

Abstract
Comparison between two or more distinct groups, such as healthy vs. disease, is necessary to determine cellular status. Mitochondria are at the nexus of cell heath due to their role in both cell metabolism and energy production as well as control of apoptosis. Therefore, direct evaluation of isolated mitochondria and mitochondrial perturbation offers the ability to determine if organelle-specific (dys)function is occurring. The methods described in this protocol include isolation of intact, functional mitochondria from HEK cultured cells and mouse liver and spinal cord, but can be easily adapted for use with other cultured cells or animal tissues. Mitochondrial function assessed by TMRE and the use of common mitochondrial uncouplers and inhibitors in conjunction with a fluorescent plate reader allow this protocol not only to be versatile and accessible to most research laboratories, but also offers high throughput.
Keywords: Cellular Biology, Issue 97, Mitochondria, TMRE, cytokines, ALS, HEK cells, fluorescence, mitochondrial dysfunction, mitochondrial membrane potential, cytochrome c
Introduction
Living cells bank metabolic energy in the form of fats and carbohydrates and use this energy for biosynthesis, membrane transport and movement. Some energy is obtained in the cytosol through the conversion of dietary sugars directly into ATP during glycolysis. However, the main source of ATP production in a cell is harnessed within the mitochondria via the mitochondrial respiratory chain1. The architecture of mitochondria provides the necessary spatial orientation for effective and efficient ATP production. Mitochondria possess a double membrane separated by an intermembrane space and together with the matrix, the innermost mitochondrial compartment, house the components and coordinate the chemical reactions involved in ATP generation. The inner membrane contains a series of membrane-bound protein complexes that comprise the respiratory chain as well as the ATP synthase, the protein complex that brings ADP and Pi together for formation of ATP. The inner membrane is folded into cristae and electrons are passed along the respiratory chain complexes via cytochrome c, a soluble electron carrier which moves between complexes within the intermembrane space. As electrons move, oxidation of reducing equivalents occurs and hydrogen ions are pumped from the matrix to the intermembrane space. As a consequence of the high ion concentration within the intermembrane space, an electrochemical gradient builds resulting in a membrane potential across the inner mitochondrial membrane (∆ψ)2. Oxygen is the final electron acceptor of the electron transport chain, and hydrogen ions flow through the ATP synthases from the intermembrane space back to the matrix, and in doing so directly cause ATP formation. This process as described in its entirety is known as oxidative phosphorylation. The folds of the cristae increase the surface area of the inner membrane, allowing for maximal electron transport and ATP production within each mitochondrion. The proteins, enzymes and other molecules involved in oxidative phosphorylation are derived from both nuclear and mitochondrial genes. Mitochondria contain their own circular DNA, encoding for 13 proteins as well as tRNAs and mRNAs necessary for ATP production3. However, many more proteins are required and thus are nuclear encoded. Most of these nuclear-encoded proteins are targeted to the mitochondrial matrix by the use of presequences at the N-terminus of the precursor protein, and their import is driven in part by ∆ψ4,5.
Beyond contributing to the bioenergetics of a cell, mitochondria also influence major metabolic processes such as TCA and beta-oxidation, cellular signaling through regulating calcium, as well as a key role in apoptosis6. Specifically, during times of cellular stress, BCL-2 family proteins that reside on or interact at the mitochondrial outer membrane can cause mitochondrial outer membrane permeability (MOMP)7,8. During MOMP, cytochrome c and other proteins are released into the cytosol, and together with several cytosolic proteins form a complex called the apoptosome9,10. The apoptosome activates caspases that go on to cleave cellular proteins and DNA during the execution phase of apoptosis. As soon as MOMP occurs, ∆Ψ is collapsed and ATP production halted. Thus, as apoptosis is initiated mitochondrial function is compromised and changes in ∆Ψ can be correlated to mitochondrial and cell health12. While apoptosis is an endpoint in many disease models, mitochondrial function and changes to ∆Ψ can also yield valuable information about disease origination and/or progression. For instance, mitochondrial structural and functional changes have been documented during the course of neurodegenerative diseases13,14.
In the first part of the protocol, isolation of intact mitochondria that retain their ΔΨ is described. HEK-293T cells were exposed to different concentrations and combinations of recombinant TNF-α, IL1-β and IFN-γ to induce apoptosis. These cytokines were chosen because they are frequently reported to be high in primary human septic samples15 and the extrinsic pathway of apoptosis can be triggered by interaction of TNF-α binding to its receptor6 . Since there are subtle variations necessary to isolate functional mitochondria from primary tissues as compared to cultured cells, and because much research utilizes animals, the protocol also describes how to isolate mitochondria from liver and spinal cords of anterior lateral sclerosis (ALS) mouse model.
The second part of the protocol was developed to monitor perturbations to the mitochondrial membrane potential using a potential-sensitive fluorescent dye with a fluorescent plate reader. Differences between cellular status (i.e., healthy vs. unhealthy) are differentiated by quantitating the strength of ΔΨ of isolated mitochondria in conjunction with uncoupling agents, respiratory chain inhibitors, complex inhibitors and ionophores, all of which cause dissipation of the mitochondrial membrane potential. The healthier the mitochondria, the larger the change in ∆Ψ upon treatment with mitochondrial inhibitors, therefore the response of mitochondria can be used as an indicator of mitochondrial (dys)function.
Use of isolated mitochondria rather than in situ assessment of function offers definitive evidence that a pathology or treatment directly modulates changes to the organelle16-18. While there are methods in the literature to isolate mitochondria from cultured cells, they are vague17 and/or utilize specialized equipment16. This protocol describes in detail the isolation method and is readily adaptable to other cell lines, including primary tissue and cultures13,14,19,20. Many isolated mitochondria studies utilize the same mitochondrial uncouplers and inhibitors used in this protocol but with a Clark electrode (21 is a representative example of many papers in the literature), which again is a very specific and specialized piece of equipment. Furthermore, this traditional method has limitations such as low throughput and high complexity22,23 and requires a substantial amount of mitochondria (~500 µg/reaction). In this protocol, the fluorescent membrane-sensing potential probe TMRE is used in conjunction with a fluorescent plate reader, which is a standard machine in many laboratories. TMRE is widely regarded since it quickly enters cells and isolated mitochondria and can be used at low concentrations24. Multiple reactions can quickly be set up in tandem and batch analyzed using this protocol. Furthermore, the reactions require a much small amount of isolated mitochondria (~10 µg/reaction). By requiring less material, smaller tissue or cell culture samples can be utilized as a starting point for mitochondria isolation, more replicates or reactions can be set up, and potentially enough material for other isolated mitochondria experiments such as ATP production, oxygen consumption, or import assays are possible.
Protocol
All animal experiments conformed to National Institutes of Health guidelines and were approved by the Wake Forest University Animal Care and Use Committee. Breeding pairs for SOD1G93A [B6SJL-TgN (SOD1-G93A) 1Gur] mouse model were obtained from The Jackson Laboratory (Bar Harbor, ME). Nontransgenic wild-type (WT) females and SOD1G93A males [B6SJL-TgN (SOD1-G93A) 1Gur] were bred to generate SOD1G93A mice and nontransgenic WT littermates that were used in the experiments.
1. Models for Investigation
- Cell culture
- Maintain human embryonic kidney cells (HEK-T293) cells in DMEM supplemented with 10% heat inactivated fetal bovine serum, 1% penicillin-streptomycin, and 1% L-glutamine, at 37 °C in 5% CO2.
- Grow cells in T75 flasks and allow them to reach 90% confluence.
- Carry out cell splitting via trypsinization (0.25% Trypsin-EDTA) for 3-5 min at 37 °C in 5% CO2, inactivation of trypsin with equal volume of cell media, and centrifugation of cells for 5 min at 700 x g in 4 °C.
- Aspirate the media and resuspend the cell pellet in culture media.
- Seed cells in the appropriate flask or plate 7 x 104 cells into a T75 flask 48 hr prior to cytokine treatment. This should give ~70% density at time of treatment.
- Serum starve cells 24 hr later by aspirating the media and replacing it with the same volume of serum-free media (DMEM, sterile).
- Prepare TNF-α, IL1-β, and IFN-γ, from lyophilized powder with ultra-pure water at working concentrations of 5 pg/µl, 10 ng/µl, and 10 ng/µl, respectively and add them to the wells/flasks.
- Treat serum starved cells by the addition of solubilized cytokines directly to the cell culture media of appropriate flasks. Treatments consisted of individual cytokines, a cocktail of all 3 (referred to as “*3”), or sterile water as a control (referred to as “0”).
- Incubate treated cells for 24, 48 or 72 hr before assessment and harvesting.
- Cell viability assessment
- Make a 5 mg/ml solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in 1x PBS.
- For 12-well plates, add 100 µl of MTT solution to each well and gently swirl to assure even distribution.
- Incubate plates for 15 min–1 hr at 37 °C in 5% CO2, and check under a microscope for the presence of purple coloration. If no purple is present, swirl again and allow cells to incubate in 5 min intervals until purple is observed. The amount of time necessary should be determined by each investigator.
- Using a repeat pipettor, add 700 µl of MTT solvent (4 mM HCl and 0.1% NP40 in isopropanol) to each well and rock the plate(s) at room temperature for 5-10 min, or until all purple color had left the cells. If purple color does not come out of cells, add an additional 200 µl of solvent and continue to swirl.
- For analysis, take 100 µl from each well and place into a 96-well plate and read on a plate reader using 570 nm and a reference wavelength of 630 nm, however 595 nm is also acceptable as long as readings are taken at consistent settings.
- Animal Model of ALS
- All animal experiments conformed to National Institutes of Health guidelines and were approved by the Wake Forest University Animal Care and Use Committee. Breeding pairs for SOD1G93A [B6SJL-TgN (SOD1-G93A) 1Gur] mouse model were obtained from The Jackson Laboratory (Bar Harbor, ME). Nontransgenic wild-type (WT) females and SOD1G93A males [B6SJL-TgN (SOD1-G93A) 1Gur] were bred to generate SOD1G93A mice and nontransgenic WT littermates that were used in the experiments.
- Perform genotyping with standard primers against mutant SOD125.
2. Isolation of Mitochondria
NOTE: It is important to work quickly and keep everything on ice throughout the procedure.
- Preparation of mitochondrial isolation buffer (MIB), spinal cord isolation buffer (SC MIB) and experimental buffer (EB)
- Prepare the following stock solutions ahead of time for MIB and EB.
- Make 1 M Tris/MOPS by dissolving 12.1 g of Tris base in 70 ml of H2O. Add dry MOPS to get pH to 7.4. Adjust volume to 100 ml final. Filter sterilize and store at 4 °C.
- Make 1 M Kphos by mixing 80.2 ml of K2HPO4 and 19.8 ml of KH2PO4. Store at room temperature.
- Make 0.2 M EGTA/Tris by adding 3.8 g of EGTA to 10 ml of H2O. Add 1 M Tris/MOPS until dissolved, ~30-40 ml. Adjust to 50 ml, sterile filter and store at room temperature. Note that the pH will be ~6.7.
- Make 1 M Glutamate by making 10 ml of 1 M solution of glutamic acid. Filter sterilize and store 4 °C.
- Make 1 M Malate by making 10 ml of 1 M solution of malic acid. Add Tris/MOPS to 50 mM. Filter sterilize and store at 4 °C.
- Make MIB at a concentration of 200 mM sucrose, 10 mM Tris/MOPS, pH 7.4, and 1 mM EGTA/Tris. Filter sterilize and store at 4 °C.
- Make SC MIB at a concentration of 250 mM sucrose, 20 mM HEPES-KOH pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and protease inhibitor cocktail.
- Make EB at a concentration of 125 mM KCl, 10 mM Tris/MOPS, pH 7.4, 5 mM glutamate, 2.5 mM malate, 1 mM K phosphate, pH 7.4, and 10 mM EGTA/Tris. Filter sterilize and store at 4 °C.
- Equipment preparation prior to harvesting the cells.
- Rinse a small glass vessel and homogenization pestle three times with sterile water and place on ice.
- Gather necessary items such as a standard drill, cell scrapers, 1.5 ml, 15 ml and 50 ml tubes and solutions.
- Mitochondria Isolation
- Cultured Cells
- For each sample type, use two T175 flasks of cells.
- Ensure that cells are ~90% confluent and use in control experiments, or plate and treat as described above in step 1.2.
- On the bench top, aspirate media and wash the adherent cells twice with 15 ml of 1x PBS each time.
- Aspirate the buffer and scrape flasks to remove the adherent cells from the bottom of the flask.
- Add 15 ml of 1x PBS to each flask, swirl, and transfer to individual 15 ml conical tubes and place on ice.
- Once scraping is done, centrifuge the tubes for 5 min at 700 x g, 4 °C using a table top centrifuge using a swinging bucket rotor.
- Aspirate supernatants and resuspend each pellet in 1 ml of MIB.
- Combine both suspensions and transfer to a small glass vessel for homogenization.
- Add MIB to the vessel until buffer reaches the first line. Homogenize cells with the pestle attached to a drill at medium speed for three passes. Ensure that the vessel is on ice during this step, not to remove the pestle above the liquid and to use a steady speed with continuous passes.
- Transfer the homogenized solution to a 50 ml conical tube.
- Draw the solution into a 3 ml syringe using an 18 gauge 1 ½ inch needle and expel it back into the conical tube on ice with a 27 gauge ½ inch needle. Take care to expel the solution against the inside wall of the tube as to utilize that force for cell membrane disruption.
- Repeat the syringe steps for a total of five times.
- Transfer the solution into a 15 ml conical tube and centrifuge for 5 min at 600 x g, 4 °C in a table top centrifuge using a swinging bucket rotor.
- Carefully remove the supernatant and distribute among three 1.5 ml tubes. NOTE: Mitochondria are in the supernatant after this first, low-speed spin, while cell membranes and unbroken cells are pelleted.
- Centrifuge tubes in a fixed angle rotor at 10,000 x g, 4 °C for 5 min.
- Aspirate supernatants and combine the pellets in 100 µl MIB and immediately place on ice. NOTE: Mitochondria are in the pellet after this high-speed spin. Mitochondria will typically maintain their membrane potential for ~2-3 ion ice after isolation and are most stable as a concentrated stock solution in MIB.
- Mitochondria isolation from mouse tissue
- Anesthetize the mouse according to IACUC protocol. Set a vaporizer at 5.0% to induce anesthesia. Confirmed that the animal is anesthetized by a lack of reflex for foot pinch or blink reflex when the eye is approached or tapped with a cotton swab. Keep the mouse under anesthesia for the entire surgical procedure by keeping the vaporizer between 1.5% and 2.0%.
- Excise the liver and spinal cord from each animal and place separately in ice cold 1x PBS -/- to rinse away any blood.
- For the liver, transfer the tissue to a weigh dish on ice and chop into fine pieces using a fresh razor blade for 1 min.
- Add tissue and appropriate buffer (MIB for liver or SC MIB for the spinal cord) to the vessel until buffer reaches the first line. Homogenize tissue with the pestle by hand for five passes. Ensure that the vessel is on ice during this step, not to remove the pestle above the liquid and to use a steady speed with continuous passes.
- Transfer the homogenates to clean 15 ml tubes.
- Centrifuge tubes in a fixed angle rotor at 750 x g, 4 °C for 10 min.
- Save the supernatants into clean tubes and place them on ice. NOTE: Mitochondria are in the supernatant after this first, low-speed spin, while cell membranes and unbroken cells are pelleted.
- Re-suspend the spinal cord pellets in 500 µl of SC MIB.
- Re-homogenize each three times, only filling the vessel half way with SC MIB this time.
- Transfer the new homogenate to a fresh tube.
- Centrifuge the in a fixed angle rotor at 750 x g, 4 °C for 10 min.
- Combine these new supernatants, which contain more mitochondria, with the first supernatant from each sample.
- Centrifuge tubes in a fixed angle rotor at 10,000 x g, 4 °C for 5 min.
- Aspirate supernatants and resuspend the liver pellets in 500 µl of MIB and the spinal cord pellets in 50 µl SC MIB and immediately place on ice. NOTE: Mitochondria are in the pellet after this high-speed spin. Mitochondria will typically maintain their membrane potential for ~2-3 ion ice after isolation and are most stable as a concentrated stock solution in MIB.
- Perform a protein concentration assay to estimate the concentration of mitochondria in solution. Follow instructions for using a commercial Protein Assay Kit or similar method26. Typical concentrations of mitochondria are: 2 T175 flasks yields ~2 mg/ml, 1 mouse liver ~3-5 mg/ml, and 1 mouse spinal cord ~1-3 mg/ml.
- Cytochrome c assay using an R&D Rat/Mouse Cytochrome c Quantikine ELISA Kit (adapted from the protocol provided by the manufacturer).
- Immediately before setting up the reactions, dilute mitochondria to 0.5 mg/ml working concentration with EB and distribute diluted mitochondria in 1.5 ml tubes for reactions (typically 30-50 μl of mitochondria per tube).
- Add either EB or DMSO, at 1% of the total volume, to the diluted mitochondria and incubate the reactions on the bench top for 7-10 min. These reactions are stable for up to 30 min at room temperature if a longer time frame is needed.
- Pellet mitochondria by centrifugation in a fixed angle rotor at 10,000 x g, 4 °C for 5 min and carefully separate the supernatant and place each in a separate 1.5 ml tubes. NOTE: Tubes can either be frozen at -20 °C for analysis later or used immediately.
- Determine release of cytochrome c by a comparing the concentration of cytochrome c in the pellet and supernatant27.
- Prepare the samples by solubilizing the pellets in the original volume of the reaction with 0.5% Tx-100 in 1x PBS.
- For each sample, prepare 2 wells on the ELISA plate, one for the now solubilized pellet and one for the supernatant from the reaction by addition of 100 μl of cytochrome c conjugate (use straight out of bottle) plus 100 μl of 0.5% Tx-100 in 1x PBS, and then all of the pellet or supernatant sample.
- Gently vortex the plate a low setting (level 2-4) for 20 sec to mix, cover and incubate for 1 hr, 37 °C.
- Next, wash the plate four times with wash buffer diluted according to the manufacturer’s instructions. Remove any excess solution by tapping the wells on paper towel.
- Next, add 150 μl of 1:1 A+B developing solution to each well and incubate the plate for 20-30 min in the dark at room temperature.
- Finally, add 50 μl of stop solution to each well and take absorbance readings at 540 nm using a microplate reader. Calculate the amount of cytochrome c release by comparing the amount of cytochrome c in the pellet vs. supernatant for each reaction.
3. Assessment of Mitochondrial (Dys)function
Immediately before reactions are set up, dilute mitochondria to 0.5 mg/ml working concentration with EB and placed in 1.5 ml tubes for reactions (typically 30-50 μl of mitochondria are used per tube).
- Mitochondrial treatments
- Add appropriate treatments (EB control, 1 μM FCCP mixed with 50 nM Valinomycin, 10 μM rotenone, 5 μM oligomycin, 2 mM KCN, or 200 μM ADP, final concentrations) to separate reactions at 1/10th the total volume of diluted mitochondria. Incubate the reactions on the bench top for 7 min. NOTE: Care should be taken when handling these substances as accidental ingestion or absorption through the skin could be harmful.
- Dilute TMRE, reconstituted in sterile water at a working concentration of 100 μM and stored at -20 °C, to 2 μM and add a volume equal to the mitochondrial reaction volume.
- Incubate the reactions on the bench top for 7 min.
- Pellet mitochondria by centrifugation in a fixed angle rotor at 10,000 x g, 4 °C for 5 min.
- Load half of the supernatant volume of each sample onto a 396-well plate and read fluorescence. NOTE: Excitation and emission wavelengths of TMRE should be optimized according to manufacturer’s instructions using the volume and plate that will be used for the assay. Using a standard 384-well black plate with 25 µl of 1 µM TMRE (final reaction concentration) the strongest signal was obtained using excitation/emission wavelengths 485 nm/535 nm.
- Calculate the fold-difference in fluorescence between control (EB) and each treatment by dividing the relative fluorescence value (RFU) obtained from the plate reader for the experimental sample by the RFU from the control sample.
Representative Results
Treatment of HEK-293T cells with 200 pg/ml TNF-α, 40 ng/ml IL1-β, and 75 ng/ml IFN-γ (*3) for 24-48 hr leads to progressive amounts of cell death (Figure 1A). Cell viability was assessed using MTT assays and consistently demonstrates that there is ~10% decrease in cell viability with 24 hr treatment and ~20% decrease with 48 hr treatment. Cells treated with similar concentrations (100 pg/ml TNF-α, 40 ng/ml IL1-β, and 75 ng/ml IFN-γ) yield similar cell death results at 48 hr, and treatment with individual cytokines reveals that the decrease in viability is due to combinatorial affects (Figure 1B). The viability data in Figure 1B represent the mean +/- standard deviation of 3 separate experiments each run in triplicate, and the tightness of the error bars demonstrate the reproducibility of the data. The amount of cytokines can be adjusted in this treatment protocol to further elucidate the contribution of each cytokine on cell death, and additional cytokines or other factors could also be included, as long as appropriate controls are executed.
Isolation of mitochondria from cells and tissues has been described in the literature since the 1940s28,29. The premise for the procedure is cell disruption followed by differential centrifugation, where crude mitochondria are known to pellet at ~10,000 x g. Many protocols that utilize isolated mitochondria require intact double membranes that can sustain their membrane potential, and therefore it is necessary to evaluate the integrity once isolation is complete. During the development of the mitochondria isolation protocol from cultured HEK cells, a cytochrome c assay was used to indicate if intact mitochondria were obtained. Using commercially available cytochrome c ELISA kits, mitochondria were quantified after isolation and diluted in EB, and then either EB or DMSO added and the reactions to incubate on the bench top. Then, by separating mitochondria from surrounding supernatant, analysis of cytochrome c content of both fractions was used as a gauge for mitochondrial membrane intactness; if the majority of cytochrome c is retained in the mitochondrial pellet then the mitochondria are considered intact, while if more than 15% of the cytochrome c is found in the supernatant, then mitochondria were considered unsuitable for functional studies. As seen in Table 1, the protocol described yields ~12% cytochrome c release and ~15% when isolated mitochondria are pre-treated with DMSO. Similar results have been reported previously for mitochondria isolated from other cell lines or animal tissues19,20,27. Alternatively, mitochondrial integrity can also be assessed with TMRE, which was used during isolation of mouse liver and spinal cord mitochondria. During initial isolation protocol development, trials evaluated mitochondria isolated from liver and spinal cords of healthy, normal mice mitochondria. For each sample, ΔΨ was evaluated by comparing the fluorescent signal from isolated mitochondria treated with EB to those incubated with FCCP + Valinomycin (Table 2). A ratio of fluorescence between treated and untreated mitochondria of >1 was used as an indication of healthy, intact mitochondria.
Once successful mitochondria isolation protocols were developed, mitochondria isolated from control, untreated cells and *3 cytokine treated cells, or normal and SOD1 mice were analyzed. HEK mitochondria from both groups were treated with various uncouplers and inhibitors and ∆Ψ measured by changes to TMRE uptake. The fold difference in control EB treated mitochondria to uncoupler/inhibitor treated mitochondria indicates a difference in the strength of ∆Ψ between the two groups (representative data and calculations in Table 3). Furthermore, the percent decrease from untreated cells to *3 treated cells signify that mitochondrial health was impacted by cytokine treatments, corroborating cell viability data (Figure 2). Evaluation of ΔΨ strength via TMRE uptake using mitochondria isolated from liver and spinal cords of four individual normal, control mice as compared to four ALS mice indicate that tissue health can also be assessed using this protocol. As illustrated by fold differences and percentage decreases in ΔΨ, comparison of EB-treatment and FCCP + Valinomycin-treatment of mitochondria from control and disease-progressed animals are significantly different (Table 4; see also references 13 & 14).
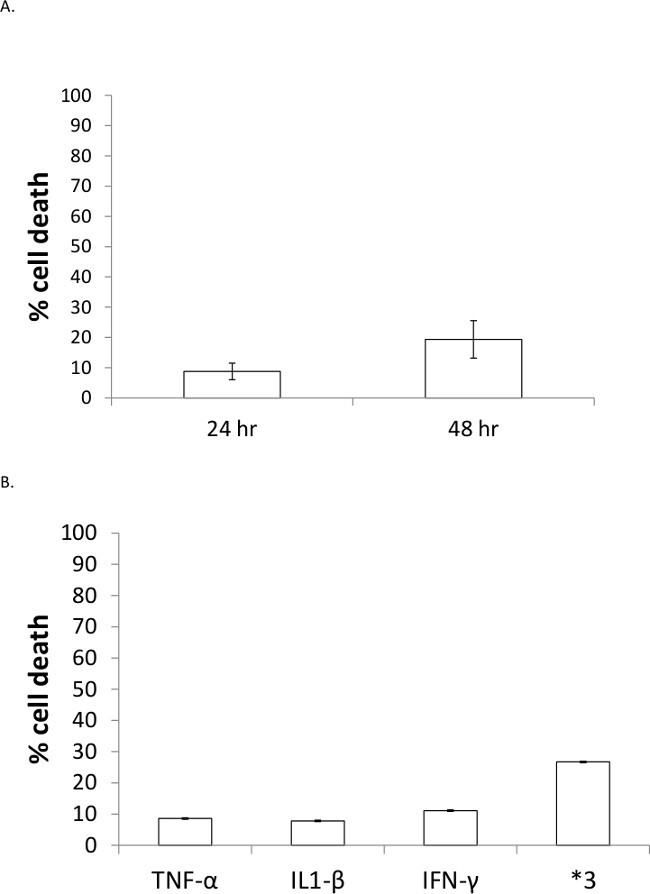 Figure 1: Cell death induced by *3 treatment is progressive over time and greater than with individual cytokine treatments. (A) HEK-293T cells were treated for 24 or 48 hr and cell death was measured using an MTT assay. Values were normalized to control, untreated samples and data represent triplicate measurements for n = 3, +/- standard deviation. (B) HEK-293T cells were treated with individual cytokines or a cocktail (*3) for 48 hr and cell death assessed via MTT assay. Data represents n = 5 for each of individual cytokine and n = 6 for *3, +/- standard deviation.
Figure 1: Cell death induced by *3 treatment is progressive over time and greater than with individual cytokine treatments. (A) HEK-293T cells were treated for 24 or 48 hr and cell death was measured using an MTT assay. Values were normalized to control, untreated samples and data represent triplicate measurements for n = 3, +/- standard deviation. (B) HEK-293T cells were treated with individual cytokines or a cocktail (*3) for 48 hr and cell death assessed via MTT assay. Data represents n = 5 for each of individual cytokine and n = 6 for *3, +/- standard deviation.
| Pellet (AU) | Supernatant (AU) | % cytochrome c release | |
| 0 | 0.818 | 0.115 | 12.6 ± 2.27 |
| DMSO | 0.793 | 0.142 | 15.3 ± 2.23 |
Table 1: Retention of cytochrome c in isolated mitochondria from cultured cells. Data represent the average of n = 4, +/- standard deviation.
| liver 1 | liver 2 | spinal cord 1 | spinal cord 2 | ||
| RFU | EB | 97,830 | 94,132 | 339,716 | 290,154 |
| F/V | 435,188 | 461,299 | 385,366 | 482,480 | |
| ratio | 4.45 | 4.9 | 1.13 | 1.66 | |
| EB = experimental buffer and F/V = FCCP + valinomycin. |
Table 2: Measure of ΔΨ from mitochondria isolated from normal mouse tissues. Two separate animals were used to isolate liver and spinal cord mitochondria in tandem (e.g. liver 1 and spinal cord 1 are from the same animal).
| EB | oligo | rot | KCN | ADP | F/V | ||
| RFU | 0 | 4,844 | 12,507 | 12,734 | 9,925 | 10,892 | 4,844 |
| *3 | 6,517 | 13,639 | 12,435 | 10,235 | 13,154 | 6,517 | |
| Fold Difference | 0 | NA | 2.58 | 2.63 | 2.05 | 2.25 | 2.65 |
| *3 | NA | 2.09 | 1.91 | 1.57 | 2.02 | 2.18 | |
| % decrease | 0 vs.*3 | 18.94 | 27.41 | 23.35 | 10.24 | 17.67 | |
| EB = experimental buffer; oligo = oligomycin; rot = rotenone; KCN = potassium cyanide, and F/V = FCCP + valinomycin. |
Table 3: Raw data and calculations from cell culture isolated mitochondria experiments.
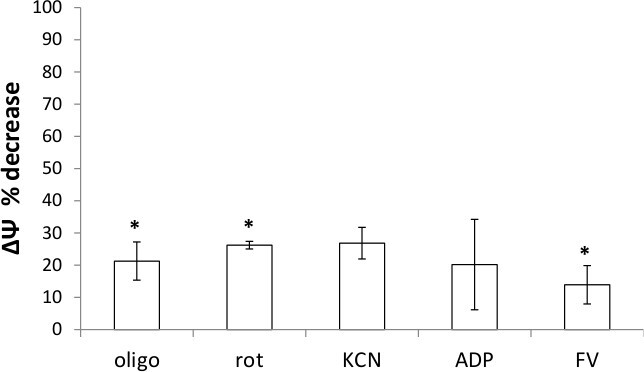 Figure 2: Mitochondria isolated from cytokine-treated cells have decreased membrane potential. Percent decrease of ∆Ψ for 0 vs. *3 for each treatment as compared to control, as calculated in Table 2. Data represent averages of duplicate reactions for n = 3 (oligomycin, rotenone, and FCCP/Valinomycin) or n = 2 (KCN and ADP) +/- standard deviation. *= statistically significant p-values based on fold difference calculations; p = 0.03, 0.01, and 0.05 for oligomycin, rotenone and FCCP/Valinomycin, respectively.
Figure 2: Mitochondria isolated from cytokine-treated cells have decreased membrane potential. Percent decrease of ∆Ψ for 0 vs. *3 for each treatment as compared to control, as calculated in Table 2. Data represent averages of duplicate reactions for n = 3 (oligomycin, rotenone, and FCCP/Valinomycin) or n = 2 (KCN and ADP) +/- standard deviation. *= statistically significant p-values based on fold difference calculations; p = 0.03, 0.01, and 0.05 for oligomycin, rotenone and FCCP/Valinomycin, respectively.
| Spinal Cord | Liver | ||
| Fold Difference | Control | 1.54 ± 0.48 | 3.20 ± 1.56 |
| SOD1 | 1.10 ± 0.30 | 3.12 ± 1.75 | |
| % decrease | 28.1 | 2.48 | |
| p-value | 0.03 | 0.41 |
Table 4: Mitochondria isolated from spinal cords of SOD1 mice have decreased membrane potential as assessed by FCCP + Valinomycin. Data represent the average of 4 animals per group.
Discussion
Treatment of HEK-293T cells with recombinant cytokines causes moderate amounts of cell death over 48 hr (Figure 1). The amount of cell death induced by TNF-alpha treatment is similar to previously reported studies30 and cell viability decreases after co-administration of multiple cytokines that are greater than summative amounts with any cytokine alone is also consistent with the literature31,32. The ability to adjust the amounts and types of cytokine treatments as well as comparison of cocktails to individual cytokines make this cell culture model attractive for studying the specific effects of cytokine exposure on kidney cells. Furthermore, the results are very reproducible and easily achieved (Figure 2). Considerations to keep in mind include the confluence of the cells. For example, cells treated at 100% confluence do not respond as well to the same concentrations of cytokines when treated at 70-80% confluence. Furthermore, the number of passages the cells have gone through should be tracked, as cell response could possibly be influenced having been in culture for too long (more than 8-12 weeks since thawing). While treatment of cultured cells with cytokines decreased cell viability and subsequently mitochondrial health, it is by no means a model for sepsis per se. However, use of a more clinically relevant model, such as employing primary kidney cells isolated from mice and/or treatment with naturally secreted cytokines from stimulated THP1 cells33, is a natural extension of this work. Given the gap in understanding of renal failure during sepsis, such cell culture models and assessment of mitochondria could provide initial insight into the mechanisms as well as a preliminary platform for testing the effectiveness of pharmaceutical compounds as preventative or intervention treatments.
Mitochondria isolated from cultured cells or animal tissues afford the capacity to rapidly evaluate the impact of cell treatments or disease progression on mitochondrial health. The isolation and evaluation protocols are not technically demanding to perform, and could easily be modified for use with other cell lines or primary tissues. The MIB and EB used for each sample type are good starting recipes that can be adjusted as necessary if a particular cell or tissue type has different requirements. For example, addition of fatty-acid free BSA is usually necessary for use with neurons, as included with the spinal cord protocol15,34. The cell disruption method can also be altered by using more syringe expulsions or changing to hand homogenization instead of using a drill or vice versa. Furthermore, re-homogenization of the first centrifugation pellet, as done in the spinal cord mitochondria procedure, may be necessary to obtain a satisfactory amount of mitochondria. A good indicator that an isolation protocol produces viable mitochondria is use of the cytochrome c ELISA. This assay is a quick and easy alternative to western blot analysis of cytochrome c release and is also a useful assessment of cytochrome c release after treatment of mitochondria with peptides or other compounds19,27. Alternatively, TMRE can be used during protocol development, but it is important to evaluate mitochondria isolated from a sample that is known to be healthy. Furthermore, when isolating mitochondria from mouse tissues that are not routinely described in the literature, liver mitochondria can serve as a positive control as well as used to set a baseline. For instance, as in this protocol, livers from every animal, both during protocol development as well as experiments were also excised and used for mitochondria isolation. Another consideration for a preparation is the amount of mitochondria obtained at the end of the isolation procedure. If the concentration of mitochondria is 1 mg/ml or less then assay results may prove unreliable (unpublished observations VDGM). Either the amount of starting material, MIB recipe or the disruption method should be optimized before moving to functional assays. Once a successful isolation protocol is attained, the mitochondria can also be used in other assays such as total ATP content and ATP generation13,14.
The mitochondria function protocol described here allows for indirect measurement of mitochondrial uptake of the potential-sensing dye TMRE using a standard fluorescent plate reader. After mitochondria have been isolated and the appropriate chemicals added, they are incubated with TMRE and then pelleted. By treating whole cells or animals and isolating mitochondria prior to ΔΨ assessment, confounding variables such as multi-drug resistant transport or ATP flux are avoided. The amount of fluorescence in the supernatant is then analyzed with the understanding that the healthier the mitochondria the stronger their ∆Ψ and therefore more uptake of TMRE. As a result, the amount of fluorescence in the supernatant should be less in healthier mitochondria and more in unhealthy mitochondria. By including an EB-only treatment, the fold difference of fluorescence between this control and uncoupled mitochondria can be calculated. Another important control with every set of reactions run is TMRE alone. The result from this control provides a maximum possible fluorescent reading. Since the TMRE added to reactions is freshly diluted, there is inherent variability. For example, TMRE-only readings of ~20,000, 25,000 and 30,000 were achieved at various times during protocol development. If the TMRE-only signal is much greater than any of the treated mitochondria samples and/or if treated mitochondria are not significantly different from untreated, then the isolation of coupled mitochondria was most likely not successful. The decrease in ΔΨ as measured by a ratio obtained when following this protocol is similar to previously reported amounts for control, healthy cells2,35. Comparison of ∆Ψ changes from mitochondria isolated from control, untreated HEK-293T cells and *3 cytokine-treated cells demonstrated that the decreased cell viability observed in MTT assays translates to decreased mitochondria function. This plate reader assay could easily be used with mitochondria isolated from any source and used to examine disease models such as ALS, as demonstrated here13,14.
Several studies have utilized mitochondria-specific fluorescent dyes to detect MOMP, which were important in the genesis of this plate reader assay. However, those studies either use whole cells to screen compounds that block or induce MOMP35,36, explore new ∆Ψ sensing dyes2 or identify other formats such as flow cytometry on a microchip37. This protocol is unique in that it describes a detailed method to isolate and use mitochondria to rapidly investigate effects of whole-cell treatment through response to known ∆Ψ-altering compounds. This protocol utilizes a variety of uncouplers and inhibitors, and indicates that any of them would be suitable for assessing mitochondrial health. Thus, while this particular investigation centers on mitochondrial changes due to cellular stress or progressive cellular dysfunction, the protocol could also be used to explore changes in mitochondrial metabolism through the use of specific compounds and/or the addition of electron sources such as succinate and NADH.
Important considerations when performing the isolated mitochondria assessment include consistency in treatment volumes, keeping the mitochondria in MIB and diluting them in EB immediately before the treatments are going to be set up, and using mitochondria within 3 hr of isolation to ensure their ∆Ψ is still being maintained. The choice of TMRE over other mitochondria-specific fluorescent dyes such as JC-1 was due to the ability to use small concentrations and simplicity of analyzing fluorescence using only one wavelength. This method could similarly be conducted using fluorescent microscopy2,38 or flow cytometry37, however the use of a plate reader takes less time and requires less optimization35. In addition, this protocol is much less technically demanding, quicker to perform, and more readily reproducible as compared to using a Clark electrode22 (Del Gaizo Moore, unpublished observations). Titrating amounts of each mitochondrial treatment may provide further insight into mitochondrial function and optimization of concentrations should be performed. The total volume of the mitochondrial reactions can be adjusted to suite the amount of sample obtained during isolation as well as the number of treatment reactions necessary. If possible, a standard amount of 50 μl is used; however, as little as 20 μl of mitochondria produces reliable results.
While ∆Ψ correlates to oxygen consumption, the assay presented does not directly measure oxygen consumption. While fluorescent oxygen sensing probe such as MitoExpress have been developed, presumably to avoid some of the downfalls of using a Clark Electrode cited above, the price per reaction and the requirement of larger amounts of mitochondria per assay remain limiting. Therefore, the use of TMRE and classical ΔΨ uncouplers or inhibitors has several advantages. Again, batch analysis of multiple samples or multiple treatments/sample, reproducibility, lower starting material requirement and/or lower amount of isolated mitochondria required per reaction render this protocol attractive.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This research was supported in part by NSF grant CHE-1229562 (VDGM), the Office of Undergraduate Research at Elon University, the Elon Chemistry Department, and the Elon Lumen Prize (TL and TAD), the Elon College Fellows Program (JAC), and the Elon College Honors Program (TAD).
References
- Madeira VM. Overview of mitochondrial bioenergetics. Methods In Molecular Biology. 2012;810:1–6. doi: 10.1007/978-1-61779-382-0_1. [DOI] [PubMed] [Google Scholar]
- Sakamuru S, et al. Application of a homogenous membrane potential assay to assess mitochondrial function. Physiological Genomics. 2012;44:495–503. doi: 10.1152/physiolgenomics.00161.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DA. Structure and function of the mitochondrial genome. Journal Of Inherited Metabolic Disease. 1992;15:439–447. doi: 10.1007/BF01799602. [DOI] [PubMed] [Google Scholar]
- Schatz G. The protein import system of mitochondria. The Journal of Biological Chemistry. 1996;271:31763–31766. doi: 10.1074/jbc.271.50.31763. [DOI] [PubMed] [Google Scholar]
- Mayer A, Neupert W, Lill R. Mitochondrial protein import: reversible binding of the presequence at the trans side of the outer membrane drives partial translocation and unfolding. Cell. 1995;80:127–137. doi: 10.1016/0092-8674(95)90457-3. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harbor Perspectives In Biology. 2013;5:a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Youle RJ. SnapShot: BCL-2 proteins. Cell. 2009;138:404. doi: 10.1016/j.cell.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochimica et Biophysica Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxidants & Redox Signaling. 2013;19:546–558. doi: 10.1089/ars.2012.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Molecular Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Joshi DC, Bakowska JC. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. Journal Of Visualized Experiments. 2011. [DOI] [PMC free article] [PubMed]
- Vinsant S, et al. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part I, background and methods. Brain and Behavior. 2013;3:335–350. doi: 10.1002/brb3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinsant S, et al. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain and Behavior. 2013;3:431–457. doi: 10.1002/brb3.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse N, Shimizu K, Payne M, Busija D. Isolation and characterization of intact mitochondria from neonatal rat brain. Brain research. Brain Research Protocols. 2001;8:176–183. doi: 10.1016/s1385-299x(01)00108-8. [DOI] [PubMed] [Google Scholar]
- Schmitt S, et al. A semi-automated method for isolating functionally intact mitochondria from cultured cells and tissue biopsies. Analytical Biochemistry. 2013;443:66–74. doi: 10.1016/j.ab.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nature Protocols. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Debatin KM, Poncet D, Kroemer G. Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene. 2002;21:8786–8803. doi: 10.1038/sj.onc.1206039. [DOI] [PubMed] [Google Scholar]
- Del Gaizo Moore V, et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. The Journal of Clinical Investigation. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–2309. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. The Journal of Biological Chemistry. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Hynes J, et al. Investigation of drug-induced mitochondrial toxicity using fluorescence-based oxygen-sensitive probes. Toxicological Sciences : An Official Journal Of The Society of Toxicology. 2006;92:186–200. doi: 10.1093/toxsci/kfj208. [DOI] [PubMed] [Google Scholar]
- Papkovsky DB. Methods in optical oxygen sensing: protocols and critical analyses. Methods in enzymology. 2004;381:715–735. doi: 10.1016/S0076-6879(04)81046-2. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, et al. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis : An International Journal On Programmed Cell Death. 2007;12:803–813. doi: 10.1007/s10495-007-0720-1. [DOI] [PubMed] [Google Scholar]
- Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Smith PK, et al. Measurement of protein using bicinchoninic acid. Analytical Biochemistry. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Certo M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Hogeboom GH, Schneider WC, Pallade GE. Cytochemical studies of mammalian tissues; isolation of intact mitochondria from rat liver; some biochemical properties of mitochondria and submicroscopic particulate material. The Journal Of Biological Chemistry. 1948;172:619–635. [PubMed] [Google Scholar]
- Hogeboom GH, Claude A, Hotch-Kiss RD. The distribution of cytochrome oxidase and succinoxidase in the cytoplasm of the mammalian liver cell. The Journal of Biological Chemistry. 1946;165:615–629. [PubMed] [Google Scholar]
- Marques-Fernandez F, et al. TNFalpha induces survival through the FLIP-L-dependent activation of the MAPK/ERK pathway. Cell Death & Disease. 2013;4:e493. doi: 10.1038/cddis.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain, Behavior, And Immunity. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- Downen M, Amaral TD, Hua LL, Zhao ML, Lee SC. Neuronal death in cytokine-activated primary human brain cell culture: role of tumor necrosis factor-alpha. Glia. 1999;28:114–127. [PubMed] [Google Scholar]
- Schildberger A, Rossmanith E, Weber V, Falkenhagen D. Monitoring of endothelial cell activation in experimental sepsis with a two-step cell culture model. Innate Immunity. 2010;16:278–287. doi: 10.1177/1753425909341885. [DOI] [PubMed] [Google Scholar]
- Brown MR, et al. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. Journal Of Neuroscience Methods. 2004;137:299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Wong A, Cortopassi GA. High-throughput measurement of mitochondrial membrane potential in a neural cell line using a fluorescence plate reader. Biochemical and Biophysical Research Communications. 2002;298:750–754. doi: 10.1016/s0006-291x(02)02546-9. [DOI] [PubMed] [Google Scholar]
- Blattner JR, He L, Lemasters JJ. Screening assays for the mitochondrial permeability transition using a fluorescence multiwell plate reader. Analytical Biochemistry. 2001;295:220–226. doi: 10.1006/abio.2001.5219. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Fukura Y, Shinohara Y, Baba Y. Analysis of mitochondrial membrane potential in the cells by microchip flow cytometry. Electrophoresis. 2005;26:3025–3031. doi: 10.1002/elps.200410402. [DOI] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Payne RM. Transactivator of transcription fusion protein transduction causes membrane inversion. The Journal of Biological Chemistry. 2004;279:32541–32544. doi: 10.1074/jbc.M405930200. [DOI] [PubMed] [Google Scholar]