89 equiv) was then added, and the reaction was stirred at room temperature for 24 h. The reaction was diluted with CH2Cl2 and saturated NaHCO3 (aq). The layers were separated, and the aqueous solution was extracted 2× with CH2Cl2. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography using hexanes/EtOAc 90/10 → 87/13, then hexanes/EtOAc/NH4OH 87/13/0.5 → 80/20/0.5, as the eluent, to afford separated diastereomers 1 (100 mg, 50%) and 2 (40 mg, 20%). These diastereomers were converted to their HCl salts by addition of 2 M HCl in ether. For 1: 1H NMR (HCl salt, 300 MHz, MeOD-d6) δ 7.15–7.19 (1H, m), 6.73–6.78 (2H, m), 6.65–6.67 (1H, m), 3.37–3.52 (3H, m), 2.40–2.48 (1H, m), 1.94–2.09 (5H, m), 1.63–1.82 (4H, m), 1.53 (2H, t, J = 10.7 Hz), 1.43 (3H, s), 1.22–1.29 (3H, m), 0.94–0.97 (6H, m), 0.83 (3H, d, J = 5.7 Hz). MS (ESI) 330.3 m/z (M + H)+. Anal. Calcd for C22H35NO·1.0HCl·0.5H2O: C, 70.47; H, 9.95; N, 3.74. Found: C, 70.56; H, 10.01; N, 3.51. For 2: 1H NMR (HCl salt, 300 MHz, MeOD-d6) δ 7.13–7.19 (1H, m), 6.63–6.78 (3H, m), 3.35–3.52 (3H, m), 2.39–2.53 (2H, m), 2.27–2.31 (1H, m), 2.10–2.20 (2H, m), 1.90–1.98 (4H, m), 1.47–1.68 (3H, m), 1.42 (3H, s), 1.11–1.23 (3H, m), 0.91 (6H, d, J = 6 Hz), 0.82 (3H, d, J = 6 Hz). MS (ESI) 330.3 m/z (M + H)+. Anal. Calcd for C22H35NO·1.00HCl·0.6H2O·0.5diethyl ether: C, 69.65; H, 10.28; N, 3.38. Found: C, 69.63; H, 10.07; N, 3.13.
3-((3R,4R)-1-Cyclooctyl-3,4-dimethylpiperidin-4-yl)phenol (3)
A solution of (3R,4R)-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine I-1 (1.00 g, 4.87 mmol, 1.00 equiv) in tetrahydrofuran (THF) (20.0 mL) was treated with cyclooctanone (1.77 g, 14.0 mmol, 2.88 equiv) and Ti(OiPr)4 (3.68 mL, 12.4 mmol, 2.55 equiv), and the mixture was stirred at 40 °C for 21 h. NaBH(OAc)3 (2.97 g, 14.0 mmol, 2.88 equiv) was added, and the reaction was stirred at 40 °C for 1 h and then allowed to cool to room temperature. MeOH (3.00 mL) was added, and the solution was stirred for 1 h at room temperature and then diluted with EtOAc and saturated NaHCO3 (aq). The layers were separated, and the aqueous solution was extracted 2× with EtOAc. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography using DCM/EtOAc 100/0 → 50/50 as the eluent to afford 3 in 71% yield. 1H NMR (HCl salt, 300 MHz, MeOD-d6) δ 7.17 (1H, t, J = 6 Hz), 6.72–6.78 (2H, m), 6.65–6.67 (1H, m), 3.36–3.53 (4H, m), 2.43–2.48 (3H, m), 2.09 (2H, br s), 1.91–1.97 (5H, m), 1.51–1.72 (8H, m), 1.44 (3H, s), 0.82 (3H, d, J = 5.4 Hz). MS (ESI) 316.3 m/z (M + H)+. Anal. Calcd for C21H33NO·1.0HCl·0.9H2O: C, 68.51; H, 9.80; N, 3.80. Found: C, 68.51; H, 10.02; N, 3.72.
General Procedure for the Preparation of 4 and 5
To an ice-chilled solution of (3R,4R)-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine I-1 (1.00 equiv) in ACN (0.16 M) was added the appropriate carboxylic acid (1.20 equiv), HATU (1.20 equiv), and Et3N (2.50 equiv), and the reaction was stirred at room temperature for 21 h. The solution was diluted with EtOAc and H2O. The layers were separated, and the aqueous solution was extracted 2× with EtOAc. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated. In the case of II-4, LiOH (19 mg) was added to a solution of the crude material in MeOH (6.00 mL), and the reaction was stirred at room temperature for 3 h in order to hydrolyze the overcoupled product. The solution was diluted with EtOAc and saturated NH4Cl(aq). The layers were separated, and the aqueous solution was extracted 2× with EtOAc. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated. The crude residue was purified via flash chromatography (for II-3: hexane/DCM/EtOAc 50/50/0→ 35/35/30; for II-4: hexane/EtOAc 100/0 → 20/80) to afford the desired material, which was used directly in the next reaction.
The latter coupled product (1.00 equiv) was dissolved in a 1/1 v/v CH2Cl2/trifluoroacetic acid (TFA) (0.14 M) and stirred at room temperature for 2 h. The reaction was concentrated to dryness, then partitioned between EtOAc and saturated NaHCO3(aq). The layers were separated, and the aqueous solution was extracted 2× with EtOAc. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated to afford the deprotected material, which was used directly in the next reaction.
Borane dimethyl sulfide complex, 10 M (12.5–25 equiv), was added to a stirred solution of the latter deprotected product (1.00 equiv) in THF(ah) (0.07 M) under Ar(g) at −40 °C. The reaction was heated to reflux for 5 h. The mixture was then transferred to a 1 L flask and cooled in an ice bath. MeOH was added cautiously (note: vigorous bubbling), and the mixture was stirred at room temperature for 1 h, followed by addition of 2 M HCl/ether and stirring at room temperature for 0.5 h. The solution was concentrated to dryness, then partitioned between CH2Cl2/THF 3/1 and saturated NaHCO3(aq). Solid NaCl was added. The layers were separated, and the aqueous solution was extracted 3× with CH2Cl2/THF 3/1. The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified via flash chromatography (for 4: CH2Cl2/iPrOH 100/0 → 87/13; for 5: CH2Cl2/MeOH 100/0 → 91/9) to afford the desired materials, which were converted to their HCl salts by addition of 2 M HCl/ether.
(R)-3-(((3R,4R)-4-(3-Hydroxyphenyl)-3,4-dimethylpiperidin-1-yl)methyl)-1,2,3,4-tetrahydroisoquinolin-7-ol (4)
Prepared from the general procedure using (3R,4R)-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine I-1 (400 mg, 1.95 mmol) and (3R)-2-(tert-butoxycarbonyl)-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid II-1 to yield 100 mg of 4 in 14% overall yield. 1H NMR (HCl salt, 300 MHz, MeOD-d6) δ 7.05–7.14 (2H, m), 6.73–6.81 (3H, m), 6.59–6.64 (2H, m), 4.23–4.36 (2H, m), 3.80 (1H, br s), 3.05–3.09 (2H, m), 2.92 (1H, d, J = 8.4 Hz), 2.58–2.75 (4H, m), 2.45 (2H, br s), 2.09 (1 H, br s), 1.66 (1H, br s), 1.34 (3H, s), 0.88 (3H, d, J = 5.1 Hz). MS (ESI) 367.2 m/z (M + H)+. Anal. Calcd for C23H30N2O2·1.5HCl·0.7diethyl ether: C, 65.50; H, 8.20; N, 5.92. Found: C, 65.34; H, 8.03; N, 5.92.
3-((3R,4R)-3,4-Dimethyl-1-(((R)-1,2,3,4-tetrahydroisoquinolin-3-yl)methyl)piperidin-4-yl)phenol (5)
Prepared from the general procedure using (3R,4R)-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine I-1 (225 mg, 1.10 mmol) and (3R)-2-(tert-butoxycarbonyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid II-2 to yield 170 mg of 5 in 47% overall yield. 1H NMR (HCl salt, 300 MHz, MeOD-d6) δ 7.29–7.33 (4H, m), 7.18 (1H, t, J = 6 Hz), 6.66–6.80 (3H, m), 4.48–4.61 (2H, m), 4.37 (1H, br s), 3.98 (1H, br s), 3.67–3.69 (3H, m), 3.52 (1H, br s), 3.24 (2H, br s), 3.12 (1H, br s), 2.73 (1H, br s), 2.43 (1H, br s), 1.96 (1H, br s), 1.48 (3H, s), 0.98 (app s, 3H). MS (ESI) 351.1 m/z (M + H)+. Anal. Calcd for C23H30N2O·2.00HCl·1.3H2O: C, 61.82; H, 7.81; N, 6.27. Found: C, 61.89; H, 7.82; N, 6.05.
tert-Butyl (S)-(1-(4-(3-Hydroxyphenyl)piperidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate (III-2)
To a mixture of 4-(3-hydroxyphenyl)piperidine III-1 (3.00 g, 16.9 mmol, 1.00 equiv) in THF (170 mL) was added Boc-l-valine (3.68 g, 16.9 mmol, 1.00 equiv), BOP (7.49 g, 16.9 mmol, 1.00 equiv), and Et3N (5.19 mL, 16.9 mmol, 2.20 equiv), and the reaction was stirred for 2 h at room temperature. The reaction was diluted with CH2Cl2 and saturated NaHCO3(aq). The layers were separated, and the aqueous solution was extracted 3× with CH2Cl2. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography using hexane/EtOAc 90/10 → 20/80 as the eluent, to afford 6.20 g of III-2 in 97% yield. 1H NMR (300 MHz, MeOD-d6) δ 7.09 (1H, sep, J = 3.3 Hz), 6.60–6.70 (3H, m), 4.66 (1H, d, J = 12.4 Hz), 4.40 (1H, d, J = 5.4 Hz), 4.24 (1H, d, J = 10.2 Hz), 3.16–3.26 (1H, m), 2.72–2.78 (2H, m), 1.83–2.04 (3H, m), 1.70–1.72 (1H, m), 1.53–1.59 (1H, m), 1.46 (s, 9H), 0.92–1.00 (6H, m). MS (ESI) m/z 377.0 (M + H)+.
(S)-3-(1-(2-Amino-3-methylbutyl)piperidin-4-yl)phenol (III-3)
Intermediate III-2 (6.20 g, 16.5 mmol, 1.00 equiv) was dissolved in 25.0 mL of CH2Cl2, cooled in an ice bath, and 25.0 mL of TFA was added. The mixture was stirred in an ice bath for 0.5 h and then allowed to warm to room temperature for 0.5 h. The solution was concentrated and then partitioned between CH2Cl2/THF 3/1 and saturated NaHCO3(aq). Solid NaCl was added. The layers were separated, and the aqueous solution was extracted 3× with CH2Cl2/THF 3/1. The combined organic layers were washed with satd. NaCl(aq), dried over Na2SO4, filtered, and concentrated. Additional recovery was performed by concentration of the aqueous layers, and trituration with CH2Cl2 to afford 4.16 g of deprotected product in 91% yield. 1H NMR (300 MHz, MeOD-d6) δ 7.07–7.12 (1H, m), 6.61–6.71 (3H, m), 4.70 (1H, d, J = 9.9 Hz), 4.11 (1H, d, J = 10.2 Hz), 3.71 (1H, dd, J = 19.8, 3.9 Hz), 3.20–3.25 (1H, m), 2.72–2.79 (2H, m), 1.84–2.16 (3H, m), 1.53–1.71 (2H, m), 0.91–1.04 (6H, m). MS (ESI) m/z 277.2 (M + H)+.
The latter deprotected product (2.00 g, 7.24 mmol, 1.00 equiv) was dissolved in THF(ah) (90.0 mL) under Ar(g), and cooled to −40 °C. Borane·dimethyl sulfide, 10 M (7.24 mL, 10.0 equiv) was added, and the reaction was heated to reflux for 4 h. The mixture was transferred to a 1L flask, and cooled in an ice bath. MeOH (80.0 mL) was added cautiously (note: vigorous bubbling), and the mixture was stirred at room temperature for 1 h, followed by addition of 2 M HCl/ether (15.0 mL) and stirring at room temperature for 0.5 h. The solution was concentrated to dryness, then partitioned between CH2Cl2/THF 3/1 and satd. NaHCO3(aq). Solid NaCl was added. The layers were separated, and the aqueous solution was extracted 3X with CH2Cl2/THF 3/1. The combined organic layers were dried over Na2SO4, filtered and concentrated. The borane reduction was repeated on a 2.14 g scale and combined with the 2.00 g scale reaction as described above. The crude was purified via flash chromatography using CH2Cl2/MeOH 100/0 → 80/20 as the eluent, then CH2Cl2/MeOH/NH4OH(aq) 80/20/0.5 → 70/30/0.5 to afford 2.78 g of III-3 in 71% yield. 1H NMR (300 MHz, MeOD-d6) δ 7.08 (1H, t, J = 6 Hz), 6.67–6.71 (2H, m), 6.60 (1H, dd, J = 5.7, 1.2 Hz), 3.11 (1H, d, J = 8.4 Hz), 2.93 (1H, d, J = 7.8 Hz), 2.83–2.88 (1H, m), 2.42–2.46 (2H, m), 2.28–2.35 (2H, m), 1.99 (1H, dt, J = 6.9,1.8 Hz), 1.78–1.82 (3H, m), 1.66–1.75 (2H, m), 0.96–1.03 (6H, m). MS(ESI) m/z 263.1 (M+H)+.
General Procedure for the Preparation of III-4 and III-5
To a stirred solution of intermediate III-3 (1.00 equiv) in THF (0.1 M) was added the appropriate carboxylic acid (1.20 equiv), BOP (1.20 equiv) and Et3N (5.00 equiv), and the reaction was stirred at room temperature for 3–4 h. The reaction was diluted with EtOAc and satd. NaHCO3(aq). The layers were separated, and the aqueous solution was extracted 2× with EtOAc. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered and concentrated. The crude residue was purified via flash chromatography (for III-4: CH2Cl2/iPrOH/NH4OH(aq) 100/0/0 → 90/10/0.5, for III-5: CH2Cl2/iPrOH 100/0 → 93/7) to afford the desired material.
tert-Butyl (R)-7-Hydroxy-3-(((S)-1-(4-(3-hydroxyphenyl)piperidin-1-yl)-3-methylbutan-2-yl)carbamoyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (III-4)
Prepared from the general procedure using intemediate III-3 (750 mg, 2.85 mmol) and (3R)-2-(tert-butoxycarbonyl)-7-hydroxy-1, 2, 3,4-tetrahydroisoquinoline-3-carboxylic acid II-1 (1.00 g, 3.43 mmol) to afford 1.52 g of III-4 in 93% yield. 1H NMR (300 MHz, MeOD-d6) δ 7.08 (1H, t, J = 5.7 Hz), 6.97 (1H, d, J = 6 Hz), 6.58–6.68 (5H, m), 4.72 (1H, s), 4.51–4.54 (2H, m), 3.84 (1H, s), 3.03–3.15 (2H, m), 2.79 (2H, s), 2.35–2.46 (3H, m), 1.94–2.03 (2H, m), 1.60–1.80 (5H, m), 1.50–1.52 (9H, m), 0.825 (6H, s). MS (ESI) m/z 538.2 (M + H)+.
tert-Butyl (R)-3-(((S)-1-(4-(3-Hydroxyphenyl)piperidin-1-yl)-3-methylbutan-2-yl)carbamoyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (III-5)
Prepared from the general procedure using intemediate III-3 (150 mg, 0.572 mmol) and (3R)-2-(tert-butoxycarbonyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid II-2 (190 mg, 0.686 mmol) to afford 200 mg of III-5 in 67% yield. 1H NMR (300 MHz, CDCl3) δ 7.14–7.23 (5H, m), 6.65–6.75 (3H, m), 4.69–4.73 (2H, m), 4.49 (1H, d, J = 11.7 Hz), 3.99 (1H, s), 3.74 (1H, s), 3.16–3.23 (6H, m), 2.88 (2H, d, J = 33.3 Hz), 2.64 (1H, s), 1.70–1.87 (5H, m), 1.50 (9H, s), 1.37 (1H, t, J = 5.4 Hz), 0.87 (6H, br s). MS (ESI) m/z 522.3 (M + H)+.
(R)-7-Hydroxy-N-((S)-1-(4-(3-hydroxyphenyl)piperidin-1-yl)-3-methylbutan-2-yl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (6, AT-076)
Intermediate III-4 (1.52 g, 2.83 mmol, 1.00 equiv) was dissolved in CH2Cl2 (76.0 mL) and cooled in an ice bath. TFA (41.0 mL) was added, and the reaction was stirred at room temperature for 1.5 h. The solution was concentrated to dryness and then partitioned between CH2Cl2/THF 3/1 and saturated NaHCO3(aq). The layers were separated, and the aqueous solution was extracted 3× with CH2Cl2/THF 3/1. The combined organic layers were washed with minimal saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated. Additional recovery was performed by concentration of the aqueous layers, and trituration with CH2Cl2/THF 3/1. The crude residue was purified via flash chromatography using CH2Cl2/MeOH/NH4OH(aq) 100/0/0 → 93/7/0.5 as the eluent to afford 690 mg of 6 in 56% yield, which was converted to the HCl salt by addition of 2 M HCl/ether. 1H NMR (300 MHz, MeOD-d6) δ 7.08 (1H, t, J = 5.7 Hz), 6.95 (1H, d, J = 6.3 Hz), 6.65–6.69 (2H, m), 6.58–6.62 (2H, m), 6.51 (1H, d, J = 1.8 Hz), 4.03–4.06 (1H, m), 3.92–3.99 (2H, m), 3.59 (1H, dd, J = 7.8, 3.6 Hz), 3.11–3.14 (1H, m), 2.93–3.01 (2H, m), 2.85–2.88 (1H, m), 2.51–2.52 (2H, m), 2.40–2.44 (1H, m), 2.21–2.22 (1H, m), 2.07–2.08 (1H, m), 1.72–1.86 (5H, m), 0.95 (6H, app t, J = 5.4 Hz). MS (ESI) m/z 438.4. Anal. Calcd for C26H35N3O3·2.00HCl·0.9H2O: C, 59.29; H, 7.43; N, 7.98. Found: C, 59.32; H, 7.30; N, 7.85.
(R)-N-((S)-1-(4-(3-Hydroxyphenyl)piperidin-1-yl)-3-methylbutan-2-yl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (7)
TFA (3.00 mL) was added to a solution of intermediate III-5 (170 mg, 0.326 mmol, 1.00 equiv) in CH2Cl2 (3.00 mL), and the reaction was stirred at room temperature for 2 h. The mixture was concentrated, and excess TFA was removed by azeotroping with hexane. Trituration with diethyl ether afforded 120 mg of 7 as a TFA salt in 57% yield. 1H NMR (TFA salt, 300 MHz, MeOD-d6) δ 7.29–7.30 (3H, m), 7.18–7.20 (1H, m), 7.12 (1H, t, J = 5.7 Hz), 6.65–6.74 (3H, m), 4.35–4.49 (3H, m), 3.99 (1H, br s), 3.62 (1H, t, J = 8.4 Hz), 3.40–3.44 (2H, m), 3.18–3.28 (3H, m), 3.08 (1H, br s), 2.79 (1H, br s), 2.18–2.21 (1H, m), 1.87–2.05 (4H, m), 1.31 (1H, t, J = 5.4 Hz), 1.03 (6H, app t, J = 4.8 Hz). HRMS (ESI) Calcd for C26H36N3O2 (M + H)+ 422.2802; found 422.2802. LCMS RT = 4.43 min; m/z (M + H)+ = 422.3.
tert-Butyl (S)-(3-Methyl-1-oxo-1-(4-phenylpiperidin-1-yl)butan-2-yl)carbamate (IV-2)
To a mixture of 4-phenylpiperidine IV-1 (2.50 g, 15.5 mmol, 1.00 equiv) in THF (55.0 mL) was added Boc-l-valine (3.37 g, 15.5 mmol, 1.00 equiv), BOP (6.86 g, 15.5 mmol, 1.00 equiv), and Et3N (4.75 mL, 34.1 mmol, 2.20 equiv), and the reaction was stirred for 20 h at room temperature. The reaction was diluted with CH2Cl2 and satd. NaHCO3 (aq). The layers were separated, and the aqueous solution was extracted 2X with CH2Cl2. The combined organic layers were washed with satd. NaCl(aq), dried over Na2SO4, filtered and concentrated. The residue was purified by flash chromatography using CH2Cl2/MeOH 100/0 → 95/5 as the eluent to afford 4.73 g of IV-2 in 85% yield. 1H NMR (300 MHz, MeOD-d6) δ 7.15–7.30 (5H, m), 4.67 (1H, d, J = 15 Hz), 4.40 (1H, d, J = 6 Hz), 4.24 (1H, d, J = 12 Hz), 3.21–3.25 (1H, m), 2.72–2.88 (2H, m), 1.54–2.04 (5H, m), 1.45 (9H, s), 0.92–1.00 (6H, m). MS (ESI) m/z 361.3 (M + H)+.
(S)-3-Methyl-1-(4-phenylpiperidin-1-yl)butan-2-amine (IV-3)
Intermediate IV-2 (2.48 g, 0.880 mmol, 1.00 equiv) was dissolved in 17.0 mL of CH2Cl2, cooled in an ice bath, and 17.0 mL of TFA was added. The mixture was stirred in an ice bath for 0.5 h and then allowed to warm to room temperature for 1 h. The solution was concentrated to dryness and then partitioned between CH2Cl2 and saturated NaHCO3(aq). The layers were separated, and the aqueous solution was extracted 2× with CH2Cl2. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated to afford the deprotected product in quantitative yield. 1H NMR (300 MHz, MeOD-d6) δ 7.18–7.31 (5H, m), 4.69 (1H, d, J = 12 Hz), 4.18 (1H, d, J = 6 Hz), 4.05–4.11 (1H, m), 3.23–3.27 (1H, m), 2.76–2.90 (2H, m), 2.08–2.14 (1H, m), 1.88–2.00 (2H, m), 1.50–1.65 (2H, m), 0.98–1.09 (6H, m). MS (ESI) m/z 261.3 (M + H)+.
The latter deprotected product (1.87 g, 7.18 mmol, 1.00 equiv) was dissolved in THF(ah) (15.0 mL) under Ar(g), and cooled to −40 °C. Borane·dimethyl sulfide, 10 M (7.24 mL, 10.0 equiv) was added, and the reaction was heated to reflux for 3.5 h. The mixture was transferred to a 1 L flask and then cooled in an ice bath. MeOH (80.0 mL) was added cautiously (note: vigorous bubbling), and the mixture was allowed to stir at room temperature for 1 h, followed by addition of 2 M HCl/ether (10.0 mL) and stirring at room temperature for 0.5 h. The solution was concentrated to dryness and then partitioned between CH2Cl2/THF 3/1 and saturated NaHCO3(aq). The layers were separated, and the aqueous solution was extracted 3× with CH2Cl2/THF 3/1. The combined organic layers were washed with NaCl(aq), dried over Na2SO4, filtered, and concentrated. The residue was purified via flash chromatography using CH2Cl2/MeOH 100/0 → 50/50 then CH2Cl2/MeOH/NH4OH 49.75/49.75/0.5 as the eluent to afford 1.3 g of IV-3 in 73% yield. 1H NMR (300 MHz, MeOD-d6) δ 7.22–7.29 (3H, m), 7.14–7.18 (2H, m), 3.11 (1H, d, J = 7.8 Hz), 2.94 (1H, d, J = 8.4 Hz), 2.73–2.78 (1H, m), 2.50–2.55 (1H, m), 2.40 (1H, dd, J = 9.3, 2.4 Hz), 2.23–2.31 (2H, m), 1.98 (1H, dt, J = 8.7, 2.1 Hz), 1.73–1.85 (4H, m), 1.60 (1H, sep, J = 5.1 Hz), 0.96 (6H, dd, J = 4.8, 3.9 Hz). MS (ESI) m/z 247 (M + H)+.
tert-Butyl (R)-7-Hydroxy-3-(((S)-3-methyl-1-(4-phenylpiperidin-1-yl)butan-2-yl)carbamoyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (IV-4)
To a stirred solution of intermediate IV-3 (140 mg, 0.568 mmol, 1.00 equiv) in 6.00 mL of THF was added (3R)-2-(tert-butoxycarbonyl)-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid II-1 (200 mg, 0.682 mmol, 1.20 equiv), BOP (300 mg, 0.682 mmol, 1.20 equiv), and Et3N (0.40 mL, 2.84 mmol, 5.00 equiv), and the reaction was stirred at room temperature for 4 h. The reaction was diluted with EtOAc and saturated NaHCO3 (aq). The layers were separated, and the aqueous solution was extracted 2× with EtOAc. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated. The residue was purified via flash chromatography using CH2Cl2/iPrOH 100/0 → 90/10 as the eluent to afford IV-4 in quantitative yield. 1H NMR (300 MHz, CDCl3) δ 7.29–7.33 (2H, m), 7.19–7.22 (3H, m), 7.02 (1H, d, J = 6 Hz), 6.65 (1H, dd, J = 6, 1.5 Hz), 6.52–6.58 (1H, m), 5.91 (1H, br s), 4.83 (1H, d, J = 54.9 Hz), 4.52 (2H, q, J = 12 Hz), 3.85 (1H, br s), 3.27 (1H, dd, J = 11.7, 2.4 Hz), 2.97 (1H, J = 11.7, 4.8 Hz), 2.67–2.88 (2H, m), 2.40 (2H, br s), 1.66–2.20 (8H, m), 1.52 (9H, s), 1.26 (1H, s), 0.86 (6H, dd, J = 15.6, 5.1 Hz). MS (ESI) m/z 522 (M + H)+.
(R)-7-Hydroxy-N-((S)-3-methyl-1-(4-phenylpiperidin-1-yl)butan-2-yl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (8)
HCl/ether 2 M ( 2.00 mL) was added to a stirred solution of intermediate IV-4 (110 mg, 0.211 mmol, 1.00 equiv) in diethyl ether and MeOH, and the reaction was stirred at room temperature for 5 h. The mixture was concentrated, and excess HCl was removed by azeotroping 3× with ether. Trituration with ether (3×) afforded 90 mg of 8 as an HCl salt in 87% yield. 1H NMR (300 MHz, CDCl3) δ 7.25–7.29 (2H, m), 7.16–7.20 (4H, m), 6.91 (1H, d, J = 6 Hz), 6.61 (1H, d, J = 5.7 Hz), 6.46 (1H, s), 4.23–4.26 (1H, m), 3.70 (2H, q, J = 12.3 Hz), 3.45 (1H, d, J = 7.5 Hz), 3.27 (1H, dd, J = 8.4, 3.9 Hz), 3.10 (1H, d, J = 6.6 Hz), 2.94 (1H, dd, J = 12.3, 3.9 Hz), 2.74 (1H, br s), 2.53–2.59 (1H, m), 2.36 (2H, d, J = 9 Hz), 2.27–2.31 (1H, m), 2.06–2.09 (1H, m), 1.84–1.95 (6H, m), 1.26 (1H, s), 0.96 (6H, d, J = 5.1 Hz). HRMS (ESI) Calcd for C26H36N3O2 (M+H)+ 422.2802, found 422.2802. LCMS RT = 4.44 min; m/z (M + H)+ = 422.3.
In Vitro Pharmacology
Cell Culture
All receptors were in CHO cells transfected with human receptor cDNA. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum, in the presence of 0.4 mg/mL G418 and 0.1% penicillin/streptomycin, in 100 mm plastic culture dishes. For binding assays, the cells were scraped off the plate at confluence. For determination of inhibition of cAMP accumulation, cells were subcultured onto 24-well plates and used at confluence.
Receptor Binding
Binding to cell membranes was conducted in a 96-well format, as described previously.14 Cells were removed from the plates by scraping with a rubber policeman, homogenized in Tris buffer using a Polytron homogenizer, then centrifuged once, and washed by an additional centrifugation at 27 000g for 15 min. The pellet was resuspended in 50 mM Tris, pH 7.5, and the suspension incubated with [3H]nociceptin, [3H]DAMGO, [3H]DPDPE, or [3H]U69593, for binding to NOP, μ-, δ-, or κ-opioid receptors, respectively. The total volume of incubation was 1.0 mL, and samples were incubated for 60–120 min at 25 °C. The amount of protein in the binding reaction varied from approximately 15 to 30 μg. The reaction was terminated by filtration using a Tomtec 96 harvester (Orange, CT) with glass fiber filters. Bound radioactivity was counted on a Pharmacia Biotech beta-plate liquid scintillation counter (Piscataway, NJ) and expressed in counts per minute. IC50 values were determined using at least six concentrations of test compound, and calculated using Graphpad/Prism (ISI, San Diego, CA). Ki values were determined by the method of Cheng and Prusoff.28
Functional Activity [35S]GTPγS Binding Assay
[35S]GTPγS binding was conducted basically as described by Traynor and Nahorski.29 Cells were scraped from tissue culture dishes into 20 mM HEPES, 1 mM EDTA and then centrifuged at 500g for 10 min. Cells were resuspended in this buffer and homogenized using a Polytron homogenizer. The homogenate was centrifuged at 27 000g for 15 min, and the pellet resuspended in Buffer A, containing 20 mM HEPES, 10 mM MgCl2, 100 mM NaCl, pH 7.4. The suspension was recentrifuged at 27 000g and suspended once more in Buffer A. The pellet was sometimes frozen at −70 °C prior to use. For the binding assay, membranes (8–15 μg protein) were incubated with [35S]GTPγS (50 pM), GDP (10 μM), and the appropriate compound, in a total volume of 1.0 mL, for 60 min at 25 °C. Samples were filtered over glass fiber filters and counted as described for the binding assays. Statistical analysis was conducted using the program Prism.
Acknowledgments
The authors would like to thank Lawrence Toll for helpful suggestions with the in vitro biological characterization, and Pankaj Daga for assistance with the molecular docking experiments.
Glossary
Abbreviations
- SAR
structure–activity relationship
- JDTic
(3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide
- NOP
nociceptin opioid receptor
- KOP
kappa opioid receptor
- DOP
delta opioid receptor
- MOP
mu opioid receptor
- CHO
Chinese hamster ovary
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- BOP
(benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
This work was supported by grants from the National Institutes of Health R01DA027811 and R01DA014026 to NZ.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- a Zimmerman D. M.; Nickander R.; Horng J. S.; Wong D. T. (1978) New structural concepts for narcotic antagonists defined in a 4-phenylpiperidine series. Nature 275, 332–334. [DOI] [PubMed] [Google Scholar]; b Zimmerman D. M.; Leander J. D.; Cantrell B. E.; Reel J. K.; Snoddy J.; Mendelsohn L. G.; Johnson B. G.; Mitch C. H. (1993) Structure-activity relationships of trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine antagonists for mu- and kappa-opioid receptors. J. Med. Chem. 36, 2833–2841. [DOI] [PubMed] [Google Scholar]; c Carroll F. I.; Dolle R. E. (2014) The discovery and development of the N-substituted trans-3,4-dimethyl-4-(3′-hydroxyphenyl)piperidine class of pure opioid receptor antagonists. ChemMedChem 9, 1638–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitch C. H.; Leander J. D.; Mendelsohn L. G.; Shaw W. N.; Wong D. T.; Cantrell B. E.; Johnson B. G.; Reel J. K.; Snoddy J. D.; Takemori A. E.; et al. (1993) 3,4-Dimethyl-4-(3-hydroxyphenyl)piperidines: Opioid antagonists with potent anorectant activity. J. Med. Chem. 36, 2842–2850. [DOI] [PubMed] [Google Scholar]
- Zimmerman D. M.; Gidda J. S.; Cantrell B. E.; Schoepp D. D.; Johnson B. G.; Leander J. D. (1994) Discovery of a potent, peripherally selective trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonist for the treatment of gastrointestinal motility disorders. J. Med. Chem. 37, 2262–2265. [DOI] [PubMed] [Google Scholar]
- a Thomas J. B.; Atkinson R. N.; Rothman R. B.; Fix S. E.; Mascarella S. W.; Vinson N. A.; Xu H.; Dersch C. M.; Lu Y.; Cantrell B. E.; Zimmerman D. M.; Carroll F. I. (2001) Identification of the first trans-(3R,4R)- dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J. Med. Chem. 44, 2687–2690. [DOI] [PubMed] [Google Scholar]; b Thomas J. B.; Atkinson R. N.; Vinson N. A.; Catanzaro J. L.; Perretta C. L.; Fix S. E.; Mascarella S. W.; Rothman R. B.; Xu H.; Dersch C. M.; Cantrell B. E.; Zimmerman D. M.; Carroll F. I. (2003) Identification of (3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)- 3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro- 3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J. Med. Chem. 46, 3127–3137. [DOI] [PubMed] [Google Scholar]
- Munro T. A.; Huang X. P.; Inglese C.; Perrone M. G.; Van’t Veer A.; Carroll F. I.; Beguin C.; Carlezon W. A. Jr.; Colabufo N. A.; Cohen B. M.; Roth B. L. (2013) Selective kappa opioid antagonists nor-BNI, GNTI and JDTic have low affinities for non-opioid receptors and transporters. PLoS One 8, e70701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho G. D.; Bercovici A.; Tulshian D.; Greenlee W. J.; Fawzi A.; Smith Torhan A.; Zhang H. (2007) Synthesis and structure-activity relationships of 4-hydroxy-4-phenylpiperidines as nociceptin receptor ligands: Part 1. Bioorg. Med. Chem. Lett. 17, 3023–3027. [DOI] [PubMed] [Google Scholar]
- Ho G. D.; Bercovici A.; Tulshian D.; Greenlee W. J.; Fawzi A.; Fernandez X.; McLeod R. L.; Smith Torhan A.; Zhang H. (2007) Synthesis and structure-activity relationships of 4-hydroxy-4-phenylpiperidines as nociceptin receptor ligands: Part 2. Bioorg. Med. Chem. Lett. 17, 3028–3033. [DOI] [PubMed] [Google Scholar]
- Zaveri N.; Jiang F.; Olsen C.; Polgar W.; Toll L. (2005) Small-molecule agonists and antagonists of the opioid receptor-like receptor (ORL1, NOP): ligand-based analysis of structural factors influencing intrinsic activity at NOP. AAPS J. 7, E345–E352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto H.; Ozaki S.; Itoh Y.; Miyaji M.; Arai S.; Nakashima H.; Kato T.; Ohta H.; Iwasawa Y. (1999) Discovery of the first potent and selective small molecule opioid receptor-like (ORL1) antagonist: 1-[(3R,4R)-1-cyclooctylmethyl-3- hydroxymethyl-4-piperidyl]-3-ethyl-1, 3-dihydro-2H-benzimidazol-2-one (J-113397). J. Med. Chem. 42, 5061–5063. [DOI] [PubMed] [Google Scholar]
- Zaratin P. F.; Petrone G.; Sbacchi M.; Garnier M.; Fossati C.; Petrillo P.; Ronzoni S.; Giardina G. A.; Scheideler M. A. (2004) Modification of nociception and morphine tolerance by the selective opiate receptor-like orphan receptor antagonist (−)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9- tetrahydro-5H-benzocyclohepten-5-ol (SB-612111). J. Pharmacol. Exp. Ther. 308, 454–461. [DOI] [PubMed] [Google Scholar]
- Kormos C. M.; Cueva J. P.; Gichinga M. G.; Runyon S. P.; Thomas J. B.; Brieaddy L. E.; Mascarella S. W.; Gilmour B. P.; Navarro H. A.; Carroll F. I. (2014) Effect of the 3- and 4-methyl groups on the opioid receptor properties of N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidines. J. Med. Chem. 57, 3140–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. B.; Fix S. E.; Rothman R. B.; Mascarella S. W.; Dersch C. M.; Cantrell B. E.; Zimmerman D. M.; Carroll F. I. (2004) Importance of phenolic address groups in opioid kappa receptor selective antagonists. J. Med. Chem. 47, 1070–1073. [DOI] [PubMed] [Google Scholar]
- Davies R. J., Xu J. (2008) Preparation of substituted spiro[benzoazepine-piperidine] as modulators of muscarinic receptors. WO 2008021545A2, Feb 21, 2008.
- Thomas J. B.; Fall M. J.; Cooper J. B.; Rothman R. B.; Mascarella S. W.; Xu H.; Partilla J. S.; Dersch C. M.; McCullough K. B.; Cantrell B. E.; Zimmerman D. M.; Carroll F. I. (1998) Identification of an opioid kappa receptor subtype-selective N-substituent for (+)-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine. J. Med. Chem. 41, 5188–5197. [DOI] [PubMed] [Google Scholar]
- Zaveri N. T.; Jiang F.; Olsen Cris M.; Deschamps Jeffrey R.; Parrish D.; Polgar W.; Toll L. (2004) A novel series of piperidin-4-yl-1,3-dihydroindol-2-ones as agonist and antagonist ligands at the nociceptin receptor. J. Med. Chem. 47, 2973–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Adapa I. D.; Toll L. (1997) Relationship between binding affinity and functional activity of nociceptin/orphanin FQ. Neuropeptides 31, 403–408. [DOI] [PubMed] [Google Scholar]; b Toll L.; Berzetei-Gurske I. P.; Polgar W. E.; Brandt S. R.; Adapa I. D.; Rodriguez L.; Schwartz R. W.; Haggart D.; O’Brien A.; White A.; Kennedy J. M.; Craymer K.; Farrington L.; Auh J. S. (1998) Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res. Monogr. 178, 440–466. [PubMed] [Google Scholar]
- a Spagnolo B.; Calo G.; Polgar W. E.; Jiang F.; Olsen C. M.; Berzetei-Gurske I.; Khroyan T. V.; Husbands S. M.; Lewis J. W.; Toll L.; Zaveri N. T. (2008) Activities of mixed NOP and mu-opioid receptor ligands. Br. J. Pharmacol. 153, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dooley C. T.; Spaeth C. G.; Berzetei-Gurske I. P.; Craymer K.; Adapa I. D.; Brandt S. R.; Houghten R. A.; Toll L. (1997) Binding and in vitro activities of peptides with high affinity for the nociceptin/orphanin FQ receptor, ORL1. J. Pharmacol. Exp. Ther. 283, 735–741. [PubMed] [Google Scholar]
- Arunlakshana O.; Schild H. O. (1959) Some quantitative uses of drug antagonists. Br. J. Pharmacol. Chemother. 14, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T.; Jenkinson S.; Watson C. (2006) Determining the potency and molecular mechanism of action of insurmountable antagonists. J. Pharmacol. Exp. Ther. 319, 710–723. [DOI] [PubMed] [Google Scholar]
- a Zaveri N. (2003) Peptide and nonpeptide ligands for the nociceptin/orphanin FQ receptor ORL1: research tools and potential therapeutic agents. Life Sci. 73, 663–678. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mustazza C.; Bastanzio G. (2011) Development of nociceptin receptor (NOP) agonists and antagonists. Med. Res. Rev. 31, 605–648. [DOI] [PubMed] [Google Scholar]; c Bignan G. C.; Connolly P. J.; Middleton S. A. (2005) Recent advances towards the discovery of ORL-1 receptor agonists and antagonists. Expert Opin. Ther. Pat. 15, 357–388. [Google Scholar]
- McElvain S. M.; Clemens D. H. (1958) Piperidine Derivatives. XXX. 1,4-Dialkyl-4-arylpiperidines. J. Am. Chem. Soc. 80, 3915–3923. [Google Scholar]
- Wu H.; Wacker D.; Mileni M.; Katritch V.; Han G. W.; Vardy E.; Liu W.; Thompson A. A.; Huang X. P.; Carroll F. I.; Mascarella S. W.; Westkaemper R. B.; Mosier P. D.; Roth B. L.; Cherezov V.; Stevens R. C. (2012) Structure of the human kappa-opioid receptor in complex with JDTic. Nature 485, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. A.; Liu W.; Chun E.; Katritch V.; Wu H.; Vardy E.; Huang X. P.; Trapella C.; Guerrini R.; Calo G.; Roth B. L.; Cherezov V.; Stevens R. C. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll I.; Thomas J. B.; Dykstra L. A.; Granger A. L.; Allen R. M.; Howard J. L.; Pollard G. T.; Aceto M. D.; Harris L. S. (2004) Pharmacological properties of JDTic: a novel kappa-opioid receptor antagonist. Eur. J. Pharmacol. 501, 111–119. [DOI] [PubMed] [Google Scholar]
- a M?llereau C.; Parmentier M.; Mailleux P.; Butour J. L.; Moisand C.; Chalon P.; Caput D.; Vassart G.; Meunier J. C. (1994) ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 341, 33–38. [DOI] [PubMed] [Google Scholar]; b Zaveri N.; Polgar W. E.; Olsen C. M.; Kelson A. B.; Grundt P.; Lewis J. W.; Toll L. (2001) Characterization of opiates, neuroleptics, and synthetic analogs at ORL1 and opioid receptors. Eur. J. Pharmacol. 428, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Meng F.; Taylor L. P.; Hoversten M. T.; Ueda Y.; Ardati A.; Reinscheid R. K.; Monsma F. J.; Watson S. J.; Civelli O.; Akil H. (1996) Moving from the orphanin FQ receptor to an opioid receptor using four point mutations. J. Biol. Chem. 271, 32016–32020. [DOI] [PubMed] [Google Scholar]; b Meng F.; Ueda Y.; Hoversten M. T.; Taylor L. P.; Reinscheid R. K.; Monsma F. J.; Watson S. J.; Civelli O.; Akil H. (1998) Creating a functional opioid alkaloid binding site in the orphanin FQ receptor through site-directed mutagenesis. Mol. Pharmacol. 53, 772–727. [DOI] [PubMed] [Google Scholar]
- a Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. (2012) Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Granier S.; Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Weis W. I.; Kobilka B. K. (2012) Structure of the delta-opioid receptor bound to naltrindole. Nature 485, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Traynor J. R.; Nahorski S. R. (1995) Modulation by mu-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 47, 848. [PubMed] [Google Scholar]