

Abstract
This protocol outlines the steps required to perform ex vivo validation of in vivo near-infrared fluorescence (NIRF) xenograft imaging experiments in mice using fluorophore labelled nanobodies and conventional antibodies.
First we describe how to generate subcutaneous tumors in mice, using antigen-negative cell lines as negative controls and antigen-positive cells as positive controls in the same mice for intraindividual comparison. We outline how to administer intravenously near-infrared fluorophore labelled (AlexaFluor680) antigen-specific nanobodies and conventional antibodies. In vivo imaging was performed with a small-animal NIRF-Imaging system. After the in vivo imaging experiments the mice were sacrificed. We then describe how to prepare the tumors for parallel ex vivo analyses by flow cytometry and fluorescence microscopy to validate in vivo imaging results.
The use of the near-infrared fluorophore labelled nanobodies allows for non-invasive same day imaging in vivo. Our protocols describe the ex vivo quantification of the specific labeling efficiency of tumor cells by flow cytometry and analysis of the distribution of the antibody constructs within the tumors by fluorescence microscopy. Using near-infrared fluorophore labelled probes allows for non-invasive, economical in vivo imaging with the unique ability to exploit the same probe without further secondary labelling for ex vivo validation experiments using flow cytometry and fluorescence microscopy.
Keywords: Medicine, Issue 98, Nanobody, antibody, VHH, fluorescence imaging, molecular imaging, xenograft, animal model
Introduction
In the present report, we describe the implementation of near-infrared fluorophore labelled probes for validation of in vivo xenograft imaging experiments by using ex vivo flow cytometry and fluorescence microscopy of the dissected xenograft tumors. We compare a single domain nanobody (s+16a, 17 kDa) 1 and a monoclonal antibody (Nika102, 150 kDa) 2,3 directed to the same target antigen for specific in vivo near-infrared fluorescence imaging in a lymphoma xenograft model. The target antigen ADP-ribosyltransferase ARTC2.2 is expressed as a GPI-anchored cell surface ecto-enzyme by lymphoma cells 4-9.
Nanobodies derived from camelid heavy-chain-only antibodies are the smallest available antigen-binding fragments 10,11. With only ~15 kDa, these small antibody fragments are soluble, very stable and are renally cleared from circulation 8,10. These properties make them particularly suited for specific and efficient targeting of tumor antigens in vivo 12-20. Common antigen targets of available nanobodies are the epidermal growth factor receptor (EGFR1 or HER-1), human epidermal growth factor type 2 (HER-2 or CD340), carcinoembryonic antigen (CEA) and vascular cell adhesion molecule-1 (VCAM-1) 21. Nanobody conjugates are promising tools for cancer immunotherapy and treatment of inflammatory diseases 22.
Recent studies have shown that nanobodies allow higher tumor-to-background (T/B)-ratios than conventional antibodies in in vivo molecular imaging applications 8,17,19. This is explained mainly by the relatively poor and slow tissue penetration of conventional antibodies, slow clearance from circulation, and long retention in non-targeted tissues 23. Moreover, excess of conventional antibodies leads to non-specific accumulation in target antigen-negative tumors caused by the enhanced permeability and retention (EPR) effect 24,25. This means that higher doses of conventional antibodies may increase not only specific signals but also nonspecific signals, thus reducing the maximum achievable tumor-to-background ratio. In contrast, increasing the dose of nanobodies increases the signals of antigen-positive tumors but not of normal tissue or antigen-negative tumors (unpublished data).
Beyond the comparison of nanobodies and conventional antibodies, we outline an intraindividual assessment of antigen-positive and -negative xenografts in the same mice for direct comparison of specific and nonspecific signals due to the EPR effect. The near-infrared fluorophore conjugated probes allowed us to exploit a single probe in vivo and ex vivo using near-infrared fluorescence imaging, flow cytometry, and fluorescence microscopy. Applying our protocols allows for nonradioactive, highly sensitive, and inexpensive optimization of in vivo molecular imaging experiments such as evaluation of new antibody constructs for specific tumor targeting.
The aim of this tutorial study is to highlight the use of NIRF-imaging for evaluation of new antibody constructs in preclinical molecular imaging.
In this protocol, all experiments were performed with a small-animal NIRF-Imaging system, a fluorescence activated cell sorter (FACS) flow cytometer, and a confocal microscope.
Protocol
NOTE: Experiments were performed in accordance with international guidelines on the ethical use of animals and were approved by the local animal welfare commission of the University Medical Center, Hamburg.
1. Preparation of Tumor Cells, Mice, and Antibody Constructs
- Preparation of Lymphoma Cells and Aliquoting of Basement Matrix (Matrigel).
- The day before injection of tumor cells put a sterilized tip box (1,000 µl tips) and the appropriate pipette in -20 °C freezer.
- Thaw the bottle with the basement matrix on ice in the 4 °C fridge O/N.
- On the day of injection fill an ice bucket and place the basement matrix along with pipette, tips, and 1 ml syringes with 30 G needles on ice.
- Aliquot lymphoma cells in a volume of 100 µl of RPMI medium in 1.5 ml microcentrifuge tubes and mix carefully with 100 µl of the basement matrix. Draw up into pre-cooled syringes and put on ice until injection. NOTE: Use good sterile technique and to work on ice the whole time to prevent clogging of the basement matrix.
- Mice Preparation
- Use 8-10 week old athymic nude mice (NMRI-Foxn1nu).
- To reduce autofluorescence of the intestine keep mice on an alfalfa-free diet for 1 week prior to in vivo imaging.
- For injection of lymphoma cells anesthetize mice to effect with 2% isoflurane in an induction chamber. Maintain 1-2% isoflurane for the duration of the procedure using an isofluorane manifold.
- Inject lymphoma cells subcutaneously into the shoulder flanks. For direct intra-individual comparison inject antigen-positive and antigen-negative cells on the right and left side, respectively.
- Use a thumb and index finger to pinch the skin of the mouse and pull it away from the body of the mouse. Inject slowly and evenly into the pouch created by the fingers, creating a single cluster of cells beneath the skin. The basement matrix helps keeping injected cells in place.
- Preparation of Antibody Constructs
- Label monoclonal antibodies and single domain nanobodies with a commercially available protein labelling kit to the fluorescent dye AlexaFluor-680 (AF680) (excitation wavelength = 679 nm, emission wavelength = 702 nm) according the manufacturer’s instructions. Calculate the number of dyes per nanobody and conventional antibody using molar extinction coefficients of 15,720 mol-1 cm-1 and 203,000 mol-1 cm-1, respectively. Assess the purity of antibody constructs by SDS-PAGE size fractionation and Coomassie brilliant blue stain.
- Ensure the specificity of the labeled conjugates by conducting a series of in vitro experiments before the actual imaging studies. Test the specificity of labeled antibody constructs in vitro through blocking studies and unspecific isotype controls 8,20. NOTE: In step 1.2.4 cell numbers for antigen-negative and antigen-positive cells (or different cells lines) may have to be adapted according to different growth rates. For these experiments, 0.5x106 DC27.10 ARTC2 negative and 1.5x106 DC27.10 ARTC2 positive cells were used to obtain tumors of similar size. In step 1.3.1 the process of labeling and labeling efficacy of antibody probes with fluorophores may differ by the labeling kit used.
2. In Vivo Imaging
After 7-9 days, when tumors reach ~8 mm in diameter, inject 50 µg of AlexaFluor680 labelled antibody constructs in a volume of 200 µl saline intravenously into the tail vein of the mouse (mAb-AF680: 50 µg with 2 dyes/molecule ≈ ~4^14 fluorochromes ≈ ~0.8 µg fluorochromes; Nanobody-AF680: 50 µg with 0.3 dyes/molecule ≈ ~5.6^14 fluorochromes ≈ ~1.1 µg fluorochromes).
- Initialize the imaging system and anesthetize mice to effect with 3% isoflurane using an XGI-8 anesthesia system in the induction chamber prior to imaging. Maintain 1-2% isoflurane for the duration of the imaging procedure using the isoflurane manifold housed in the imaging chamber.
- Position mice on heated imaging stage with tumors directed towards the camera. Monitor the proper anesthetization by pinching the toe or tail of the animal; any reaction of the animal indicates that anesthesia is too light.
- Monitor the respiration rate; the anesthesia is too light if the respiration rate is increased and too deep if the respiration rate is decreased, deep or irregular. Protect animal’s corneas with an eye ointment to prevent dryness while under anesthesia.
- Choose fluorescent filter sets of 615-665 nm for excitation, 695-770 nm for emission, and 580-610 nm excitation for background subtraction with a 51 2x 512 pixel matrix size. Set exposure time to auto, pixel binning to medium and F/Stop to 2. NOTE: Shorter exposure times enable faster frame rates; longer exposure times provide greater sensitivity. Binning controls the pixel size on the CCD camera. Increasing the binning increases the pixel size, sensitivity, and frame rate, but reduces spatial resolution. F/Stop sets the size of the camera lens aperture. The aperture size controls the amount of light detected and the depth of field.Note that settings can differ by imaging device and experimental set up. Consult the manufacturer’s manual for optimal results.
- In the imaging software optimize exposure time, F/stop and pixel binning based on the expression level of the cell line. Change these settings at any time during an experiment without impacting the quantitative result. Alternatively, let the Imaging Wizard software automatically determine the parameters.
Image mice before injection and 6 hr after injection of antibody constructs. NOTE: The labelling efficacy may affect the required dose needed for optimal imaging results. Therefore the amount of required antibody-construct for optimal imaging results has to be determined empirically. It may vary depending on labelling efficacy and size of the construct as well as the tumor model and the target-antigen expression.
3. Harvesting and Preparation of Tumors
Fill an ice bucket and place 4%, paraformaldehyde (PFA) phosphate buffered saline (PBS)/protease inhibitor (AEBSF) and PBS/0.2% bovine serum albumin (BSA) on ice.
Once the imaging session is finished, increase the concentration of isoflurane to 4%. After animal ceases breathing, remove it from the imaging stage and perform cervical dislocation.
Mount mouse on a Styrofoam block and spray with 70% ethanol.
Use scissors and forceps to cut the outer skin and pull it back carefully to expose tumors. Remove tumors with scalpel to ensure tumor tissue remains intact.
Cut tumors in half using a scalpel. Place one half in a collection tube with PBS/AEBSF for FACS analyses and the other half in a 50 ml tube with 4% PFA for immunohistochemistry.
- Preparation for immunohistochemistry
- Keep tumors in 4% PFA in the fridge at 4 °C for 24 hr. Transfer tumors to a tube with PBS/30% sucrose and keep in the fridge at 4 °C until the tumor sinks to the bottom of the tube.
- Cut the tumors in appropriate pieces for the cryomolds. Put the tumors in cryomolds and fill with OCT compound so that the tumors are covered.
- Put molds on dry ice and wait until the compound is completely frozen. Transfer frozen tumors to -80 °C freezer and store for immunohistochemistry.
- Cut frozen tumors in sections of 8 μm with a microtome. Use standard immunohistochemistry protocol to stain with antibody against CD31-AF488 to visualize blood vessels and diamidino-phenylindole (DAPI) to visualize nuclei. NOTE: Tumor sections are very sensitive to light as they already contain fluorescently labelled antibodies. Minimize exposure to light as much as possible.
- Preparation for FACS analysis
- Place petri dish on ice and remove the plunger from a 2.0 ml syringe. Put the tumor in the cell strainer and cut it gently into 3-4 pieces using a scalpel. Pour the initial PBS in the petri dish and use the flat end of the plunger to mash the tumor within the cell strainer.
- Flush the cell strainer with the initial PBS to wash out all cells with a 10 ml pipette. Transfer cell suspension in a new tube and spin at 500 x g for 5 min. Discard the supernatant and resuspend the cells in 10 ml PBS/0.2% BSA. Count cells.
- Aliquot 1-5 x 106 lymphoma cells in a 5 ml FACS tube. Spin cells again (500 x g), discard supernatant and resuspend in 100 µl PBS/0.2% BSA.
- Optionally, block FcR using an anti-CD16/CD32-mAb (FcgR3/2) that binds to FcγR on ice for 10 min. Wash cells once with PBS/0.2% BSA.
- Add anti-CD45-mAb to discriminate leucocytes from other cells. Incubate for 20 min on ice in the dark, followed by two washes with PBS/0.2% BSA.
- Right before FACS analysis stain with propidium iodide for 15 min on ice to discern dead cells, followed by two washes with plain PBS. Resuspend in 150 µl for FACS analysis. NOTE: In step 3.7.4 and step 3.7.5 other antibodies may be used depending on the tumor entity.
4. FACS Analysis
- Use a series of gating tools to gate the population of interest in the form of large scatter, doublet discrimination, exclusion of dead cells, gating for CD45 positive cells, and gating for antigen-positive cells.
- At first, gate out cell debris in a forward scatter (FSC-A) versus side scatter (SSC-A) using a two-parameter plot. Next discard cell doublets. Finally, gate out non CD45-antigen-positive (CD45) and dead/dying cells (LD – live/dead-stain).
Record samples using the same template for all experiments. NOTE: Refer to the manufacturer’s protocol for technical advice regarding the use of hardware and analytic software.
5. Microscopic Analysis
Analyze stained tumor cryosections using a confocal microscope with an oil immersion lens (40X objective). Use a He-Neon 633 nm laser excitation of AF680, an Argon Laser for AF488, and a 405-Diode for DAPI.
- Process the raw image data using a software compatible with the microscope. Perform background-correction and noise filtering if necessary. Perform additional image adjustments such as sectioning, cropping as well as brightness and contrast adjustments.
- Finally generate a single composite overlay image from all detection channels. Adjust the brightness and contrast of the individual layers. The composite images will show localization of cells (DAPI-stain, blue), distribution of labelled antibody constructs (AF680-stain, red) and blood vessels (AF488-stain, green).
6. In Vivo Imaging Analysis
Open image files in imaging software and create an overlay image by combining photograph image data with fluorescence image data. Optimize image display by removing tissue autofluorescence background signal from the specific fluorescent signal. This can be done by subtracting the image acquired with a background filter set from the image acquired with the filter set specific for the tracer.
- Draw identical circular measurement regions of interest (ROI) around the antigen-positive tumor and the antigen-negative tumor. To determine background signal intensity, place a circular ROI in an area of the animal where fluorescence signal is expected to be low (e.g., hind leg).
- Use the same ROIs for all animals at all time points. For positioning, use the photographic black-and-white images to identify tumor margins.
- Display ROI data in a measurement table. Use average radiant efficiency data, which enables a more quantitative comparison of fluorescent signals for further statistical analyses.
- Display and compare data as absolute signal intensities or calculate the signal-to-background ratio using measured ROI data from targeted tissue and background tissue. Calculate the tumor-to-background ratio by dividing the tumor uptake value by the background value determined from the hind limb.
- As controls, also analyze antigen-negative and antigen-positive tumors in the same animals as well as animals injected with labelled isotype controls to assess the specificity of signals in vivo.
Representative Results
Fluorescently labelled probes allow for the combination of different NIRF-imaging techniques (Figure 1A). We aimed to perform in vivo NIRF-imaging, flow cytometry, and fluorescence microscopy sequentially in order to compare fluorescently labelled nanobodies and monoclonal antibodies for specific in vivo imaging (Figure 1B).
Mice were injected with 50 μg of nanobody and monoclonal antibody to evaluate the specificity of the fluorescently labelled constructs for in vivo imaging. The results showed specific labeling of antigen-positive tumors with both nanobody and monoclonal antibody at 6 hr after injection (Figure 2). ROI analyses of the antigen-positive tumors showed a much higher T/B ratio of ~12 for the nanobody compared to ~6 for the monoclonal antibody. Moreover, the nanobody showed no nonspecific signal in the antigen-negative tumors, whereas the monoclonal antibody showed nonspecific confounding signals in the antigen-negative tumors.
Besides the nonspecific signal of the negative tumors, the monoclonal antibody also induced nonspecific background signals in the entire animal. This is likely due to excessive free circulating antibodies, which are too large to be renally excreted. Contrariwise, animals injected with nanobodies showed nonspecific signals only in the kidneys due to the renal elimination of the small nanobodies.
Flow cytometry analyses of tumor cell suspensions showed specific labeling of antigen-positive tumor cells with both AF680-conjugates 6 hr after injection. The stronger fluorescence signal of the nanobody labelled cells compared to monoclonal antibody labelled cells reflects the in vivo NIRF-imaging results. Importantly, the flow cytometric analyses reveal that there is no nonspecific labelling of antigen-negative cells with either of the two constructs (Figure 3).
Fluorescence microscopy of tumor cryosections showed a strong and almost homogenous labeling of antigen-positive cells with the nanobody 6 hr after injection. Contrariwise, the monoclonal antibody showed a much weaker and rather inhomogeneous staining (Figure 4A). Antigen-negative tumors show no staining 6 hr after injection of the nanobody, whereas antigen-negative tumors injected with the conventional antibody show nonspecific scattered staining in the interstitial space (Figure 4B).
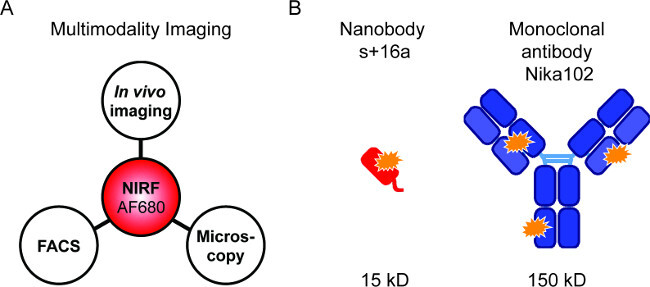 Figure 1: Flourescence imaging and antibody constructs. (A) Imaging setup for evaluation of AF680-conjugates: In vivo NIRF-imaging followed by flow cytometry and fluorescence microscopy. (B) Schematic of AlexaFluor680 labelled nanobody s+16a (red) and monoclonal antibody Nika102 (blue). Orange stars indicate the AlexaFluor680 fluorochromes.
Figure 1: Flourescence imaging and antibody constructs. (A) Imaging setup for evaluation of AF680-conjugates: In vivo NIRF-imaging followed by flow cytometry and fluorescence microscopy. (B) Schematic of AlexaFluor680 labelled nanobody s+16a (red) and monoclonal antibody Nika102 (blue). Orange stars indicate the AlexaFluor680 fluorochromes.
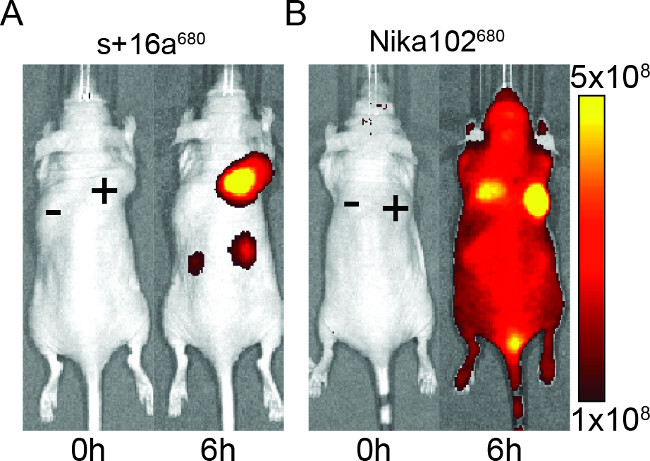 Figure 2: In vivo NIRF-imaging. Images of the fluorescence signal of antigen-positive (+) and antigen-negative (-) tumors in mice that have been injected with nanobody s+16a (A) and monoclonal antibody Nika102 (B). In vivo imaging was performed before (0 h) and 6 h after injection. Signal intensities are displayed as radiant efficiency (p/sec/cm2/sr) / (µW/cm2).
Figure 2: In vivo NIRF-imaging. Images of the fluorescence signal of antigen-positive (+) and antigen-negative (-) tumors in mice that have been injected with nanobody s+16a (A) and monoclonal antibody Nika102 (B). In vivo imaging was performed before (0 h) and 6 h after injection. Signal intensities are displayed as radiant efficiency (p/sec/cm2/sr) / (µW/cm2).
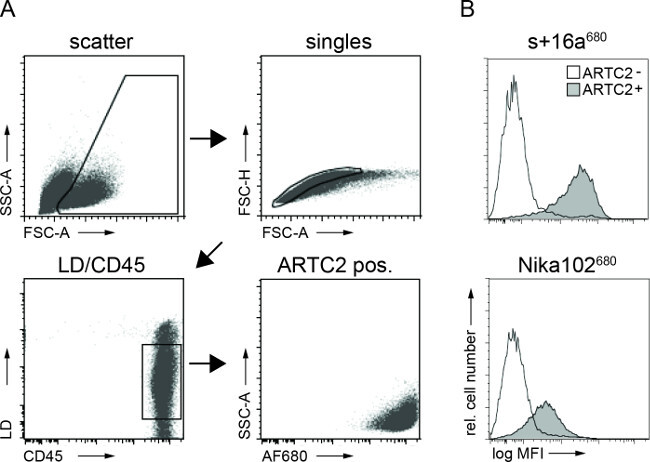 Figure 3: Ex vivo FACS analyses of cell bound antibody constructs from tumor cell suspensions. (A) Gating strategy for FACS analyses of tumor cells. (B) Histograms display the amount of the intravenously injected AF680-conjugated nanobody s+16a and antibody Nika102 specifically bound to the tumor cells in vivo. Antigen-negative tumors are displayed as unfilled histograms and antigen-positive tumors are displayed as filled histograms.
Figure 3: Ex vivo FACS analyses of cell bound antibody constructs from tumor cell suspensions. (A) Gating strategy for FACS analyses of tumor cells. (B) Histograms display the amount of the intravenously injected AF680-conjugated nanobody s+16a and antibody Nika102 specifically bound to the tumor cells in vivo. Antigen-negative tumors are displayed as unfilled histograms and antigen-positive tumors are displayed as filled histograms.
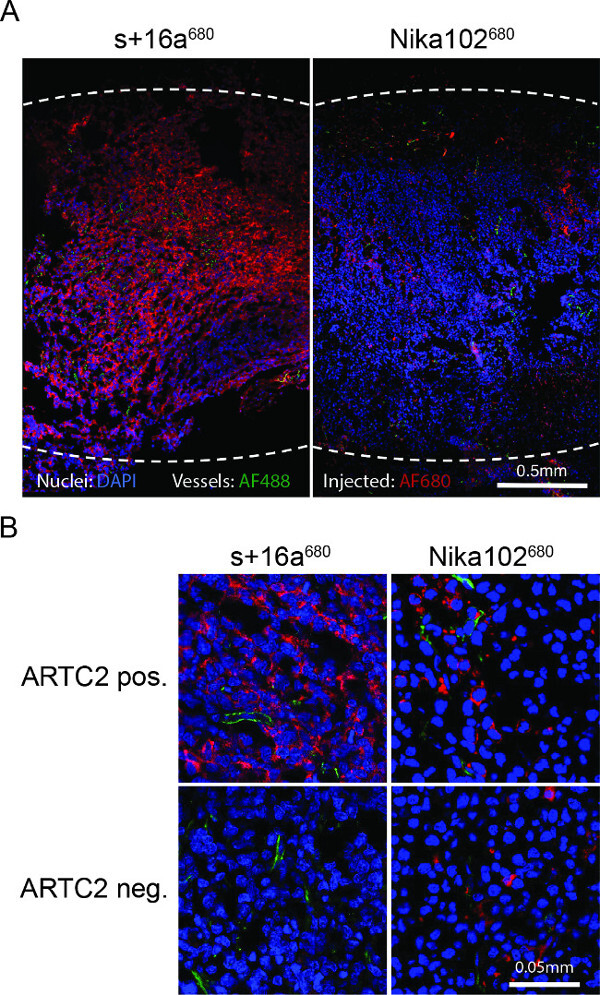 Figure 4: Ex vivo fluorescence microscopy. (A) Overview fluorescence microscopy of entire antigen-positive tumor cryosections 6 h after injection of s+16a680 or Nika102680. Signal intensities of the in vivo intravenously injected AF680-conjugates without any secondary-labelling agents are displayed in red. Ex vivo counterstained nuclei are displayed in blue and vessels in green. Dotted lines indicate outer margins of the entire tumors. (B) Close-up fluorescence microscopy of antigen-positive and antigen-negative tumors.
Figure 4: Ex vivo fluorescence microscopy. (A) Overview fluorescence microscopy of entire antigen-positive tumor cryosections 6 h after injection of s+16a680 or Nika102680. Signal intensities of the in vivo intravenously injected AF680-conjugates without any secondary-labelling agents are displayed in red. Ex vivo counterstained nuclei are displayed in blue and vessels in green. Dotted lines indicate outer margins of the entire tumors. (B) Close-up fluorescence microscopy of antigen-positive and antigen-negative tumors.
Discussion
We used near-infrared fluorophore labelled nanobodies and conventional monoclonal antibodies directed against the same target on lymphoma cells for a multimodal comparison of in vivo and ex vivo analyses. We showed that nanobodies are well suited as diagnostic tools for rapid and specific in vivo detection of lymphomas.
In vivo, s+16a680 allowed a fast and more specific detection of ARTC2-positive xenografts. Apart from the different kinetics for best tumor visualization in vivo, the major drawback of Nika102680 was the high nonspecific signal from ARTC2-negative tumors and nonspecific background signals.
Ex vivo flow cytometric analyses of dispersed cells from dissected tumors showed no nonspecific binding to ARTC2-negative lymphoma cells of injected AF680-conjugates. Ex vivo fluorescence revealed strong and almost homogenous staining of cells in ART2C-positive tumor sections in case of nanobody s+16a, confirming that the nanobody was able to reach even remote areas within the tumor after 6 h. In contrast, the monoclonal antibody showed weaker and inhomogeneous staining of cells in ARTC2-positive tumors after 6 hr. Better imaging results with the conventional antibody can be achieved after 24 hr or 48 hr (data not shown). In order to perform a thorough comparison of two differently sized constructs, imaging at different time points (serial-imaging) has to be performed to identify the optimal imaging time point for each construct.
Like other previous studies, the results reported here emphasize that in vivo molecular imaging with labelled nanobodies allows rapid and specific same-day tumor imaging with high tumor-to-background ratios 12-15,17-19. Contrariwise, conventional antibodies result in low tumor-to-background ratios and nonspecific signals from antigen-negative tumors early after injection due to their slow clearance from the body. In order to obtain optimal imaging results with conventional antibodies, imaging time points 24 hr or even 48 hr after injection are commonly needed. These findings are in accord with previous studies that have suggested that conventional antibodies with proven therapeutic benefit have limited utility in molecular imaging 17,19,26. Therefore conventional antibodies might be rather suited for therapeutic purposes due to their long plasma half-life while nanobodies are rather suited for imaging purposes due to their rapid clearance from the circulation. These differences are due to the fact that any excess of the smaller nanobodies (15-17 kDa) is rapidly cleared via renal elimination while excess of larger conventional antibodies (150 kDa) is retained in the circulation. So the major advantage of nanobodies for molecular imaging is the low background signal at early imaging time points regardless of the injected dose. This allows same day imaging and could be translatable to the clinical setting. Contrariwise, conventional antibodies have to be exactly titrated to minimize nonspecific background signals, while maintaining enough specific signal from the targeted tissue (unpublished data).
One of the limitations of the in vivo NIRF-imaging technique is the low penetration depth which generally allows only imaging of subcutaneous but not of orthotopic tumor models. However, this limitation might be overcome in an experimental setting by the recently developed tomographic photo-acoustic techniques that allow whole-body imaging of living mice 27. Another limitation of the NIRF-imaging technique is the assessment of the tissue dose as compared to radionuclide-mediated imaging. However, the nanobodies may be radiolabelled for positron emission tomography (PET) imaging of xenograft models and exact quantitative assessment of tracer biodistribution. Indeed, our NIRF-imaging results are in accordance with a recent study that compared nanobodies and conventional antibodies for PET imaging. The authors also came to the conclusion that nanobodies allow same-day imaging with high tumor-to-background ratios 15.
However, only the labelling of antibody constructs with the near-infrared fluorescent dye AF680 allowed us the comprehensive in vitro, in vivo and ex vivo near-infrared fluorescence imaging comparison using flow cytometry, fluorescence microscopy, and NIRF-imaging. For this reason, and because it is nonradioactive, highly sensitive, inexpensive, and uses comparatively easy-to-produce targeted probes, we advocate the use of the NIRF-imaging technique for evaluation of new antibody constructs in preclinical molecular imaging.
Disclosures
Friedrich Koch-Nolte and Friedrich Haag receive a share of antibody and protein sales via MediGate GmbH, a wholly owned subsidiary of the University Medical Center Hamburg-Eppendorf.
Acknowledgments
This work was supported by the graduate school ‘Inflammation and regeneration’ of the Collaborative Research Centre 841 of the Deutsche Forschungsgemeinschaft (Alexander Lenz, Valentin Kunick, William Fumey), by the Collaborative Research Centre 877 of the Deutsche Forschungsgemeinschaft (Friedrich Koch-Nolte), by the Werner Otto Foundation (Peter Bannas), by the Wilhelm Sander Foundation (Peter Bannas, Friedrich Koch-Nolte), and by the Deutsche Forschungsgemeinschaft (Martin Trepel, Friedrich Haag and Friedrich Koch-Nolte). We thank the University Cancer Center Hamburg (UCCH) In Vivo Optical Imaging Core Facility and staff at UKE for consultation and their high quality service. The Core Facility was supported in part by grants from Deutsche Krebshilfe (German Cancer Aid).
References
- Koch-Nolte F, et al. Single domain antibodies from llama effectively and specifically block T cell ecto-ADP-ribosyltransferase ART2.2 in vivo. FASEB J. 2007;21:3490–3498. doi: 10.1096/fj.07-8661com. [DOI] [PubMed] [Google Scholar]
- Koch-Nolte F, et al. A new monoclonal antibody detects a developmentally regulated mouse ecto-ADP-ribosyltransferase on T cells: subset distribution, inbred strain variation, and modulation upon T cell activation. J Immunol. 1999;163:6014–6022. [PubMed] [Google Scholar]
- Koch-Nolte F, et al. Use of genetic immunization to raise antibodies recognizing toxin-related cell surface ADP-ribosyltransferases in native conformation. Cell Immunol. 2005;236:66–71. doi: 10.1016/j.cellimm.2005.08.033. [DOI] [PubMed] [Google Scholar]
- Bannas P, et al. Activity and specificity of toxin-related mouse T cell ecto-ADP-ribosyltransferase ART2.2 depends on its association with lipid rafts. Blood. 2005;105:3663–3670. doi: 10.1182/blood-2004-08-3325. [DOI] [PubMed] [Google Scholar]
- Bannas P, et al. Quantitative magnetic resonance imaging of enzyme activity on the cell surface: in vitro and in vivo monitoring of ADP-ribosyltransferase 2 on T cells. Mol Imaging. 2010;9:211–222. [PubMed] [Google Scholar]
- Bannas P, et al. Transgenic overexpression of toxin-related ecto-ADP-ribosyltransferase ART2.2 sensitizes T cells but not B cells to NAD-induced cell death. Mol Immunol. 2011;48:1762–1770. doi: 10.1016/j.molimm.2011.04.015. [DOI] [PubMed] [Google Scholar]
- Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35:208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Bannas P, et al. In vivo near-infrared fluorescence targeting of T cells: comparison of nanobodies and conventional monoclonal antibodies. Contrast Media Mol Imaging. 2014;9:135–142. doi: 10.1002/cmmi.1548. [DOI] [PubMed] [Google Scholar]
- Scheuplein F, et al. NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J Immunol. 2009;182:2898–2908. doi: 10.4049/jimmunol.0801711. [DOI] [PubMed] [Google Scholar]
- Hamers-Casterman C, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- Wesolowski J, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. 2009;198:157–174. doi: 10.1007/s00430-009-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaneycken I, et al. Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J. 2011;25:2433–2446. doi: 10.1096/fj.10-180331. [DOI] [PubMed] [Google Scholar]
- Xavier C, et al. Synthesis, preclinical validation, dosimetry, and toxicity of 68Ga-NOTA-anti-HER2 Nanobodies for iPET imaging of HER2 receptor expression in cancer. J Nucl Med. 2013;54:776–784. doi: 10.2967/jnumed.112.111021. [DOI] [PubMed] [Google Scholar]
- Tchouate Gainkam LO, et al. Correlation Between Epidermal Growth Factor Receptor-Specific Nanobody Uptake and Tumor Burden: A Tool for Noninvasive Monitoring of Tumor Response to Therapy. Mol Imaging Biol. 2011;13(5):940–948. doi: 10.1007/s11307-010-0428-4. [DOI] [PubMed] [Google Scholar]
- Vosjan MJ, et al. Facile labelling of an anti-epidermal growth factor receptor Nanobody with 68Ga via a novel bifunctional desferal chelate for immuno-PET. Eur J Nucl Med Mol Imaging. 2011;38:753–763. doi: 10.1007/s00259-010-1700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira S, Heukers R, Sornkom J, Kok RJ, van Bergen En Henegouwen PM. Targeting tumors with nanobodies for cancer imaging and therapy. Journal Of Controlled Release. 2013;172(3):607–617. doi: 10.1016/j.jconrel.2013.08.298. [DOI] [PubMed] [Google Scholar]
- Kijanka M, et al. Rapid optical imaging of human breast tumour xenografts using anti-HER2 VHHs site-directly conjugated to IRDye 800CW for image-guided surgery. Eur J Nucl Med Mol Imaging. 2013;40:1718–1729. doi: 10.1007/s00259-013-2471-2. [DOI] [PubMed] [Google Scholar]
- Zaman MB, et al. Single-domain antibody bioconjugated near-IR quantum dots for targeted cellular imaging of pancreatic cancer. J Nanosci Nanotechnol. 2011;11:3757–3763. doi: 10.1166/jnn.2011.4167. [DOI] [PubMed] [Google Scholar]
- Oliveira S, et al. Rapid visualization of human tumor xenografts through optical imaging with a near-infrared fluorescent anti-epidermal growth factor receptor nanobody. Mol Imaging. 2012;11:33–46. [PubMed] [Google Scholar]
- Gainkam LO, et al. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT. J Nucl Med. 2008;49:788–795. doi: 10.2967/jnumed.107.048538. [DOI] [PubMed] [Google Scholar]
- Chakravarty R, Goel S, Cai W. Nanobody: the 'magic bullet' for molecular imaging. Theranostics. 2014;4:386–398. doi: 10.7150/thno.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siontorou CG. Nanobodies as novel agents for disease diagnosis and therapy. International journal of nanomedicine. 2013;8:4215–4227. doi: 10.2147/IJN.S39428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, et al. Recent trends in antibody-based oncologic imaging. Cancer Lett. 2012;315:97–111. doi: 10.1016/j.canlet.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. Journal Of Controlled Release. 2012;161:175–187. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nature reviews. Clinical Oncology. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisy MR, et al. In vivo near-infrared fluorescence imaging of carcinoembryonic antigen-expressing tumor cells in mice. Radiology. 2008;247:779–787. doi: 10.1148/radiol.2472070123. [DOI] [PubMed] [Google Scholar]
- Herzog E, et al. Optical imaging of cancer heterogeneity with multispectral optoacoustic tomography. Radiology. 2012;263:461–468. doi: 10.1148/radiol.11111646. [DOI] [PubMed] [Google Scholar]