

Abstract
Optical mapping has proven to be a valuable technique to detect cardiac electrical activity on both intact ex vivo hearts and in cultured myocyte monolayers. HL-1 cells have been widely used as a 2-Dimensional cellular model for studying diverse aspects of cardiac physiology. However, it has been a great challenge to optically map calcium (Ca) transients and action potentials simultaneously from the same field of view in a cultured HL-1 atrial cell monolayer. This is because special handling and care is required to prepare healthy cells that can be electrically captured and optically mapped. Therefore, we have developed an optimal working protocol for dual channel optical mapping. In this manuscript, we have described in detail how to perform the dual channel optical mapping experiment. This protocol is a useful tool to enhance the understanding of action potential propagation and Ca kinetics in arrhythmia development.
Keywords: Cellular Biology, Issue 97, Cellular optical mapping, cultured atrial monolayer, action potential, calcium transient, voltage-sensitive dye, calcium dye
Introduction
A unique calcium (Ca) and voltage (Vm) dual channel optical mapping technique1-5 is emerging as an efficient tool to simultaneously record Vm and Ca signals in both intact hearts and cultured cell monolayers. This technique makes it possible to obtain powerful information regarding the relationship between calcium transients and action potentials to better understand the underlying electrophysiological mechanisms of cardiac arrhythmias.
Cultured cell monolayers have proven to be a useful cellular model to study cardiac electrophysiology and the underlying mechanism of arrhythmias.4,6-8 HL-1 cells are a well-characterized atrial myocyte culture line. HL-1 cells also maintain a uniquely differentiated genotype and phenotype that includes morphologic, electrophysiologic, and pharmacologic characteristics of adult atrial myocytes. These cells express cardiac genes and proteins, including important cardiac ion channels (i.e., L- and T-type calcium channels)9 and mature isoforms of sarcomeric contractile proteins normally found in adult atrial myocytes as others and we have previously reported.10-13 In addition, HL-1 cells can be cultured to form a 2-dimensional (2-D) myocyte monolayer. Thus, the advantages of using cultured HL-1 monolayers include: 1) relatively lower cost and easier to maintain a myocyte culture line than isolating and culturing primary neonatal myocytes; 2) a confluent monolayer of cells reduces the structural complexities that result from the 3-D structure of the heart; 3) a cell monolayer can eliminate the interference of interstitial fibrosis that occurs in the intact heart. This can be used to dissect specific electrophysiological functions of a group of myocytes without interference of fibroblasts and interstitial matrix; 4) assessment of functional consequences from pharmacological or genetic manipulation in cultured cell monolayers can be effectively achieved. Therefore, HL-1 cells have become a widely used cellular model for studying diverse aspects of myocyte physiology as well as pacing-induced abnormal electrical activities.13-16 However, special handling and care is required to culture healthy cell monolayers that respond to external electrical pacing for optical mapping studies. In addition, dual florescent dye staining procedure may easily damage the integrity of cultured confluent cell monolayers. Thus, performing Vm/CA dual channel optical mapping in cultured HL-1 monolayers has been a great challenge.
The goal of this method is to provide the key steps for successfully performing dual channel optical mapping in cultured HL-1 monolayers. Here we have provided extensive details on an optimized protocol for HL-1 cell monolayer preparation, dual channel optical mapping of a cultured cell monolayer, and mapping data processing.
Protocol
1. Solution Preparation
- Norepinephrine (NP) solution
- Dissolve 40 mg of NP into 25 ml of 30 mM ascorbic acid (0.1475 g ascorbic acid in 25 ml of purified Milli-Q (MQ) water. Avoid direct light exposure during this solution preparation because norepinephrine (NP) is light sensitive.
- Filter the NP solution with a 0.22 µm syringe filter. Aliquot the solution into 1 ml sterile microtubes and store at -20 °C. Once frozen, NP is stable for one month.
- Supplemented Wash and Final Claycomb media
- Start with 43.5 ml of Claycomb medium and supplement with 5 ml of fetal bovine serum (FBS), 0.5 ml of penicillin/streptomycin, 0.5 ml of NP and 1:100 diluted L-Glutamine to make 50 ml of final supplemented Claycomb medium. Avoid direct light exposure during this solution preparation because Claycomb medium is light sensitive.
- Prepare the Wash Claycomb medium using the same recipe as the supplemented Claycomb medium except add 5% FBS and omit the NP and L-Glutamine components. NOTE: The supplemented Final and Wash Claycomb media are good for two weeks when stored at 4 °C without direct light exposure.
- Soybean Trypsin inhibitory solution
- Dissolve 25 mg of soybean trypsin inhibitor into 100 ml of Ca-free and Mg-free Dulbecco’s phosphate buffered saline (PBS).
- Filter the solution using a 0.22 µm syringe filter. NOTE: The sterilized solution is stable for one month when stored at 4 °C.
- Gelatin/Fibronectin dish coating solution
- Dissolve 0.1 g of gelatin into 500 ml of MQ water to make 0.02% gelatin.
- Autoclave, then dilute 1 ml of fibronectin into 199 ml of 0.02% gelatin and mix gently.
- Aliquot 6 ml of gelatin solution into separate tubes and store at -20 °C.
- Hanks’ Salt solution
- Using a stir plate, dissolve the following salts in MQ water (mmol/L), NaCl (136.9), KCl (5.366), KH2PO4 (0.4409), NaH2PO4 (0.4000), MgSO4 (0.8142), D-Glc (5.051), NaHCO3 (4.162), HEPEs (5.042), CaCl2 (1.261).
- Calcium sensitive dye (Rhod2) solution
- Dilute 1 mg of Rhod2 dry powder in 400 μl of 20% Pluronic F-127 in DMSO to make a Rhod2 dye stock solution and store at -20 °C.
- Dilute this stock solution 1:1,000 with Hanks' Salt solution to make a final working solution before starting the experiment.
- Voltage sensitive dye (Rh237) solution
- Dilute 5 mg of Rh237 with 2 ml of DMSO to make stock solution and store at -20 °C. Dilute the stock solution 1:1,000 with Hanks' Salt solution to make a final working solution before starting the experiment.
NOTE: 1-4 solutions are based on the Claycomb HL-1 cell culture protocol with minor modification.
2. Passaging and Culturing HL-1 Atrial Myocytes onto Coverslips
NOTE: The HL-1 cells can be obtained from Dr. Claycomb (Louisiana State University).
Grow HL-1 cells in T25 flasks and feed with 5 ml of supplemented Claycomb Medium daily. Passage the cells in T25 flasks every five days according to the Claycomb HL-1 cell culture protocol (described below)10.
Place round coverslips (20 mm diameter) into 12-well culture plates 24 hr before the cells in the T25 flask are fully confluent and ready to be passaged, and then cover the coverslips with gelatin/fibronectin overnight at 37 °C.
The next day, aspirate and discard the gelatin/fibronectin from the culture plates and replace with 1.5 ml of supplemented Final Claycomb medium in each well.
Split cells from a T25 culture flask after reaching full confluence at day 5. Aspirate the medium from the T25 flask containing the confluent culture and rinse the culture briefly with PBS at 37 °C.
Aspirate PBS and add 3 ml of 0.05% trypsin/EDTA at 37 °C by pipetting the trypsin/EDTA onto the bottom of the T25 flask to avoid direct contact with the cells. Incubate at 37 °C for 1 min.
Remove trypsin/EDTA by aspiration and add another 1.3 ml of trypsin/EDTA per T25 flask. Incubate at room temperature for 1 min. Examine under a microscope and if cells are still attached to the flask, tap to fully dislodge remaining cells.
Add 1.3 ml of soybean trypsin inhibitor to the cells and transfer cells from the flask into a 15 ml centrifuge tube. Rinse the empty flask with 5 ml of Wash Claycomb medium. Add the rinse to the cells in the 15 ml centrifuge tube. Centrifuge at 500 x g for 5 min.
After centrifuging, aspirate the supernatant and gently re-suspend the cell pellet in 6 ml of supplemented Final Claycomb medium.
Seed each well of a 12-well plate with 0.5 ml of cell suspension from cell passaging. Feed the cells with fresh supplemented Final Claycomb medium daily. NOTE: Closely monitor the growth of the cells, typically the cell monolayers reach confluent status on day 4 and the spontaneous activities remain visible. Then, the cultured monolayer will be ready for optical mapping on day 4-5.
3. Loading Calcium and Voltage Sensitive Dyes to the Cells
For optical mapping studies, gently remove the coverslip from the 12-well cell culture plate and rinse with Hanks' Salt solution.
To stain cells with Ca sensitive dye Rhod2, incubate the cell monolayer on the coverslip with 3 ml of the oxygenated (bubbling with 5% CO2/95% O2 gas) Rhod2 working solution at 37 °C in a water bath for 30 min.
To stain the cells with Vm sensitive dye Rh237, immerse the Rhod2 loaded cell monolayer into 3 ml of oxygenated Rh237 working solution at 37 °C in a water bath for another 15 min.
4. Dual Channel Optical Mapping
- Place the cell monolayer, which is double-stained with Vm and Ca dyes, into a circulation chamber on an inverted fluorescence microscope. Use a heated circulation system to keep the temperature of the cell chamber at 35 ± 1 °C. Carefully control the temperature of the perfusate by adjusting the speed of the circulating flow through the chamber to avoid a gradient of more than 0.3 °C across the chamber.
- Before recording Vm/Ca signals, wash the cell monolayer with fresh Hanks solution for approximately 5 min to remove the remaining dye from the previous staining steps.
Pace healthy cell monolayers using a bipolar electrode composed of a glass pipette filled with Hanks' Salt solution and a silver wire coiled around the pipette tip as previously described12,17.
Pace healthy cell monolayers at a cycle length of 200-500 msec and pulse width of 2 msec, expecting a pacing threshold at approximately 1 mV. NOTE: For each batch of cultured monolayers, at least two non-treated monolayers should be used to test the condition of the cell batch. Healthy control monolayers should be able to be captured by electrical pacing at cycle lengths of 200-500 msec. If the control monolayers fail to be electrically paced, this suggests that the entire cell batch may not be suitable for optical mapping experiments.
- To excite both Vm and Ca sensitive dyes, use an LED light source (520 nm wavelength) coupled with an excitation filter (510 nm/40) and a dichroic mirror (561 nm).
- Collect the fluorescent signals emitted by the cells via a 40X objective and split with a dichroic mirror (594 nm) into a Vm component and a Ca component. Filter the emitted light in the Vm channel with a long pass filter (700 nm) and the emitted light in the Ca channel with a band pass filter (585/40 nm).
- Collect the filtered fluorescent light using two CCD cameras (80 x 80 pixels, 1,000 fps; RedShirt Imaging).
- To avoid dye photobleaching, make recordings for less than 2 sec on each field of view to limit the time of light exposure. Here, typically image four different areas for each monolayer.
5. Data Processing of Vm or Ca Recordings
For both Vm and Ca signals (80 x 80 pixels), average the intensity values of twenty-five (5 x 5) neighboring pixels into one intensity value at different recording time points to reduce background noise. NOTE: After the data averaging, each recording time point of an individual action potential or Ca transient yields a data matrix containing 16 x 16 averaged intensity values (averaged data channels) with a spatial resolution of 50 μm.
- Define activation time (AT) of recorded Vm or Ca signals.
- To define an activation time (AT) of an individual action potential or Ca transient, determine the time of 50% of peak amplitude of the Vm or Ca signals using a custom-made MATLAB-based algorithm as previously described.12
- Manually proofread the analyzed ATs to eliminate any errors that may occur during the program analysis.
- Store the defined AT matrix (16 x 16 averaged data channels) of recorded action potential and calcium transient, respectively, in the software used.
- Calculate action potential or calcium transient conduction velocity (CV) vectors on the AT matrix using the vector field analysis method as previously described with minor modifications12,18.
- Calculate the CV vector of a data channel relative to its surrounding AT data points (5 × 5 neighboring averaged data channels) to simulate a smooth polynomial surface of activation times (AT surface) using a least-square algorithm within the MATLAB program12,18.
- Calculate conduction vector using the formulas below Formula 1:
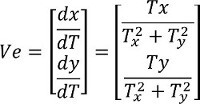 Formula 2: θ = arctan [(dy/dT)/(dx/dT)] NOTE: Ve is a CV vector projected at x- and y-axes; dx/dT is the projection of the CV vector in x-axis; dy/dT is the projection of the CV vector in y-axis. T is the AT surface; x, y refer to the 2-D coordinates; Tx = ∂T/∂x is the partial derivative of T with respect to x; Ty = ∂T/∂y is the partial derivative of T with respect to y; θ is the final direction of a CV vector.
Formula 2: θ = arctan [(dy/dT)/(dx/dT)] NOTE: Ve is a CV vector projected at x- and y-axes; dx/dT is the projection of the CV vector in x-axis; dy/dT is the projection of the CV vector in y-axis. T is the AT surface; x, y refer to the 2-D coordinates; Tx = ∂T/∂x is the partial derivative of T with respect to x; Ty = ∂T/∂y is the partial derivative of T with respect to y; θ is the final direction of a CV vector. - Group all the vectors of the AT matrix into 36 pathways, each of which covers a ten-degree sector on the compass.
- Select the pathway containing the largest population of the CV vectors among the 36 conduction pathways. Calculate the average CV of this pathway to represent the CV of the monolayer.
6. Visualization of the Wave Front Propagation
Normalize the signal intensity of an individual action potential or Ca transient at different recording time points relative to its peak value using MATLAB software.
Temporarily store the normalized intensity data of every recorded time point into a 16 x 16 intensity matrix in the MATLAB software used.
Construct a color-coded intensity map from the stored intensity matrix of each recording time point using the surf.m function. NOTE: In the intensity map, red represents high intensity of the Vm or Ca signals, and blue reflects low intensity of the fluorescence signals.
Create an .avi file using the avifile.m function.
Save all the intensity maps at different recording time points of the action potential or Ca transient using the addframe.m function. NOTE: At this point, two separate avi files of Vm and Ca propagation will be generated.
Representative Results
A cultured confluent monolayer exhibits a regular intrinsic rhythm as demonstrated in Movie 1. We then performed Vm/Ca dual channel optical mapping in a fully confluent HL-1 monolayer. Figure 1A shows example traces of Vm and Ca signals from a recorded single beat. Representative isochronal maps of uniformly propagated Vm and Ca signals using the dual channel optical mapping system are shown in Figures 1B and 1C. Representative chronological images of uniformly propagated action potentials and Ca transients were obtained from the same HL-1 culture monolayer. Finally, Movies 2 and 3 provide animation examples of a uniformly propagated action potential (Movie 2) and calcium transient (Movie 3) in the HL-1 confluent monolayer.
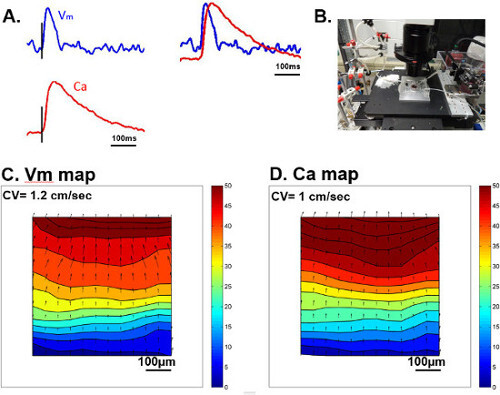 Figure 1: (A) Example traces of Vm (blue) and Ca (red) from an electrically paced beat at a cycle length (CL) of 500 msec. (B) An image of our optical mapping system for HL-1 monolayers. (C) An isochronal map of action potential propagation from the same paced beat throughout the whole vision field. (D) An isochronal map of Ca transient propagation from the same paced beat throughout the vision field. In both isochronal maps, time lapse is represented with colors, starting with dark blue as initiation and dark red at 50 msec. Please click here to view a larger version of this figure.
Figure 1: (A) Example traces of Vm (blue) and Ca (red) from an electrically paced beat at a cycle length (CL) of 500 msec. (B) An image of our optical mapping system for HL-1 monolayers. (C) An isochronal map of action potential propagation from the same paced beat throughout the whole vision field. (D) An isochronal map of Ca transient propagation from the same paced beat throughout the vision field. In both isochronal maps, time lapse is represented with colors, starting with dark blue as initiation and dark red at 50 msec. Please click here to view a larger version of this figure.
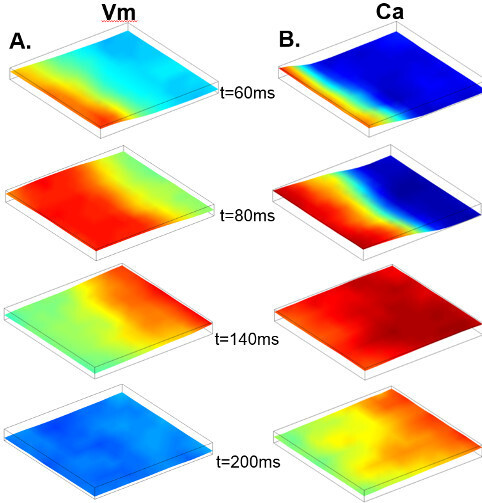 Figure 2: (A) A series of maps of Vm distribution at 60, 80, 140, and 200 msec after the start of recording. (B) Maps of Ca distribution at 60, 80, 140, and 200 msec after the start of recording.
Figure 2: (A) A series of maps of Vm distribution at 60, 80, 140, and 200 msec after the start of recording. (B) Maps of Ca distribution at 60, 80, 140, and 200 msec after the start of recording.
Movie 1: A representative movie recorded from a confluent HL-1 monolayer at culture day five, under a light microscope with a 40X objective.
Movie 2: A representative movie of a propagated action potential from a paced beat at a CL of 500 msec.
Movie 3: A representative movie of a propagated Ca transient from a paced beat at a CL of 500 msec.
Discussion
This article describes the key aspects of optical mapping in a cultured HL-1 atrial myocyte monolayer stained with calcium and voltage sensitive fluorescent dyes. It includes culturing an optimal HL-1 cell monolayer, setup of the mapping equipment, mapping a cultured monolayer, and data analysis.
To successfully map cultured cells, the key is to prepare a uniformly distributed cell monolayer. When seeding the cells on to the coverslips, be sure to evenly disperse the cells. Always map fully-grown and completely confluent cell monolayers, as these will best respond to electrical pacing. It is also important to keep the cells oxygenated while loading the dyes, but the oxygen bubbles should not come into direct contact with the cells as this may kill or dislodge them from the coverslip. Lowering dye concentration and reducing light intensity will decrease signal magnitude and reduce signal to noise ratio, while increasing dye concentration and enhancing light intensity will increase signal magnitude but also increase noise. Therefore, an optimal balance of fluorescent dye concentration and excitation light strength must be determined to enhance signal to noise ratio of recording signals. Moreover, during optical mapping recording, be sure to avoid exposing the cell monolayer to long exposures of concentrated light as this may cause photodynamic cell damage within the light exposed area. Finally, an optical mapping system that provides high temporal and spatial resolution of recording signals is required for obtaining high quality data.
While dual channel optical mapping in intact hearts has great potential for studying cardiac electrophysiology due to its ability to simultaneous collect voltage and calcium signals, one limitation is that this technique collects information from a 3-D surface as a 2-D projection map. Obtained 2-D data from intact hearts with ignored information of 3-D curvature of the heart could result in errors in data analyses, especially in conduction velocity analysis. With the advantages of using a cultured 2-D HL-1 cellular model, we are able to visualize regular and irregular rhythms, assess Ca transient and action potential kinetics, determine conduction velocity and characterize Ca and action potential propagation patterns (uniform or non-uniform propagation) without interfering from the 3-D structure of an intact heart. Therefore, successfully mapping both calcium transients and action potentials in a cultured HL-1 atrial myocyte monolayer as a complimentary approach can greatly enhance the understanding of the electrophysiologic properties of atrial myocytes and the underlying mechanisms of atrial arrhythmogenesis.
Disclosures
No conflicts of interest declared.
Acknowledgments
We would like to thank Dr. Claycomb for providing HL-1 cells and detailed maintenance protocol. We would also like to thank Ms. Elena Carrillo and Dr. Seth Robia for their assistance with generating the cell movie and Mr. Pete Caron for his assistance with making some accessories for our dual channel optical mapping system.
This work was supported by American Heart Association (10GRNT3770030 & 12GRNT12050478 to XA), National Institutes of Health (HL113640 to XA), and Loyola University Research Development Fund (to XA).
References
- Efimov IR, Nikolski VP, Salama G. Optical imaging of the heart. Circ Res. 2004;95(1):21–33. doi: 10.1161/01.RES.0000130529.18016.35. [DOI] [PubMed] [Google Scholar]
- Salama G, Hwang SM, et al. Simultaneous optical mapping of intracellular free calcium and action potentials from Langendorff perfused hearts. Current protocols in cytometry / editorial board, J. Paul Robinson, managing editor ... [et al.] 2009;12(Unit 12 17) doi: 10.1002/0471142956.cy1217s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurita KR, Singal A. Mapping action potentials and calcium transients simultaneously from the intact heart. Am J Physiol Heart Circ Physiol. 2001;280(5):H2053–H2060. doi: 10.1152/ajpheart.2001.280.5.H2053. [DOI] [PubMed] [Google Scholar]
- Fast VG. Simultaneous optical imaging of membrane potential and intracellular calcium. J Electrocardiol. 2005;38(4 Suppl):107–112. doi: 10.1016/j.jelectrocard.2005.06.023. [DOI] [PubMed] [Google Scholar]
- Sowell B, Fast VG. Ionic mechanism of shock-induced arrhythmias: role of intracellular calcium. Heart Rhythm. 2012;9(1):96–104. doi: 10.1016/j.hrthm.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fast VG, Kleber AG. Microscopic conduction in cultured strands of neonatal rat heart cells measured with voltage-sensitive dyes. Circ Res. 1993;73(5):914–925. doi: 10.1161/01.res.73.5.914. [DOI] [PubMed] [Google Scholar]
- Hou L, et al. A major role for HERG in determining frequency of reentry in neonatal rat ventricular myocyte monolayer. Circ Res. 2010;107(12):1503–1511. doi: 10.1161/CIRCRESAHA.110.232470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp P, et al. Relative contributions of connexins 40 and 43 to atrial impulse propagation in synthetic strands of neonatal and fetal murine cardiomyocytes. Circ Res. 2006;99(11):1216–1224. doi: 10.1161/01.RES.0000250607.34498.b4. [DOI] [PubMed] [Google Scholar]
- Xia M, et al. Functional expression of L- and T-type Ca2+ channels in murine HL-1 cells. J Mol Cell Cardiol. 2004;36(1):111–119. doi: 10.1016/j.yjmcc.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Claycomb WC, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95(6):2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenbach JP, Ai X, Banach K. Decreased intercellular coupling improves the function of cardiac pacemakers derived from mouse embryonic stem cells. J Mol Cell Cardiol. 2008;45(5):642–649. doi: 10.1016/j.yjmcc.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, et al. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc Res. 2013;97(3):589–597. doi: 10.1093/cvr/cvs366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286(3):H823–H829. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- Yang Z, Shen W, Rottman JN, Wikswo JP, Murray KT. Rapid stimulation causes electrical remodeling in cultured atrial myocytes. J Mol Cell Cardiol. 2005;38(2):299–308. doi: 10.1016/j.yjmcc.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Brundel BJ, Kampinga HH, Henning RH. Calpain inhibition prevents pacing-induced cellular remodeling in a HL-1 myocyte model for atrial fibrillation. Cardiovasc Res. 2004;62(3):521–528. doi: 10.1016/j.cardiores.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Umapathy K, et al. Electrogram fractionation in murine HL-1 atrial monolayer model. Heart Rhythm. 2008;5(7):1029–1035. doi: 10.1016/j.hrthm.2008.03.022. [DOI] [PubMed] [Google Scholar]
- Fast VG, Cheek ER. Optical mapping of arrhythmias induced by strong electrical shocks in myocyte cultures. Circ Res. 2002;90(6):664–670. doi: 10.1161/01.res.0000013403.24495.cc. [DOI] [PubMed] [Google Scholar]
- Bayly PV, et al. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans Biomed Eng. 1998;45(5):563–571. doi: 10.1109/10.668746. [DOI] [PubMed] [Google Scholar]