

Abstract
Salinity tolerance in rice is highly desirable to sustain production in areas rendered saline due to various reasons. It is a complex quantitative trait having different components, which can be dissected effectively by genome-wide association study (GWAS). Here, we implemented GWAS to identify loci controlling salinity tolerance in rice. A custom-designed array based on 6,000 single nucleotide polymorphisms (SNPs) in as many stress-responsive genes, distributed at an average physical interval of <100 kb on 12 rice chromosomes, was used to genotype 220 rice accessions using Infinium high-throughput assay. Genetic association was analysed with 12 different traits recorded on these accessions under field conditions at reproductive stage. We identified 20 SNPs (loci) significantly associated with Na+/K+ ratio, and 44 SNPs with other traits observed under stress condition. The loci identified for various salinity indices through GWAS explained 5–18% of the phenotypic variance. The region harbouring Saltol, a major quantitative trait loci (QTLs) on chromosome 1 in rice, which is known to control salinity tolerance at seedling stage, was detected as a major association with Na+/K+ ratio measured at reproductive stage in our study. In addition to Saltol, we also found GWAS peaks representing new QTLs on chromosomes 4, 6 and 7. The current association mapping panel contained mostly indica accessions that can serve as source of novel salt tolerance genes and alleles. The gene-based SNP array used in this study was found cost-effective and efficient in unveiling genomic regions/candidate genes regulating salinity stress tolerance in rice.
Keywords: genome-wide association study, infinium genotyping assay, rice, salt tolerance, single-nucleotide polymorphism
1. Introduction
Rice, commonly known to be salt sensitive,1 is greatly affected by soil salinity.2 Salinity affects rice growth in varying degree at all stages starting from germination through maturation.3–6 Excess salts adversely affect all major metabolic activities in rice, and cause overall decline in germination and seedling growth,4–7 leading ultimately to reduced growth and diminished grain yield. The reduction in major yield components including tiller numbers in plant and spikelet numbers per panicle has been reported to be the major cause of yield loss (27–50%) in rice cultivars during early reproductive panicle initiation stage under salinity stress.5,8,9 Millions of hectares in the humid regions of South and Southeast Asia are technically suited for rice production but are left uncultivated or are grown with very low yields because of salinity and problem soils. Salt-affected soils in arid and semi-arid regions of Asia, Africa and South America cause considerable agronomic problems. In Asia, 12 million ha of land area is thought to be salinity affected with India having >50% salinity affected area. Considerable variation for different yield contributing agronomic traits has been observed in diverse rice genotypes under salinity stress.10,11 Therefore, to maximize productivity of rice under saline soils, there is an urgent need to look for discovery of genes imparting salt tolerance and their introduction in salt-sensitive rice cultivars.12
The dissection of salt tolerance traits has been carried out by quantitative trait loci (QTLs) mapping approach using bi-parental populations. These studies have led to identification of both major and minor QTLs for various traits on different rice chromosomes.13–22 For example, Koyama et al.13 identified 11 QTLs for Na+ and K+ content related to salinity stress tolerance. Bonilla et al.14 mapped Saltol locus linked to major QTLs for Na+ and K+ uptake and Na+/K+ ratio on chromosome 1 explaining 64.3% phenotypic variance. Lin et al.15 mapped QTLs for root and shoot Na+/K+ concentration and transport on five rice chromosomes. Ammar et al.16 reported 25 QTLs for salt ion concentrations (Na+, K+ and Cl− measured in the leaf tissues at the reproductive stage) on rice chromosomes 1, 2, 3 and 8. Pandit et al.17 reported eight QTLs for salt ion concentrations on rice chromosomes 1, 8 and 12, and Cheng et al.23 reported 12 QTLs for salt ion concentrations on rice chromosomes 1, 2, 3, 4, 7 and 11, respectively. Most of these QTL mapping studies have used diverse indica and japonica intercross bi-parental mapping population to correlate with different measures of salinity stress tolerance and mapped hundreds of QTLs on 12 rice chromosomes. Many of these QTLs are specific to populations used for QTL mapping, since in bi-parental mapping populations, discovery of QTLs is limited by the germplasm used and therefore, use of a divergent set of germplasm resource is a prerequisite for identification of most of the agriculturally relevant loci. Association mapping that is based on genotyping of diverse germplasm set is an attractive approach for identification and mapping of such QTLs in plants.24–29 New statistical methods have been developed to analyse structured core/mini-core germplasm collections,30–32 and these methods have been efficiently applied to plants for candidate gene-based and genome-wide association mapping studies.33–35 Genome-wide association study (GWAS) has great potential for identifying valuable natural variations in trait-associated loci, as well as allelic variations in candidate genes underlying quantitative and complex traits, including those related to growth, development, stress tolerance and nutritional quality. Some previous studies in rice have been conducted to dissect genetic architecture of complex traits through GWAS.36,37 An extra advantage of the GWAS design for rice is the homozygous nature of most rice varieties, which makes it possible to employ a ‘genotype or sequence once and phenotype many times over’ strategy, whereby once the lines are genomically characterized, the genetic data can be reused many times over across different phenotypes and environments.37 GWAS thus has tremendous significance for identification of genes particularly for complex quantitative traits in rice. However, limited studies have been carried out in rice to identify genes/QTLs for abiotic stress tolerance using GWAS. This includes identification and mapping of genes/QTLs for aluminium stress tolerance28 using a genotyping array of 44,000 single-nucleotide polymorphisms (SNPs)37 scanned in 383 diverse rice association panel. More recently, employing candidate gene-based association mapping approach, 11 SNPs mined in five known cloned salt-tolerance genes through EcoTILLING have been shown to be associated with salinity tolerance (Na+/K+ ratio equilibrium) in rice.38 However, there is no such report till now on identification of genes particularly for reproductive stage salinity tolerance using GWAS in rice.
In the current assignment, we performed GWAS for identification of potential genes associated with salinity stress tolerance in rice by correlating the genotyping information obtained by using an SNP chip based on 6,000 stress-responsive genes with the phenotypic expression of 220 diverse rice (Oryza sativa) accessions for 14 morphological, biochemical (Na+ and K+ content) and agronomic traits.
2. Materials and methods
2.1. Phenotypic evaluation of germplasm
The experiment comprised of 220 rice accessions, which were obtained from various national and international institutes to assess the performance in normal and high saline stress conditions (Supplementary Table S1). These genotypes were evaluated in randomized complete block design with two replications under two environments, viz., normal (ECiw ∼1.0 dS/m) and high salinity stress (ECiw ∼10 dS/m) in micro plots at the Central Soil Salinity Research Institute, Karnal, Haryana, India. This experimental site is situated at 29.43°N latitude and 76.58°N longitude and 245 m above the sea level. The 35-day-old seedlings from wet bed nurseries were transplanted using two seedlings per hill at a spacing of 15 × 20 cm. Basal fertilizers for the main crop were 120-60-60 kg of NPK/ha. The recommended agronomic practices were followed to get healthy and good crop. Twenty-one days after transplanting, salinity stress was imposed using 7 NaCl : 1 Na2SO4: 2 CaCl2 on equivalent basis. Salinity was recorded once in a week and maintained at desired level (ECiw ∼10 dS/m) throughout the cropping season. Randomly five plants were tagged for each genotype in each replication, and phenotypic data were recorded for all traits. The response of the genotypes to salt stress was expressed using the stress susceptibility index (SSI) calculated according to Fischer and Maurer39 using the following formula: SSI = (1 − Ys/Yp)/D, where Ys = mean performance of a genotype under stress; Yp = mean performance of the same genotype without stress; D (stress intensity) = 1 − (mean Ys of all genotypes/mean Yp of all genotypes).
2.2. Designing of SNP chip for high-throughput genotyping
The genotyping array used in our study was designed by selecting SNPs from differentially expressed transcripts in indica rice genotypes. For this, two rice genotypes, i.e. Nagina22 and CSR30 were grown, respectively, under drought and salt stress, and the whole transcriptome was sequenced (unpublished results). A set of 6,500 transcripts, differentially expressed in both genotypes and 8,884 non-transposable elements-related annotated rice genes (MSU, version 6.1) distributed over 12 rice chromosomes, were selected, and these gene sequences from whole-genome sequencing data of six contrasting rice genotypes (unpublished) generated using Next Generation ABI-SOLiD platform were co-aligned. One SNP from each of the selected gene was identified from these co-aligned sequences and from previously reported genome-wide Perlegen re-sequencing information among 20 rice varieties.40 The identified 15,384 genic SNPs were further mapped physically (bp) on 12 rice chromosomes (Pseudomolecule 6.1). Finally, 6,000 SNPs were selected for array designing, which were distributed at physical interval of <100 kb on 12 rice chromosomes. The physical distance (kb) between SNP loci varied from 16 kb in chromosome 3 to 98 kb in chromosome 11 with an average of 51 kb (Fig. 1). The 6,000 SNP chip array included 2,456 SNPs from the previously reported genome-wide Perlegen re-sequencing data, and remaining 3,544 SNPs were identified from our transcriptome sequencing data. Functional annotation of 6,000 SNPs carrying stress-related rice genes (Fig. 2) revealed maximum correspondence to abiotic stress-responsive genes (73.5%) followed by biotic stress-related genes (16.3%), unknown expressed genes (6%) and minimum correspondence to hypothetical proteins (4%). The SNPs from abiotic stress-related genes were associated mostly with metabolism and protection (26%) followed by zinc finger, Myb, Zip, heat shock, ethylene response element-activator protein-2 (AP-2) and dehydration-responsive element-binding transcription factors (15%). Of these 6,000 SNPs, 5,246 had satisfactory custom design score amenable for multiplexing and development of high-throughput assay in Illumina Infinium platform (Illumina Inc., San Diego, CA, USA), and this 5,246 SNP array was used for genotyping of 220 rice accessions.
Figure 1.

Distribution of 6,000 SNPs in known abiotic and biotic stress-responsive and unknown expressed rice genes on 12 rice chromosomes. Physical distance (kb) between adjacent SNP loci varied from 16 kb in chromosome 3 to 98 kb in chromosome 11 with an average of 51 kb.
Figure 2.

Functional annotation of 6,000 SNPs in known abiotic and biotic stress-responsive and unknown expressed rice genes distributed over 12 rice chromosomes.
2.3. Genomic DNA isolation and array hybridization
Genomic DNA was extracted from pooled young green leaf tissue from five plants of each accession following cetyl trimethylammonium bromide (CTAB) method described by Doyle and Doyle.41 DNA was quantified using NanoDrop 8000 spectrophotometer (Thermo Scientific, USA). Concentrations were adjusted to a minimum of 50 ng/µl, and 4 µl aliquots were used for the Illumina Infinium® genotyping assay. Approximately 200 ng of unlabelled rice genomic DNA was used for hybridization. The customized array, employing exclusively Illumina Infinium® II design probes and dual color channel assays (Infinium HD Assay Ultra, Illumina), was used for genotyping, following the manufacturer’s recommendations.
2.4. SNP genotype calling
SNP genotyping data were analysed using Genome Studio V2010.1 (Illumina Inc.). SNP genotypes were called using genotyping module integrated in the software where individual SNP is viewed as GenoPlots. Data quality was rapidly confirmed with internal controls and other QC functions such as GenTrain and GenCall scores.42 After calling the data automatically, the SNPs were re-scored and checked for their presence in a canonical cluster to get a GenTrain score >0.7. We removed the samples from analysis that had call rates <0.89 and the SNPs with norm R values <0.2. Initially, using these criteria, 220 accessions and 4,929 SNPs with valid and robust genotyping data were selected for association analysis.
2.5. Population structure
To estimate the number of subgroups in the panel, both principal components analysis (PCA) and STRUCTURE 2.3.4 program were used.33–35 Only those SNPs, having both missing data <20% and minor allele frequency (MAF) ≥ 5%, were included for STRUCTURE program. For PCA analysis, genomic association and prediction integrated tool (GAPIT)43 was used. The final number of principal components (PCs) that adequately explain population structure was determined through scree plot generated by GAPIT. The Efficient Mixed-Model Association (EMMA)44 was used to correct for confounding effects due to subpopulation structure and relatedness between individuals. Genetic divergence among subpopulations was detected through net nucleotide distance estimated by STRUCTURE.
2.6. Estimation of LD decay in rice
Genome-wide linkage disequilibrium (LD) analysis was performed among the 220 accessions (association panel) and subpopulations (determined by population structure) by pairwise comparisons among the 4,191 SNP markers (with MAF > 0.02) using r2, the correlation in frequency among pairs of alleles across a pair of SNP loci. For all pairs of SNPs, r2 was calculated using the --r2 --ld-window-kb 2,000 --ld-window 99999 --ld-window-r2 0 command in PLINK.45 The command was used to calculate LD association among SNP pairs to a distance of 2,000 kb. After excluding SNPs with MAF < 0.02, we obtained 4,191 (∼85%) SNPs across the O. sativa panel. For these high-quality SNPs, missing genotypes were imputed using fastPHASE.46 Within each subpopulation, we calculated r2 for all pairs of SNPs. For LD decay analysis, 2,000 kb LD region covered between SNP pairs was classified into bins of 20 kb, and r2 values were plotted against distance in kb. To graphically view the LD (r2) between SNP pairs in the entire panel, LD was calculated between 50 subsequent SNPs using sliding window option of Analysis by Association, Evolution, and Linkage (TASSEL 3.0) software.47 The Solid Spine of LD algorithm48 was also used to graphically find out the regions with minimum D′ value of 0.8, as suggested by Duggal et al.49
2.7. Genome-wide association analysis
For association analysis of our data set (4,929 SNP genotyping and phenotyping information of 220 accessions), GAPIT was used that involves EMMA (executed by R package) and CMLM (compressed mixed linear model)/P3D (population parameters previously defined) interfaces to conduct GWAS, and predicts genomic regions associated with traits. For CMLM, the relatedness matrix, measured as the genetic similarity between individuals and IBS (identical by state) values, was used to estimate random effects. The kinship matrix (K) estimated from SNP genotyping data was used jointly with population structure (Q value) to improve statistical power of association. For fixed effects, PCs through PCA analysis and Q-matrix through STRUCTURE were used as covariates for association analysis. Scree plot of PCA analysis suggested first three components as most informative, which were used as covariate matrix in K + P (kinship + PCA) model. For K + Q (kinship + Q-matrix) model, a file generated through run K = 3 in STRUCTURE program was used as covariate matrix. The critical P-values for assessing the significance of SNPs were calculated based on a false discovery rate (FDR) separately for each trait,50 which was found to be highly stringent. An FDR cut-off of 0.05 was used for determining significance.
3. Results
3.1. Success of genotyping array
The array designed with 5,246 SNPs in our study was successfully used to genotype all the 220 rice accessions. The data generated through Illumina Infinium platform were loaded in Genome Studio software where, after cluster refinement with optimum GenTrain (>0.7) and GenCall score (>0.3), 4,929 polymorphic SNPs (∼94%) were identified. After excluding the SNPs with MAF < 0.02, finally, 4,191 (∼80%) high-quality SNPs genotyped across 220 rice accessions were utilized for GWA mapping.
3.2. Genetic structure of rice accessions
Using PCA, most of the genetic variation (66%) in the accessions was explained by first two PCs. Based on 4,191 SNPs, the first PC explained almost 52% of genetic variation, whereas PC II explained 14%. The scree plot generated through GAPIT recommended the first three components as informative, where descent changes gradually (Fig. 3A). When we plotted the first two components against each other, three subpopulations were identified. A total of 130 accessions were clustered as a single large subpopulation (I) (Fig. 3B). For inferring the most likely number of populations among 220 accessions in STRUCTURE, the transformation method51 was used, and similar to PCA, three subpopulations were identified (Fig. 3C). Forty-four per cent of the accessions (97/220) did not show any admixture, 46% accessions (101/220) showed up to 20% admixture, while the remaining 10% (22/220) were found to be highly admixed.
Figure 3.

Population structure of current association panel which consisted mostly of the indica accessions. (A) Scree plot from GAPIT showing the selection of PCs for association study. (B) PCA plot of first two components. (C) Bayesian clustering of 220 rice accessions using STRUCTURE program.
We examined allele sharing across the accessions by calculating IBS coefficients among all pairs of accessions. Most of the admixture was shared between accessions of Subpopulations II and III in IBS sharing heatmap (Supplementary Fig. S1). Except some of the accessions from Panvel, Raigarh, India, the mean IBS sharing was the highest between the other accessions from this region (0.96), and they were placed in a group of accessions from Haryana and IARI, New Delhi, with little IBS sharing. The second highest IBS sharing (0.86) subgroup was found in the Subpopulation III, having all accessions from Kerala, India. The allele frequency divergence among the three putative subpopulations was found maximum between the Subpopulations I and III followed by II and III. The lowest differentiation was between the Subpopulations I and II. Genetic diversity, measured as the average distances (expected heterozygosity) between individuals in the same cluster, was found low in all the clusters (0.18).
3.3. Patterns of LD in association panel
The LD decay rate was measured as the chromosomal distance (kb) at which the average pairwise correlation coefficient (r2) dropped to half its maximum value.36 The binned r2 values were mapped against the physical distance (kb) across the genome. Overall, the LD for SNPs at 20 kb distance from each other was 0.50 (r2), which decayed to its half value (∼0.25) at around 300 kb. However, further decay in r2 was found very slow and paralleled at 0.15 up to 2 Mb (Fig. 4). Out of total pairwise LD events, only 14% pairs showed r2 above 0.5. When LD was calculated population-wise, the maximum average (r2 < 0.70) was observed for Subpopulation I, which was maintained at r2 > 0.21 up to 2 Mb. The average LD for Subpopulation III was observed comparatively low (0.44), and decayed half to its initial value at 240 kb. The average minimum LD was also found very low (0.12) for Subpopulation III. The maximum and minimum LD for Subpopulation II were 0.50 and 0.15, respectively. PLINK was also used to calculate chromosome-wise LD between SNP pairs, which was found maximum (0.40) for chromosomes 3 and 7, and minimum (r2 < 0.23) for chromosome 11. Except chromosomes 8, 10 and 11, all other chromosomes showed LD above 0.30. On an average, 84 kb distance was observed between SNP pairs before applying MAF < 0.02, which increased to 95 kb after correcting for MAF. The average maximum distance (∼125 kb) was observed for chromosomes 8 and 12 while minimum distance (∼69 kb) was observed for chromosome 3. When LD heatmap generated through TASSEL was studied, the regions of high LD with significant P-values were found to be interspersed between the low LD regions throughout the genome.
Figure 4.
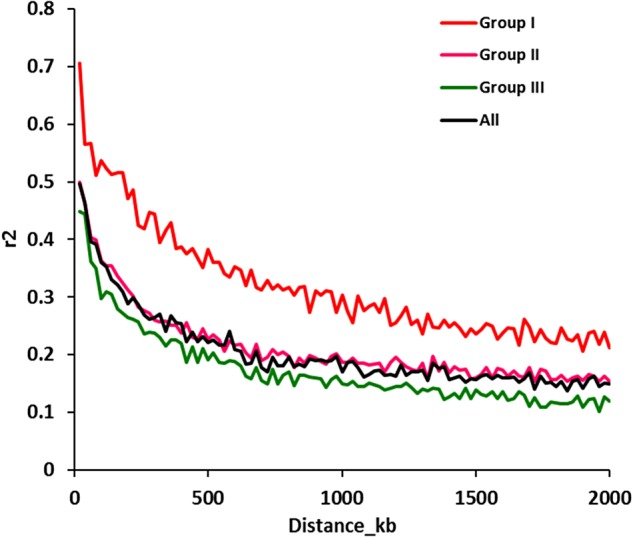
Comparison of LD patterns and LD decay in the whole panel and subgroups. The whole genome r2 values from PLINK are first sorted considering distance, and then divided into 100 blocks of 20 kb. The r2 values in each block are averaged and plotted against the genetic distance for different subgroups.
3.4. Phenotypic variation among accessions for salinity tolerance traits
Frequency distributions of traits observed under normal and salt stress among the 220 accessions are presented in the Supplementary Figs S2a and b, respectively. Some of the traits such as yield were the cumulative outcome of its component traits as determined by correlation and path analysis (Supplementary Fig. S3). The simple correlation coefficients were found positive for yield and its component traits such as productive tillers, filled grains, seed weight and spikelet fertility under normal and stress condition. Under stress condition, yield was found negatively correlated with Na+ content (−0.24) and Na+/K+ ratio (−0.22), while it was positive with K+ content. Na+ content was found negatively correlated with K+ content (−0.12). Under stress, Na+/K+ ratio was found in positive correlation with Na+ content (0.75) while negatively correlated with K+ content (−0.48), as expected. The spikelet fertility was also negatively correlated with Na+ content (−0.22) and Na+/K+ ratio (−0.23) while positively correlated with K+ content (0.13) under stress. In contrast, all the susceptibility indices were found positively correlated with Na+ content and Na+/K+ ratio while negatively correlated with K+ content. Grain weight also showed high correlation with grain length, but its association with grain width was found low but positive. The traits observed under normal and stress conditions were found independent of each other except for grain length. The ratio of filled grains/unfilled grains reduced maximally under salt stress in the third largest subpopulation. The average Na+ content and Na+/K+ ratio were the highest in the Subpopulation II at normal condition, but it was maintained low in the population after salt stress. The drop in yield under salt stress was observed comparatively more in the largest subpopulation as spikelet fertility was found minimum (50%) in this population under stress condition. Spikelet fertility was found to be 56 and 57% for Subpopulations I and II, respectively. Overall, under control condition, the spikelet fertility varied from 62 to 99%, while under salt stress, it decreased and varied from 1 to 95%. The average grain length was found maximum in Subpopulation III under both normal and salinity conditions. The canonical correlation coefficients were found high for phenotypic traits under salt stress conditions compared with normal condition. For instance, the canonical correlation in stress category was estimated 0.81 between productive tillers and yield, while it was only 0.29 under normal condition. The heritability calculated for each trait among accessions ranged from 0.61 to 0.89 with an average of 0.72. The heritability for the flowering time was observed to be low. A significant level of phenotypic variation thus was recorded among the accessions used in the study for salinity response.
3.5. GWAS
All the 220 rice accessions in our study were phenotyped for 12 agronomic traits, and two assays of Na+ and K+ accumulation were also conducted. Using 4,191 SNPs, GAPIT produced association signals for various traits under control as well as stress condition. The association signals were more in number and potent under stress condition. SNPs were considered significant only after adjusting for multiple testing at 0.05 threshold level. The associated loci in this study for different traits were interspersed on all the chromosomes. Using the CMLM approach, we successfully identified both known associations (for example, enrichment in a priori candidate genes and previously reported QTLs from rice) as well as new candidate loci in the rice genome. The results of our genome-wide association scans are summarized in Supplementary Figs S4–11 where we showed the SNP trait associations discovered in the association panel using CMLM approaches.
Under control condition, 2, 9 and 7 SNPs were significantly associated with unfilled grains, percent spikelet fertility and plant height, respectively (Supplementary Table S2). For unfilled grains, both of the significant signals were located on chromosome 9 (10154727 and 10223755). All the top SNPs from this region were found in high LD. For percent spikelet fertility, out of nine significant SNPs, four were detected on chromosome 2, one on chromosome 5 (6251237), two on chromosome 7 (22665019 and 22725607) and the remaining two on chromosome 9 (10154727 and 10223755). However, association peaks were detected only on chromosomes 2, 7 and 9 while chromosome 5 SNP was reported significant without any associated SNPs. Some of the results regarding unfilled grains and spikelet fertility were found identical, as both the significant SNPs from chromosome 9 for unfilled grains were also found significant for spikelet fertility. A GWAS peak on chromosome 3 between the coordinates, 14123489 and 14156542 for unfilled grains, was also detected for spikelet fertility, but in both the cases, the top SNP (P = 0.081) from this peak was found below the FDR-adjusted P-value. For plant height, seven SNPs on chromosome 4 covering a region of 2 Mb (8559661–12215278) were found significant with same level of FDR correction (0.011) and phenotypic variation (15%) explained by each SNP in the block. The region was found as a large LD region having high r2 with significant P-values.
Compared with control condition, the number of significant SNPs increased for stress condition. Under stress condition, the significant signals were detected for yield/plant, filled grains, productive tillers, spikelet fertility, spikelet fertility SSI, Na+ content and Na+/K+ ratio. Except for Na+ content and Na+/K+ ratio, most of the significant signals were common for different traits observed under stress condition. In total, 44 SNPs were found significant for these traits except Na+/K+ ratio for which most of the signals were different. For Na+/K+ ratio, five GWAS peaks containing 20 SNPs were recorded on chromosomes 1, 4, 6 and 7 (Supplementary Table S2). Twelve of the 20 SNPs were located on chromosome 1, on or near the Saltol, a known and highly investigated region for salinity tolerance in rice. Significant association signals on chromosome 4 for Na+/K+ ratio comprised five SNPs, originating from two GWAS peaks located on two distant regions. From the first peak, three SNPs (4: 13497584, 13649124 and 14014367) achieved the FDR threshold level (P< 0.039) while two (4: 34164920 and 34292214) achieved threshold level (P < 0.040) from second peak. The locations of these GWAS peaks were tracked between the coordinates 13.4–14.0 and 34.1–34.5 Mb, respectively. Another GWAS peak was reported at one end of chromosome 6, and two SNPs (6: 902913 and 1460570) from this peak were found significant (FDR-adjusted P < 0.046). The fifth GWAS peak was observed from chromosome 7; however, a single SNP (7:19655274) from this region achieved FDR-adjusted threshold level (P = 0.026). When top SNPs from this region were arranged, the origin of the peak was tracked between the coordinates, 19655274 and 25919115. Only one SNP located on chromosome 1 (41550991) was found significant (FDR-adjusted P = 0.002) for Na+ content under stress. None of the SNPs showed significant peaks for K+ concentration under stress condition.
Out of total 44 SNPs for various traits observed under salinity, 27 were found common for all the traits. Individually, 32, 35, 33, 37 and 37 SNPs were, respectively, associated with filled grains, productive tillers, yield/plant, percent spikelet fertility under stress and percent spikelet fertility SSI (Supplementary Table S3). Excluding common ones, only 2, 2, 1 and 3 SNPs were found uniquely associated for productive tillers, filled grains, yield/plant under stress and percent spikelet fertility under stress, respectively. In our study, association peaks were observed on all the chromosomes. However, single SNP associations, one each on chromosome 6 and 8, were observed significant. A significant SNP (29212927) on chromosome 4 was found common for all the five above-discussed traits, whereas another SNP (16517190) from the same chromosome was observed only for productive tillers. An SNP (23127237) on chromosome 11 was also found significant only for productive tillers. Two SNPs (10654854 and 41550991) on chromosome 1 and one SNP (29212927) on chromosome 4 were found common for above-discussed traits as well as for Na+/K+ ratio under stress. The peak significant SNPs from chromosome 10 for all these traits were distributed between the coordinates 5864939 and 14809695. All the association signals for percent spikelet fertility and spikelet fertility stress suceptibility index (SFSSI) were found identical.
The LD heatmap generated by TASSEL across the whole genome showed that SNPs, which are in association with different salt indices, were found in LD blocks of various r2 intensities. Some of the SNPs were found in large LD blocks of high and significant r2 values. Most of the SNPs were found in small LD blocks containing 2–10 SNPs. However, three SNPs, two from chromosome 2 (6651494 and 22253258) and one from chromosome 8 (5109310), were not found in LD with other SNPs. The average r2 values for all the three SNPs were found below the background level (r2 < 0.2).
3.6. Phenotypic variance explained by the significant SNPs
For plant height under control condition, seven SNPs in a cluster on chromosome 4 (8559661–12215278) were found significant and each of the seven significant SNPs explained almost 15% of the phenotypic variation. SNPs found significant for spikelet fertility under control condition explained on an average 5% of the phenotypic variation where both the SNPs on chromosome 7 contributed maximum 6% variation. A graph summarizing the QTL regions associated with all traits, as well as the percent of the phenotypic variation explained by significant SNPs for each trait, is provided in Fig. 5. For Na+/K+ ratio under stress condition, the minimum effect locus contributed 6% while its maximum effect locus contributed 12% to the phenotypic variation with an average of 9%. Among the traits tagged under stress condition, the maximum average phenotypic variation was explained for productive tillers (15%), followed by yield/plant (12%). For SFSSI, 37 significant SNPs explained on an average 11% of the phenotypic variation.
Figure 5.

Summary of percentage of variance explained by significant loci in the study. The x-axis represents the trait, and the y-axis shows the contribution (%) of significant loci. The label on the top of average bar is the total number of significant SNPs for respective trait.
4. Discussion
Salinity is a serious constraint to rice productivity, and due to a lack of enough improved salinity-tolerant rice varieties, most of the regions affected by salinity are left barren. To improve rice productivity in such areas, we need to identify a number of novel genes and alleles associated with such complex quantitative salinity tolerance traits in diverse rice accessions that are well adapted to changing climatic conditions and pyramid them. Several earlier studies have led to identification of QTLs for salinity tolerance in rice using bi-parental mapping populations, which, due to low resolution, scan only limited number of recombination events, and do not allow identification of many evolutionarily valid and robust QTLs for salinity stress. An alternate and complementary approach is GWAS that takes advantage of historical recombination events, which have accumulated over thousands of generations in historical populations and thus enable high-resolution mapping for identification of possible target genomic regions for complex quantitative traits in rice.36,37 The sample size of 220 rice accessions is much smaller than used in human GWAS studies, where sample sizes often exceed 10,000 individuals, but is larger or similar to the number of accessions used in GWAS in Arabidopsis thaliana,25,52,53 O. sativa,36 Zea mays54,55 and Medicago truncatula56 in which phenotypic data could be collected in common environments on replicated genotypes. The differences in genetic balance (homozygosity in self-pollinated species or inbreds of cross-pollinated species against heterozygosity in humans) and the use of germplasm resources including landraces, improved and obsolete varieties, make GWAS work for such crop species even with a small panel size.
The 5,246 SNP array designed for Illumina Infinium assay was efficient enough to rapidly genotype SNP loci in 220 rice accessions with high precision and success rate. The estimated SNP genotyping success rate (∼94%) and missing genotype information (∼5%) were comparable to that documented previously using Illumina GoldenGate assay in rice,57,58 Affymatrix custom array (44K)37 and with a recently developed Illumina Infinium RiceSNP50 array.59 This suggests the reliability and utility of the SNP chip array for high-throughput SNP genotyping in rice using Infinium assay. About 67% of the SNPs in the array were annotated as ‘coding’ SNPs and the remaining in the 5′UTRs (33%) as ‘regulatory’ SNPs. SNPs within genic regions have greater potential to affect gene function and are usually more informative.59 Also, the enrichment of array with plenty of such genic SNPs is worthwhile in genome-wide studies focussed to map genes for biotic/abiotic stress tolerance.
So far, ∼70 salt tolerance QTLs had been located in rice using bi-parental mapping populations,60,61 but fine mapping and narrowing down reports are limited. Only, Saltol QTL region14 has been inspected widely, and three genes (SKC1, SalT and pectinesterase) are annotated and functionally characterized in this region.62,63 The Na+/K+ ratio is an important ion balancing parameter for salt tolerance in rice, and our GWA mapping results identified this important region as a dense GWAS peak covering the Saltol region, which helps control K+ homeostasis under salinity.62 Previous studies claimed that Saltol controls salinity only at seedling stage and suggested that tolerance at seedling and reproductive stages is regulated by a different set of genes and QTLs.11 However, in our study, where all the traits were measured at reproductive stage, the Saltol region was mapped with a set of highly significant SNPs on chromosome 1. In our study, the results for Na+/K+ ratio were found more important than other traits as all the significant SNPs for this trait were found in complete LD. The results of mixed model considering Na+/K+ ratio are shown in Figs 6 and 7. The significant SNPs detected on chromosome 1 on or near the Saltol region were novel in our study and were not reported earlier from the three known genes of Saltol. All the 12 SNPs were scattered between the coordinates 9.6 and 14.5 Mb, and the top one was located at 12.8 Mb. The top hit from this region was from the coordinate 1:12854366 (LOC_Os01g22900; neutral/alkaline invertase), the other one was from the coordinate 1:12831995 (LOC_Os01g22870; expressed protein). Two significant SNPs (1: 11608731 and 12054948) from this region were between the two Saltol specific genes (SKC1 and pectinesterase). The region was found with the most prominent GWAS peak in the study with very high LD (average r2 = 0.56). Its relevance for salt tolerance in seedling stage has already been explored, and it has been widely used for introgression into a wide range of recipient elite cultivars. The mapping of novel functional allelic variants detected in the genes underlying Saltol QTL in current study at reproductive stage is a significant finding, which once validated can effectively be utilized in rice breeding programs to enhance salt tolerance.
Figure 6.
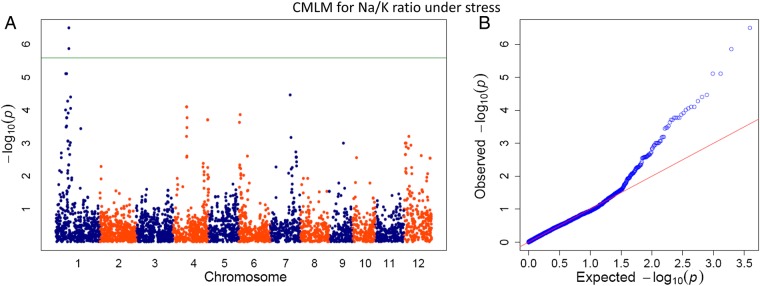
Manhattan plot (A) and corresponding quantile–quantile plots (B) of P-values analysed using CMLM approach for Na+/K+ ratio under stress.
Figure 7.

Chromosome view of significant associations, identified for Na+/K+ ratio under stress. The alongside colour-coded graphs are representing MAF, P-value and r2 of significant SNPs from the respective chromosomes.
For Na+/K+ ratio, chromosome 4 was also found important, and two different regions on this chromosome were mapped for the trait. SNPs on this chromosome showing significant associations were found in an LD block. This LD block is situated near a QTL region for Na+ concentration and Na+/K+ ratio, found by Koyama et al.13 However, this region has not yet been fine mapped. We investigated all the locus ID spanning this LD block with available GEO data set (GSM159265) for salinity tolerance and found that three genes, i.e. LOC_Os04g23550, LOC_Os04g23580 and LOC_Os04g24110, were significantly up-regulated under salt stress with fold change value of 3.6, 9.26 and 3.12, respectively, in salt-tolerant rice variety Agami. Another association peak located at the end of this chromosome with two top SNPs (34164920 and 34292214) explaining equal phenotypic variation at the same significance level showed very high LD with surrounding 13 SNPs covering a distance of 1.2 Mb. Two QTLs for salt tolerance and potassium concentration were mapped just prior to this region, respectively, by Lin et al.20 and Cai and Morishima.64 Investigation of available GEO data set (GSM159265) of the entire locus from this region identified four genes, i.e. LOC_Os04g57760, 57800, 57810 and 57850 with 7.50-, 2.10-, 4.24- and 3.90-fold change in expression, respectively.
For Na+/K+ ratio, the most significant SNP (6:1460570; LOC_Os6g03700) from an association peak located at one end of the chromosome 6 was surrounded by two dehydration-responsive element-binding protein (LOC_Os6g03670 and LOC_Os6g03750) and two calcium-dependent protein kinase (CDPK) proteins. All these proteins, including the significant one of our study, are located between the coordinates, 1435857 and 1478931, covering only 43 kb region. Out of these proteins, OsDREB1C (LOC_Os6g03670) is known and characterized for salinity stress tolerance in Arabidopsis65 and in rice.66 This region also carried the QTL for Na+ ion concentration proposed by Koyama et al.13
A single SNP (19655274) identified on chromosome 7 in this panel is in the gene, LOC_Os07g32880 (ATP synthase gamma chain, putative, expressed). When top SNPs from this region of Manhattan plot (<0.01 simple P-value) were ordered, the region was tracked between the coordinates, 18204319 and 21821714. This region was not mapped with earlier known QTLs for salinity-related parameters such as Na+ content or for Na+/K+ ratio; however, a cluster of QTLs for yield-related components under salt stress was identified in this region by Mohammadi et al.67
For sodium content, the only significant SNP (FDR-adjusted P = 0.002) was reported from chromosome 1: 41550991 (LOC_Os01g71240) in the gene calcium-transporting ATPase, plasma membrane type. Though the role of such plant Ca2+ATPases in salinity stress68,69 is known, its significant association with Na+ content observed in the study needs enrichment of the region with SNPs and further LD mapping studies.
When the traits such as yield/plant, productive tillers, filled grains, percent spikelet fertility and SFSSI observed under stress were employed for association mapping, typically same set of SNPs hit all these parameters with little difference in P-value. A very high canonical correlation was observed among all these traits under stress, which probably led to co-localization of GWAS peaks containing the same set of SNPs. Association signals were not detected as dense GWAS peaks for these traits. All these traits are very critical for reproductive stage and showed greater variability with very high range correlations under stress. Susceptibility indices were computed for yield and its related traits, of which we found associations only for SFSSI and not for yield. Similarly, Pandit et al.17 detected QTL (qSSISFH8.1) on chromosome 8 only for SFSSI and not for seed weight SSI and yield SSI. One of our significant SNPs (chr8: 5109310) for SFSSI overlapped the QTL, qSSISFH8.1, which harboured another QTL (qNaSH8.1) for straw sodium content under high salinity. This SNP also overlapped with a region for high salt concentration discovered by Islam et al.70 An SNP (chr5: 766912), significant for all the traits under stress, was found in the region of a QTL (qNar/Kr5) for Na+/K+ ratio in root discovered by Ahamdi and Fotokian.18 Two significant SNPs (7:2453894 and 7:22268862) were detected near qSNC7 and qSDS7 discovered by Lin et al.15 Another QTL (qRNC9) for root Na+ concentration was overlapped by two SNPs on chromosome 9 (16503353 and 17002144). An SNP on chromosome 9 (6030743) showed significant signal within the range of qSNC9/qSKC9 QTL for shoot Na+ and K+ concentration discovered by Wang et al.71 All the SNPs that showed significant association on chromosome 10 (5864939–14809695) were found between the markers aligned with a QTL discovered by Islam et al.70 for high salt tolerance. At reproductive stage, finding a number of regions underlying salinity stress in current panel of rice indicates complexity of genetic architecture of this trait, which cannot be unravelled by a single GWAS study or analysis method. To provide concrete evidence that they do affect the trait, the significantly associated genomic regions in the study can further be explored to find causal variants.
Apart from SNPs that had association with known QTLs for salinity tolerance, there were also a few SNPs, which hit specific genes that were known or functionally characterized for salt stress. A significant SNP on chromosome 1 (40514883) for filled grain under stress was found near two functionally characterized genes namely OsNAC6/SNAC2 for drought, salt and cold tolerance in rice during seedling development.72,73 Another SNP on chromosome 1 (7304902) showed significant signals near a salt stress related root protein (RS1/LOC_Os01g13210), functionally annotated for its role in salt resistance, and located 50 kb away from the tagged SNP.74 The tagged SNP and the subsequent region through RS1 gene were found in high LD range in our study. An SNP on chromosome 9 (16503353) that showed significant association for productive tillers under stress was found 100 kb apart from a known gene, Osdg1 (delayed seed germination) for salinity tolerance. Other SNPs, which were identified as significant, might have their direct role in salt tolerance, or alternatively, it may be that these SNPs are in LD with genes that are yet to be identified and arrayed on the chip used. The region across the tagged locus is critical to the size of the target region, but it is still unclear which r2 threshold should be set in defining LD structure, as the causal SNP potentially could be in LD with the associated SNP at an r2 of 0.2 or even less.75 Finding association in or near such candidate genes although highlights the importance of the candidate gene-based chip, such single-variant trait associations need enrichment of these genomic regions either with more SNPs or complete re-sequencing to avoid spurious associations. The use of lower density chip might be a factor in restricting amplification of these GWAS peaks. The involvement of larger panel coupled with higher density of SNPs would not only improve the stringency of GWAS peaks identified but also detect new QTLs of minor effects, which have been completely skipped in this study.
The current SNP array also generated association signals for phenotypic traits plant height, unfilled grains and spikelet fertility under normal growth conditions. Dozens of genes and QTLs regulating plant height in rice have been identified previously. Mixed model approach in our study detected strong signals of a cluster of seven significant SNPs within the range of Fh4-1, on chromosome 4, a known QTL for plant height. For spikelet fertility, five SNPs were found significant; however, the phenotypic variance explained by them was found below 7%. Signals for spikelet fertility were found very strong on chromosomes 2, 7 and 9. Significant SNPs on chromosome 2 overlapped with a known QTL (qSS2) for spikelet sterility under control that was identified by Li et al.76 However, these SNPs were not detected from peak region of qSS2 QTL, rather overlapped with end of the QTL region. Top SNPs on chromosome 7 were also found close to a known QTL for spikelet fertility under natural environment, earlier identified by He and Xu.77 However, significant SNPs detected on chromosome 9 did not overlap with any earlier known QTL for spikelet fertility and also found significant for unfilled grains. All the GWAS peaks for spikelet fertility from the novel regions in current set of association panel can be good candidates for further mapping and functional characterization.
In genome-wide association studies, SNP-trait association depends on the phenotypic variance in the population explained by the SNP. Recent GWAS in plants have explained a much greater proportion of the phenotypic variation, and the assumption that common genetic variation explains common phenotypic variation holds.78 In the current set of data, the phenotypic variance explained individually by SNPs for different traits with or without salinity stress was in complete concordance with earlier studies on plants such as rice37 and spring wheat.79 Its wider range (5–20%) for most of the traits under stress as well as without stress at reproductive stage entails genome-wide significance and high statistical power of current panel of 220 rice accessions.44 Previous GWA studies on Arabidopsis confirmed that for some of the traits, meaningful results can be obtained even with <100 accessions.25 In our study, for most of the traits, the variation was captured by a single SNP or with a few SNPs from a particular genomic region due to medium density SNP array, unlike the 44K array37 and ∼3.6 million SNPs used earlier27 where there is strong possibility of dense GWAS peaks due to a very high LD between adjacent genotyped SNPs. However, an earlier study conducted to evaluate the extent of LD within different subgroups of rice suggested that a modest number of SNPs across the genome may be sufficient for undertaking genome-wide LD mapping studies in rice.80
5. Conclusion
Genotyping of 220 indica rice genotypes using a custom-designed 6K genic SNP array successfully detected SNPs associated with a number of phenotypic traits at reproductive stage under salinity stress. The indica diversity panel selected in the study to unveil the genomic regions relevant for salinity tolerance was found diverse to the extent that SNP-trait associations were stronger under salinity compared with control treatment. The novel functional allelic variants within the genes underlying Saltol, a major QTL, known to control salinity tolerance at seedling stage are also identified. This finding helps us to delineate alleles in the genes harbouring one of the most important Saltol QTLs at the reproductive stage as against the earlier views that different gene networks are responsible for conferring salinity tolerance at seedling and reproductive stages. Identification of Saltol and other stress-related genomic regions in the study indicates precise evaluation of the phenotypic traits under salinity stress as well as the potential of the SNP array in detecting associations. Besides Saltol, we found some other GWAS peaks with small to moderate effects, which suggest that salinity is a complex trait and controlled by many genetic loci. The novel SNPs identified here are attractive candidates for functional characterization studies that could further enrich our understanding on genetic architecture of salinity stress tolerance in rice.
Supplementary Data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
The work was carried out under the project ‘Bioprospecting of genes and allele mining for abiotic stress tolerance’, funded by National Agricultural Innovation Project (NAIP), Indian Council for Agricultural Research, New Delhi. Funding to pay the Open Access publication charges for this article was provided by the Indian Council of Agricultural Research.
Supplementary Material
References
- 1.Shannon M.C., Rhoades J.D., Draper J.H., Scardaci S.C., Spyres M.D. 1998, Assessment of salt tolerance in rice cultivars in response to salinity problems in California, Crop Sci., 38, 394–8. [Google Scholar]
- 2.Fukuda A., Nakamura A., Tagiri A., et al. 2004, Function, intracellular localization and the importance in salt tolerance of a vacuolar Na+/H+ antiporter from rice, Plant Cell Physiol., 45, 146–59. [DOI] [PubMed] [Google Scholar]
- 3.Flowers T.J., Yeo A.R. 1981, Variability in the resistance of sodium chloride salinity within rice (Oryza sativa L.) varieties, New Phytol., 88, 363–73. [Google Scholar]
- 4.Lutts S., Kinet J.M., Bouharmont J. 1995, Changes in plant response to NaCl during development of rice (Oryza sativus L.) varieties differing in salinity resistance, J. Exp. Bot., 46, 1843–52. [Google Scholar]
- 5.Zeng L., Shannon M.C. 2000, Salinity effects on the seedling growth and yield components of rice, Crop Sci., 40, 996–1003. [Google Scholar]
- 6.Hoshikawa K. 1989, Growing rice plant—an anatomical monograph. Tokyo, Japan: Nosan Gyoson Bunka Kyokai (Nobunkyo). [Google Scholar]
- 7.Garcia A., Rizzo C.A., Ud-Din J., et al. 1997, Sodium and potassium transport to the xylem are inherited independently in rice, and the mechanism of sodium:potassium selectivity differs between rice and wheat, Plant Cell Environ., 20, 1167–74. [Google Scholar]
- 8.Walia H., Wilson C., Zeng L., Ismail A.M., Condamine P., Close T.J. 2007, Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage, Plant Mol. Biol., 63, 609–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao P.S., Mishra B., Gupta S.R., Rathore A. 2008, Reproductive stage tolerance to salinity and alkalinity stresses in rice genotypes, Plant Breed., 127, 256–61. [Google Scholar]
- 10.Zeng L., Shannon M.C., Grieve C.M. 2002, Evaluation of salt tolerance in rice genotypes by multiple agronomic parameters, Euphytica, 127, 235–45. [Google Scholar]
- 11.Moradi F., Ismail A.M., Gregorio G., Egdane J. 2003, Salinity tolerance of rice during reproductive development and association with tolerance at seedling stage, Indian J. Plant Physiol., 8, 105–16. [Google Scholar]
- 12.Blumwald E., Grover A. 2006, Salt tolerance, In: Plant biotechnology: current and future uses of genetically modified crops. John Wiley and Sons Ltd, UK, pp. 206–24. [Google Scholar]
- 13.Koyama L.M., Levesley A., Koebner R.M.D., Flowers T.J., Yeo A.R. 2001, Quantitative trait loci for component physiological traits determining salt tolerance in rice, Plant Physiol., 125, 406–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonilla P., Dvorak J., Mackill D., Deal K., Gregorio G. 2002, RFLP and SSLP mapping of salinity tolerance genes in chromosome 1 of rice (Oryza sativa L.) using recombinant inbred lines, Philipp. J. Agric. Sci., 85, 68–76. [Google Scholar]
- 15.Lin H.X., Zhu M.Z., Yano M., et al. 2004, QTLs for Na+ and K+ uptake of the shoots and roots controlling rice salt tolerance, Theor. Appl. Genet., 108, 253–60. [DOI] [PubMed] [Google Scholar]
- 16.Ammar M.H.M., Pandit A., Singh R.K., et al. 2009, Mapping of QTLs controlling Na+, K+ and Cl− ion concentrations in salt tolerant indica rice variety CSR27, J. Plant Biochem. Biotech., 18, 139–50. [Google Scholar]
- 17.Pandit A., Rai V., Bal S., et al. 2010, Combining QTL mapping and transcriptome profiling of bulked RILs for identification of functional polymorphism for salt tolerance genes in rice (Oryza sativa L.), Mol. Genet. Genomics, 284, 121–36. [DOI] [PubMed] [Google Scholar]
- 18.Ahmadi J., Fotokian M.H. 2011, Identification and mapping of quantitative trait loci associated with salinity tolerance in rice (Oryza sativa) using SSR markers, Iran. J. Biotechnol., 9, 21–30. [Google Scholar]
- 19.Zhang G.Y., Guo Y., Chen S.L., Chen S.Y. 1995, RFLP tagging of a salt tolerance gene in rice, Plant Sci., 110, 227–34. [Google Scholar]
- 20.Lin H.X., Yanagihara S., Zhuang J.Y., Senboku T., Zheng K.L., Yashima S. 1998, Identification of QTLs for salt tolerance in rice via molecular markers, Chin. J. Rice Sci., 12, 72–8. [Google Scholar]
- 21.Gong J.M., He P., Qian Q.A., Shen L.S., Zhu L.H., Chen S.Y. 1999, Identification of salt-tolerance QTL in rice (Oryza sativa L.), Chin. Sci. Bull., 44, 68–71. [Google Scholar]
- 22.Prasad S.R., Bagali P.G., Hittalmani S., Shashidhar H.E. 2000, Molecular mapping of quantitative trait loci associated with seedling tolerance to salt stress in rice (Oryza sativa L.), Curr. Sci., 78, 162–4. [Google Scholar]
- 23.Cheng L., Wang Y., Meng L., et al. 2012, Identification of salt-tolerant QTLs with strong genetic background effect using two sets of reciprocal introgression lines in rice, Genome, 55, 45–55. [DOI] [PubMed] [Google Scholar]
- 24.Pasam R.K., Sharma R., Malosetti M., et al. 2012, Genome-wide association studies for agronomical traits in a worldwide spring barley collection, BMC Plant Biol., 12, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atwell S., Huang Y.S., Vilhjálmsson B.J., Willems G., Horton M., Li Y. 2010, Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines, Nature, 465, 627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aranzana M.J., Kim S., Zhao K., et al. 2005, Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes, PLoS Genet., 1, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X., Wei X., Sang T., et al. 2010, Genome-wide association studies of 14 agronomic traits in rice landraces, Nat. Genet., 42, 961–7. [DOI] [PubMed] [Google Scholar]
- 28.Famoso A.N., Zhao K., Clark R.T., et al. 2011, Genetic architecture of aluminum tolerance in rice (Oryza sativa) determined through genome-wide association analysis and QTL mapping, PLoS Genet., 7, e1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cockram J., White J., Zuluaga D.L., et al. 2010, Genome-wide association mapping to candidate polymorphism resolution in the unsequenced barley genome, Proc. Natl. Acad. Sci. USA, 107, 21611–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritchard J., Stephens M., Donnelly P. 2000, Inference of population structure using multilocus genotype data, Genetics, 155, 945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. 2006, Principal components analysis corrects for stratification in genome-wide association studies, Nat. Genet., 38, 904–9. [DOI] [PubMed] [Google Scholar]
- 32.Yu J., Pressoir G., Briggs W.H., et al. 2006, A unified mixed model method for association mapping that accounts for multiple levels of relatedness, Nat. Genet., 38, 203–8. [DOI] [PubMed] [Google Scholar]
- 33.Thornsberry J.M., Goodman M.M., Doebley J., Kresovich S., Nielsen D., Buckler E.S. 2001, Dwarf8 polymorphisms associate with variation in flowering time, Nat. Genet., 28, 286–9. [DOI] [PubMed] [Google Scholar]
- 34.Flint-Garcia S.A., Thornsberry J.M., Buckler E.S. 2003, Structure of linkage disequilibrium in plants, Annu. Rev. Plant Biol., 54, 357–74. [DOI] [PubMed] [Google Scholar]
- 35.Zhao K.Y., Aranzana M.J., Kim S., et al. 2007, An Arabidopsis example of association mapping in structured samples, PLoS Genet., 3, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X., Zhao Y., Wei X., et al. 2011, Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm, Nat. Genet., 44, 32–9. [DOI] [PubMed] [Google Scholar]
- 37.Zhao K., Tung C.W., Eizenga G.C., et al. 2011, Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa, Nat. Commun., 2, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Negrão S., Almadanim M.C., Pires I.S., et al. 2013, New allelic SNPs found in key rice salt-tolerance genes: an association study, Plant Biotech. J., 11, 87–100. [DOI] [PubMed] [Google Scholar]
- 39.Fischer R.A., Maurer R. 1978, Drought resistance in spring wheat cultivars. I. Grain yield responses, Crop Pasture Sci., 29, 897–912. [Google Scholar]
- 40.McNally K.L., Childs K.L., Bohert R., et al. 2009, Genome-wide SNP variation reveals relationships among landraces and modern varieties of rice, Proc. Natl. Acad. Sci. USA, 106, 12273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doyle J.J., Doyle J.L. 1987, A rapid DNA isolation procedure for small quantities of fresh leaf tissue, Phytochem. Bull., 19, 11–15. [Google Scholar]
- 42.Fan J.B., Oliphant A., Shen R., et al. 2003, Highly parallel SNP genotyping. In: Cold Spring Harbor Symposia on Quantitative Biology, vol. 68. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp. 69–78. [DOI] [PubMed] [Google Scholar]
- 43.Lipka A.E., Tian F., Wang Q., et al. 2012, GAPIT: genome association and prediction integrated tool, Bioinformatics, 28, 2397–9. [DOI] [PubMed] [Google Scholar]
- 44.Kang H.M., Zaitlen N.A., Wade C.M., et al. 2008, Efficient control of population structure in model organism association mapping, Genetics, 178, 1709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Purcell S., Neale B., Todd-Brown K., et al. 2007, PLINK: a tool set for whole-genome association and population-based linkage analyses, Am. J. Hum. Genet., 81, 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheet P., Stephens M. 2006, A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase, Am. J. Hum. Genet., 78, 629–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bradbury P.J., Zhang Z., Kroon D.E., Casstevens T.M., Ramdoss Y., Buckler E.S. 2007, TASSEL: software for association mapping of complex traits in diverse samples, Bioinformatics, 23, 2633–5. [DOI] [PubMed] [Google Scholar]
- 48.Gabriel S., Schaffner S., Nguyen H., et al. 2002, The structure of haplotype blocks in the human genome, Science, 296, 2225–9. [DOI] [PubMed] [Google Scholar]
- 49.Duggal P., Gillanders E.M., Holmes T.N., Bailey-Wilson J.E. 2008, Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies, BMC Genomics, 9, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benjamini Y., Hochberg Y. 1995, Controlling the false discovery rate—a practical and powerful approach to multiple testing, J. Roy. Stat. Soc. B Met., 57, 289–300. [Google Scholar]
- 51.Evanno L., Nay B., Bodo B. 2005, Unexpected dehydrogenation products in the furan series arising from ruthenium-catalyzed 4-oxo-1, 6-enyne metathesis, Synthetic Commun., 35, 1559–65. [Google Scholar]
- 52.Chan E.K., Rowe H.C., Corwin J.A., Joseph B., Kliebenstein D.J. 2011, Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana, PLoS Biol., 9, e1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Derose-Wilson L., Gaut B.S. 2011, Mapping salinity tolerance during Arabidopsis thaliana germination and seedling growth, PLoS ONE, 6, e22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krill A.M., Kirst M., Kochian L.V., Buckler E.S., Hoekenga O.A. 2010, Association and linkage analysis of aluminum tolerance genes in maize, PLoS ONE, 5, e9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riedelsheimer C., Lisec J., Czedik-Eysenberg A., et al. 2012, Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize, Proc. Natl. Acad. Sci. USA, 109, 8872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stanton-Geddes J., Paape T., Epstein B., et al. 2013, Candidate genes and genetic architecture revealed by whole genome, sequence-based association genetics in Medicago truncatula, PLoS ONE, 8, e65688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parida S.K., Mukerji M., Singh A.K., Singh N.K., Mohapatra T. 2012, SNPs in stress-responsive rice genes: validation, genotyping, functional relevance and population structure, BMC Genomics, 13, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao K., Wright M., Kimball J., Eizenga G., McClung A. 2010, Genomic diversity and introgression in O. sativa reveal the impact of domestication and breeding on the rice genome, PLoS ONE, 5, e10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen H.D., Xie W.B., He H., et al. 2013, A high-density SNP genotyping array for rice biology and molecular breeding, Mol. Plant, 7, 541–53. [DOI] [PubMed] [Google Scholar]
- 60.Zang J.P., Sun Y., Wang Y., et al. 2008, Dissection of genetic overlap of salt tolerance QTLs at the seedling and tillering stages using backcross introgression lines in rice, Sci. China C Life Sci., 51, 583–91. [DOI] [PubMed] [Google Scholar]
- 61.Hu S., Tao H., Qian Q., Guo L. 2012, Genetics and molecular breeding for salt-tolerance in rice, Rice Genomics Genet., 3, 39–49. [Google Scholar]
- 62.Ren Z.H., Gao J.P., Li L.G., et al. 2005, A rice quantitative trait locus for salt tolerance encodes a sodium transporter, Nat. Genet., 37, 1141–6. [DOI] [PubMed] [Google Scholar]
- 63.Claes B., Dekeyser R., Villarroel R., van den Bulcke M., van Montagu M., Caplan A. 1990, Characterization of a rice gene showing organ-specific expression in response to salt stress and drought, Plant Cell, 2, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cai H.W., Morishima H. 2002, QTL clusters reflect character associations in wild and cultivated rice, Theor. Appl. Genet., 104, 1217–28. [DOI] [PubMed] [Google Scholar]
- 65.Yamaguchi-Shinozaki K., Shinozaki K. 1994, A novel cis-acting element in an Arabidopsis gene is involved in responsiveness to drought, low-temperature or high-salt stress, Plant Cell, 6, 251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dubouzet J.G., Sakuma Y., Ito Y., et al. 2003, DREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression, Plant J., 33, 751–63. [DOI] [PubMed] [Google Scholar]
- 67.Mohammadi R., Mendioro M.S., Diaz G.Q., Gregorio G.B., Singh R.K. 2013, Mapping quantitative trait loci associated with yield and yield components under reproductive stage salinity stress in rice (Oryza sativa L.), J. Genet., 92, 433–43. [DOI] [PubMed] [Google Scholar]
- 68.Mahajan S., Tuteja N. 2005, Cold, salinity and drought stresses: an overview, Arch. Biochem. Biophys., 444, 139–58. [DOI] [PubMed] [Google Scholar]
- 69.Huda K.M.K., Banu M.S.A., Tuteja R., Tuteja N. 2013, Global calcium transducer P-type Ca2+-ATPases open new avenues for agriculture by regulating stress signalling, J. Exp. Bot., 64, 3099–109. [DOI] [PubMed] [Google Scholar]
- 70.Islam M.R., Salam M.A., Hassan L., Collard B.C.Y., Singh R.K., Gregorio G.B. 2011, QTL mapping for salinity tolerance in at seedling stage in rice, Emir. J. Food Agric., 23, 137–46. [Google Scholar]
- 71.Wang Z., Chen Z., Cheng J., et al. 2012, QTL analysis of Na+ and K+ concentrations in roots and shoots under different levels of NaCl stress in rice (Oryza sativa L.), PLoS ONE, 7, e51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakashima K., Ito Y., Yamaguchi-Shinozaki K. 2009, Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses, Plant Physiol., 149, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu H., You J., Fang Y., Zhu X., Qi Z., Xiong L. 2008, Characterization of transcription factor gene SNAC2 conferring cold and salt tolerance in rice, Plant Mol. Biol., 67, 169–81. [DOI] [PubMed] [Google Scholar]
- 74.Kikuchi S., Satoh K., Nagata T., et al. 2003, Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice, Science, 301, 376–9. [DOI] [PubMed] [Google Scholar]
- 75.Freedman M.L., Monteiro A.N., Gayther S.A., et al. 2011, Principles for the post-GWAS functional characterization of cancer risk loci, Nat. Genet., 43, 513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Z.K., Pinson S.R.M., Paterson A.H., Parkm W.D., Stansel J.W. 1997, Epistasis for three grain yield components in rice (Oryza sativa L.), Genetics, 145, 453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He Y.Q., Xu C.G. 2000, Importance of epistasis as the genetic basis of sterility instability for photoperiod-sensitive genic male sterile rice, Acta Bot. Sin., 42, 1062–8. [Google Scholar]
- 78.Brachi B., Morris G.P., Borevitz J.O. 2011, Genome-wide association studies in plants: the missing heritability is in the field, Genome Biol., 12, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang J., Chen J., Bowman B.C., et al. 2014, Association mapping of Hagberg falling number in hard white spring wheat, Crop Sci., 54, 1243–52. [Google Scholar]
- 80.Mather K.A., Caicedo A.L., Polato N., Olsen K.M., McCouch S.R., Purugganan M.D. 2007, The extent of linkage disequilibrium in rice (Oryza sativa L.), Genetics, 177, 2223–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.