

Abstract
Immunoblotting (also known as Western blotting) combined with digital image analysis can be a reliable method for analyzing the abundance of proteins and protein modifications, but not every immunoblot-analysis combination produces an accurate result. Here, I illustrate how sample preparation, protocol implementation, detection scheme, and normalization approach profoundly affect the quantitative performance of immunoblotting. This study implemented diagnostic experiments that assess an immunoblot-analysis workflow for accuracy and precision. The results showed that ignoring such diagnostics can lead to pseudoquantitative immunoblot data that dramatically overestimate or underestimate true differences in protein abundance.
Introduction
Among the most indispensible tools in cell-signaling research is the immunoblot. The premise of immunoblotting is simple, but execution is tricky, and there are many variations in the method that can affect the outcome (1). Add quantitation to the end of an immunoblot and the complexity of implementations increases even further. Surprisingly, there are few objective studies on quantitative immunoblotting in the primary literature (2, 3). Lacking a systematic assessment of key factors, researchers are prone to repeat or reinforce mistakes that others have made before them.
Here, I analyze how various methodological choices affect the ability to perform quantitative immunoblotting accurately and precisely. The analysis revealed how seemingly minor variations affect immunoblot linearity and reproducibility, yielding pseudoquantitative numbers that are not directly proportional to the input material. After background subtraction, quantitative immunoblots should strive for zero-intercept linearity: y = bx, where y is the quantified band intensity, x is the abundance of the protein or modification state in the sample, and b is a proportionality coefficient. The value of b is flexible, but lines with nonzero intercepts indicate errors in background subtraction, and nonlinear relationships suggest problems with detection sensitivity or saturation. Either scenario will yield fold-change estimates that are skewed relative to the true differences among samples.
Throughout this work, I systematically altered several experimental parameters that are often neglected or overlooked when immunoblotting. Many other parameters were kept fixed: All gels were run as 15-well, 1.5-mm thick, Tris-glycine minigels on the Bio-Rad Protean III platform; all wet electrophoretic transfers were done onto low-autofluorescence, 0.45-µm polyvinyldifluoride (PVDF) under modified Towbin conditions (4) (25 mM Tris, 192 mM glycine, 0.0375% SDS, 10% methanol unless otherwise indicated); detection was performed on either a LI-COR Odyssey instrument (for fluorescence detection) or a Bio-Rad ChemiDoc MP gel imager (for chemiluminescence detection); and quantitation of raw 16-bit digital images was implemented with the ImageJ gel analysis plugin (5). Using film to perform quantitative immunoblotting was avoided entirely, because the dynamic range of film is so small that quantitative analysis is virtually impossible (3). Film can make small differences in abundance appear as large differences in band intensity. When saturated, film exposures can also hide sample-to-sample variations in high-abundance proteins such as loading controls. Therefore, throughout this study, all data were acquired as digital images. The diagnostic experiments shown here can be easily adapted for other hardware and reagent configurations.
Results
Sample preparation is a critical factor for quantitative immunoblotting
The conditions of cell lysis have a profound impact on the proteins that are extracted and the condition in which they are preserved. For example, lysis of cells or tissues with purely nonionic detergents (Triton X-100 or NP-40) causes some proteins to partition into the soluble and insoluble (pellet) fractions after centrifugation. Radioimmunoprecipitation assay (RIPA) buffer—containing dilute sodium dodecyl sulfate (SDS, a denaturing detergent) and deoxycholate (a disruptor of protein-protein interactions)—is widely used as a lysis buffer for whole-cell extraction. Nonetheless, RIPA buffer lysis still generates an insoluble fraction with major protein constituents from the cytoskeleton and extracellular matrix (6).
To test how RIPA lysis conditions affected immunoblotting results, I lysed HT-29 human colon adenocarcinoma cells in RIPA buffer, boiled the RIPA-insoluble pellet in an equal volume of dithiothreitol-containing Laemmli sample buffer (7), and then immunoblotted for 20 different protein targets. As expected, RIPA lysis buffer efficiently solubilized many cytoplasmic proteins [glyceraldehyde 3-phosphate dehydrogenase (GAPDH), heat shock protein 90 (Hsp90)] and signaling proteins [inhibitor of nuclear factor-κB α (Iκβα), various kinases] (Fig. 1A and B). RIPA buffer also extracted the cytoskeletal and cytoskeleton-associated proteins, actin and focal adhesion kinase (FAK). However, tubulin and intermediate filament proteins (lamin A and KRT5) showed substantial losses into the RIPA-insoluble fraction (Fig. 1C). Remarkably, RIPA insolubility was not limited to cytoskeletal proteins: The transcription factor GATA2 and the cell-cell adhesion protein β-catenin were also present in the insoluble fraction. In contrast, lysis with Laemmli sample buffer, followed by shearing of the viscous genomic DNA with a high-gauge needle, solubilized proteins that are tightly associated with DNA, such as histones (Fig. 1D). Despite rules of thumb for protein solubility in various lysis buffers (6), these results show that it is best to confirm proper solubilization of proteins of interest before embarking on an immunoblot study.
Fig. 1.

Radioimmunoprecipitation assay (RIPA) buffer solubilizes many, but not all, cellular proteins. (A) Examples of proteins that are entirely solubilized (100% in the supernatant, Sup). (B) Examples of proteins that are mostly solubilized (>90% Sup). (C) Examples of proteins that are partially solubilized (≤90% Sup). (D) Dimethyl-lysine 4 histone H3 (H3K4me2) resides almost entirely in the RIPA-insoluble pellet (Pel). Band intensities were quantified from the 16-bit digital image by densitometry in ImageJ and are shown normalized to the Sup lane for each target. n.d., not detected. Data are representative of 2–4 experiments.
If 10–30% of a protein were consistently lost in the insoluble fraction, then the choice of lysis conditions would not be critical. However, specific proteins can shift between soluble and insoluble fractions in a stimulus-dependent manner. As an example, I activated the FAS death receptor in MCF10A-5E human breast epithelial cells (8) and lysed the cells in NP-40 buffer (lacking SDS and deoxycholate), RIPA buffer, or Laemmli sample buffer. Although the cleavage of caspase-3 was readily detected in all three preparations (Fig. 2A), cleaved forms of caspase-8 were only detected in Laemmli sample buffer (Fig. 2B). Thus, quantitative measures of caspase-8 processing would require the Laemmli preparation for accurate results (9–11). My lab has found that similar precautions are required for monitoring regulated changes in intermediate-filament proteins (12), such as KRT5 (Fig. 1C). Regulated oligomeric or polymeric protein assemblies may be particularly susceptible to differential partitioning between soluble and insoluble fractions.
Fig. 2.

Posttranslational modifications can move protein into the insoluble fraction of common lysis buffers. MCF10A-5E cells were exposed to the Fas crosslinking agent anti-APO-1 (1 µg/ml) (48) for 24 hours, then floating and adherent cells were lysed in NP-40 lysis buffer, RIPA buffer, or dithiothreitol-containing Laemmli sample buffer (SB). (A) Effect of solubilization conditions on the detection of cleavage products of caspase-3. (B) Effect of solubilization conditions on the detection of cleavage products of caspase-8. Vinculin, tubulin, GAPDH, Hsp90, and p38 were used as loading controls where indicated. Data are representative of three experiments.
The stability and posttranslational modifications of lysate proteins are also affected by the activity of co-mingling cellular enzymes, such as proteases and phosphatases. These enzymes are usually blocked with inhibitors that are supplemented into nondenaturing lysis buffers, but the SDS and deoxycholate in RIPA are sometimes assumed to inactivate most cellular enzymes. I tested how effectively RIPA and NP-40 buffers inhibited protein phosphatases by omitting from both buffers the Ser-Thr phosphatase inhibitor microcystin-LR (13) and the Tyr phosphatase inhibitor orthovanadate (14). I lysed parallel cultures of AC16 ventricular cardiomyocytes (15) and immunoblotted for multiple phosphorylation sites along the Akt–glycogen synthase kinase-3 (GSK3)–glycogen synthase (GS) signaling axis (Fig. 3A to D). For these phosphoproteins, the addition of phosphatase inhibitors was more critical in RIPA buffer than in the nondenaturing NP-40 buffer. The extent of sensitivity depended strongly on the phosphorylation site, with Thr308 of Akt, Ser21 of GSK3α, and Ser9 of GSK3β showing greater lability than Ser473 of Akt and Ser641 of GS. These results collectively showed that lysis buffer composition substantially affects the results of quantitative immunoblotting.
Fig. 3.

Phosphatase inhibitors are critical to preserve certain phosphorylated residues under certain lysis conditions. (A, B) Effect of lysis buffer and presence or absence of phosphatase inhibitors (PPIs) on the detection of phosphorylated Akt (p-Akt) on Thr308 (T308) and Ser473 (S473). (C) Effect of lysis buffer and presence or absence of PPIs on detection of glycogen synthase kinase-3α/β phosphorylated on Ser21 and Ser9 (p-GSK3α/β). (D) Effect of lysis buffer and presence or absence of PPIs on detection of glycogen synthase phosphorylated on Ser641 (p-GS). AC16 cells were lysed in RIPA or NP-40 lysis buffer with or without PPIs. Vinculin, tubulin, GAPDH, and actin were used as loading controls where indicated. Total Akt, GSK3α/β, and GS were used to monitor specific changes in protein abundance. Band intensities were quantified from the 16-bit digital image by densitometry in ImageJ and are shown normalized to the average +PPI conditions for each target across both lysis conditions. Data are representative of two experiments.
Assessing immunoblot protocols by serial dilution
For immunoblotting, a single protocol that is optimal for all electrophoresis-transfer setups and detection methods does not exist. However, the quantitative accuracy and dynamic range of any protocol can be assessed using a serial dilution of cell extract and a panel of primary antibodies. As an example, I sought to determine wet-transfer conditions (specifically, methanol concentration) that enabled quantitative detection of most target proteins. Here, the transfer buffer always included 10% methanol (see Materials and Methods), but other protocols use 20% methanol according to the original conditions of Towbin (4). Although immunoblot bands are brighter and crisper with the higher methanol concentration, how methanol percentage affects quantitative accuracy and dynamic range of immunoblot band intensities is unknown.
Using HT-29 cell extracts, I performed an extended twofold serial dilution from a grossly overloaded sample (200 µg extract) to one below the limit of detection (100 ng extract). Two replicate gels were transferred in buffer containing 10% or 20% methanol, and then membranes were probed for seven different targets (file S1). For actin and p38, I found that detection was linear up to 50 µg of total protein, irrespective of the methanol concentration (Fig. 4A and B). This zero-intercept linearity is ideal, because band density is directly proportional to input material without the need to refer to a calibration curve. By contrast, Hsp90 and tubulin showed a hyperbolic saturation under both transfer conditions with less than 25 µg of total protein (Fig. 4C and D). Saturation can result from steric crowding of antibody epitopes, quenching of fluorescently labeled secondary antibodies, or oxidation of enzyme-conjugated secondary antibodies. Regardless of the source, band densities in this regime no longer provide a linear estimate of sample abundance, and calibration is required to obtain accurate measurements. Interestingly, the improved transfer of proteins in 20% methanol appeared to shift the detection of multiple targets from a linear regime to one of hyperbolic saturation (Fig. 4E to G). The results from this diagnostic study indicated that the lower methanol concentration was preferred for my immunoblot protocol.
Fig. 4.

Linearity and hyperbolic saturation of immunoblots determined by serial dilution. (A and B) Immunoblots for actin and p38 are linear under both transfer conditions. (C and D) Immunoblots for Hsp90 and tubulin are hyperbolically saturated under both transfer conditions. (E to G) Linear detection of immunoblots for E-cadherin, ERK1/2, and GAPDH occurred with tank transfer conditions containing 10% methanol. HT-29 cells were lysed in RIPA buffer, immunoblotted for the indicated targets, and imaged. Left panels show the immunoblots, middle panels show log-log plots of the quantified band intensities from the blots on the left, and the right panels show linear plots of the same data. Linear fits are gray when the hyperbolic model is no better than the linear model for that transfer condition. Linear fits are red when the linear fit of the associated transfer condition is better than the linear fit of the other transfer condition. Hyperbolic fits are green when the hyperbolic model is better than the linear model for that transfer condition. Data are in blue when neither the linear nor the hyperbolic model provides a better fit. Model comparisons were done by the F test (FDR = 5%; n = 5–8 dilutions). See file S1 for raw images and calculations.
Similar comparisons have also caused my lab to favor fluorescence-based detection over chemiluminescence whenever possible. I compared the linearity of IrDye-conjugated secondary antibodies to horseradish peroxidase conjugates that were incubated with an enhanced chemiluminescence cocktail (16) or a commercial substrate marketed for high-sensitivity applications (file S2). Under the same immunoblotting conditions, chemiluminescent exposures consistently yielded stronger band densities (Fig. 5A to C). However, the linear dynamic range was very limited, and signals often decreased at high protein inputs (Fig. 5C). This can occur when side products of the peroxidase-catalyzed reaction are oxidized and precipitated, causing the membrane to “brown out” and absorb the emitted photons. Titrating down the amount of protein or primary antibody can avoid the problem, provided that the researcher is aware of it.
Fig. 5.
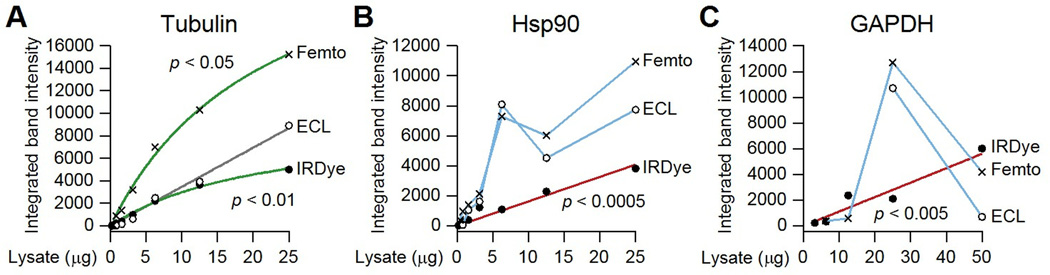
Quantitative immunoblotting is challenging when imaging by chemiluminescence. (A to C) HT-29 lysates were prepared as in Fig. 4, immunoblotted for the indicated proteins, and imaged by IRDye fluorescence, enhanced chemiluminescence (ECL), or SuperSignal West Femto chemiluminescence as described (12, 16, 45, 46). Linear fits are shown in gray when the hyperbolic model is not significantly better than the linear model for that imaging condition. Linear fits are shown in red when the linear fit of the associated imaging condition is significantly better than the linear fit of the other imaging conditions. Hyperbolic fits are shown in green when the hyperbolic model is significantly better than the linear model for that imaging condition. Data are interpolated in blue when neither the linear nor the hyperbolic model provides a better fit. All model comparisons were done by the F test at a false-discovery rate of 5% (n = 4–8 dilutions). See file S2 for raw images and calcuations.
Optimizing loading controls
Arguably the biggest source of confusion in quantitative immunoblotting is the role of protein loading and loading controls (17). Normalizing for cell numbers across samples is challenging, because it is difficult to estimate changes in cell proliferation and death among different conditions. Such estimates also do not account for variations in initial seeding density and final lysate volumes, which will affect the observed protein abundance. Consequently, immunoblot samples are typically prepared according to total cellular protein (18, 19), assuming that the average protein content per cell is constant across the different conditions.
To complement total-protein estimates, immunoblots typically include loading control proteins, which provide a secondary check that roughly equal amounts of cellular material have been added. Two key assumptions of the loading control are that (i) its abundance is roughly constant across the different conditions, and (ii) its immunoblot band intensity is linearly reflective of its abundance. However, a single loading control may not fulfill both of these assumptions (2).
If total cellular protein can be quantified accurately, then total protein is loaded equivalently across samples, and one or two loading controls suffice as a qualitative confirmation of overall protein abundance (20–22). Conversely, if the total cellular protein is not known or cannot be determined accurately, then the input must be normalized to some estimate of protein loading. A common approach found in the literature is to normalize by only one loading control, but this scaling is highly problematic. Taking one unknown quantity (the protein of interest) and dividing it by another unknown quantity (a single loading control) creates a number with very poor statistical properties, including an undefined mean. The dangers of single-variable normalization have long been recognized in data from microarrays (23) and quantitative PCR (polymerase chain reaction) (24), but not data from immunoblotting. A solution is to aggregate the band intensities from multiple loading controls, calculating a mean estimate of total cellular content that is less sensitive to the technical or biological fluctuations of a single loading control (12, 25).
To demonstrate the utility of multiprotein normalization, I immunoblotted for linker phosphorylation of Smad2 on Ser245/250/255 (p-Smad2 linker) in MCF10A-5E cells that had been stimulated with transforming growth factor-β (TGFβ) (file S3). Under these conditions, Smad2 linker phosphorylation is mediated by cyclin-dependent kinases (26), so cells were additionally pretreated with or without the pan- CDK inhibitor flavopiridol (27). The TGFβ stimulation ± flavopiridol inhibition experiment was performed in biological quadruplicate to assess reproducibility of p-Smad2 linker quantification. Lacking any total-protein normalization, p-Smad2 linker densitometry was variable, with flavopiridol producing only a marginally significant decrease in phosphorylation (Fig. 6A and B). To improve reproducibility, I blotted the same membrane for five potential loading controls: total Smad2, tubulin, Hsp90, GAPDH, and p38. The antibodies for these proteins are from various hosts and yield single immunoreactive bands under the blotting conditions used here. These properties of the antibodies and the detected control proteins enabled multiplex detection of loading controls together with p-Smad2 linker or after a single round of membrane stripping (see Materials and Methods).
Fig. 6.

Reproducibility of quantitative immunoblots across biological replicates is improved after normalization to multiple loading controls. (A) Representative immunoblot for phosphorylated Smad2 on Ser245/250/255 (p-Smad2 linker) in MCF10A-5E cells stimulated with 50 ng/ml TGFβ for 30 minutes with or without 1 hour preincubation with 300 nM flavopiridol. Tubulin, Hsp90, GAPDH, and p38 were used as loading controls. Total Smad2 was used to monitor overall changes in protein abundance and served as a fifth candidate loading control for this analysis. (B) Raw p-Smad2 linker densitometry quantified in ImageJ. (C) Decrease in the coefficient of variation among p-Smad2 biological replicates with increasing numbers of loading controls. The best (GAPDH) and worst (tubulin) single loading-control normalizations are highlighted. (D) p-Smad2 linker densitometry after normalization to the mean band intensity of tubulin, Hsp90, GAPDH, p38, and total Smad2 for each biological replicate. For B and D, data are shown as the mean ± SE. of n = 4 biological replicates, with differences in means assessed by Welch’s two-sided t test. For C, data are shown as the mean coefficient of variation ± SD. of n = 1–10 possible normalization combinations for the indicated number of loading controls. See file S3 for raw images and calculations.
Upon quantifying band densities in ImageJ, I compared the reproducibility of p-Smad2 linker after normalization to all possible combinations of loading controls: 25 = 32 combinations. For single loading-control normalization, the effect on replicate-to-replicate reproducibility heavily depended on the choice of loading control. Normalizing to GAPDH decreased the p-Smad2 linker coefficient of variation by more than twofold (21% to 9%), but normalizing to tubulin had virtually no effect (Fig. 6C). This does not imply that GAPDH is always a good loading control or that tubulin is always a bad one; rather, it emphasizes the danger of relying on a single measured variable to estimate total protein content. As higher-order combinations of loading controls were tested as normalizers, I found that the coefficient of variation of p-Smad2 linker steadily improved toward 7–8%, consistent with values reported previously (10, 11). Importantly, this reproducibility became less dependent on the specific combination of loading controls (note the shrinking error bars in Fig. 6C), indicating that I had converged on a measure of cellular content per lane that was truly representative. With all five loading controls, both the TGFβ-induced stimulation of p-Smad2 linker and its inhibition by flavopiridol were highly significant (Fig. 6D).
Others have noted that many common loading controls are abundant proteins (for example, tubulin) that result in saturated band intensities under the conditions needed to detect proteins or modification states of interest (2). On the basis of the results with the serial dilutions (Fig. 4D), saturation is probably the reason why tubulin worked poorly as a loading control for p-Smad2 linker in this setting (Fig. 6B). However, in the context of multiple loading controls that are averaged, a saturated loading control negligibly affects normalization because of its reduced sample-to-sample variation. An alternative strategy is to prepare a separate set of immunoblot lanes with decreased protein content for loading controls (2). However, normalizing from different lanes will miss lane-specific irregularities in sample preparation or electrophoretic transfer, which can be minimized with good technique but not eliminated.
A possible alternative to multiple loading controls is to use reversible total-protein stains that are compatible with PVDF membranes (such as, Memcode or Ponceau S). For such a stain to be useful, it must be quantitative for total cellular protein (directly proportional or, at least, hyperbolically saturating), and it must not interfere with the subsequent immunoblot. I tested two total-protein stains that can be reversibly applied to PVDF membranes: Memcode Reversible Protein Stain (commercially available from Thermo Scientific) and Ponceau S (28). Memcode yielded a strong blue banding pattern that was readily detected by white-light epi-illumination and a CCD camera (fig. S1A). Using a blank region of the PVDF membrane to define a background for subtraction, I found that total Memcode band intensity increased hyperbolically with zero intercept over a relevant range of lysate amounts (fig. S1B). However, the staining procedure markedly increased the 700-channel background fluorescence of the membrane, and this background was not removed by the recommended stain removal (erasing) procedure (fig. S1C). Although potentially useful for chemiluminescence immunoblots, Memcode is not suitable to two-color fluorescence detection.
I uncovered a different set of problems with Ponceau S. Compared to Memcode, the red Ponceau S stain was not as efficiently detected by the CCD camera (fig. S1A and D). However, its image densitometry was linear as a function of lysate amount, and there was no background fluorescence caused by Ponceau S staining or erasure (fig. S1E to G). Ponceau S appeared to fill all the requirements for a total-protein stain, except for one major drawback—the zero-intercept of its densitometry could not be accurately estimated from a blank region of the PVDF membrane (fig. S1D, lane B), causing a negative bias of ~10,000 intensity units (fig. S1E). Without a lysate calibration curve to estimate this bias on a PVDF membrane, I concluded that Ponceau S cannot be used for relative protein quantification. When immunoblotting for phosphoproteins, there are additional complications with erasing Ponceau S from PVDF membranes, because the alkaline conditions for erasure will chemical modify phosphorylated Ser or Thr residues (29).
Given these data with the reversible total-protein stains, accurate quantitation of immunoblot data should adopt the best practices of quantitative PCR (24) and use an assortment of three or more loading controls spanning a range of abundances when direct total-protein measures are lacking. To minimize cost and effort, my lab detects constitutively produced proteins with high-affinity antibodies that work reliably at low concentrations (25–100 ng/ml), yield single immunoreactive bands, and thus are ideal for multiplexing (see Materials and Methods; Table 1).
Table 1.
List of antibodies used and proteins or epitopes detected.
| Protein | Epitope | Host | Vendor | Catalog number | Dilution |
|---|---|---|---|---|---|
| Actin* | Total | Mouse | Ambion | AM4302 | 1:10,000 |
| Akt | Total | Rabbit | CST | 9272 | 1:1000 |
| p-Thr380 | Rabbit | CST | 2965 | 1:1000 | |
| p-Ser473 | Rabbit | CST | 4060 | 1:1000 | |
| β-catenin | Total | Mouse | BD | 610154 | 1:2000 |
| Caspase-3 | Total | Rabbit | CST | 9662 | 1:1000 |
| Caspase-8 | Total | Mouse | CST | 9746 | 1:1000 |
| E-cadherin | Total | Mouse | BD | 610182 | 1:2000 |
| ErbB3 | Total | Rabbit | CST | 4754 | 1:1000 |
| ERK | Total | Rabbit | CST | 9102 | 1:2000 |
| Total | Mouse | CST | 4696 | 1:1000 | |
| p-Thr202/Tyr204 | Rabbit | CST | 4370 | 1:1000 | |
| FAK | Total | Rabbit | SCBT | sc-558 | 1:1000 |
| GAPDH* | Total | Mouse | Ambion | AM4300 | 1:40,000 |
| GATA2 | Total | Rabbit | SCBT | sc-9008 | 1:1000 |
| GS | Total | Rabbit | CST | 3893 | 1:1000 |
| p-Ser641 | Rabbit | CST | 3891 | 1:1000 | |
| GSK3α/β | Total | Rabbit | CST | 5676 | 1:1000 |
| p-Ser21/Ser9 | Rabbit | CST | 9327 | 1:1000 | |
| Histone H3 | Dimethyl-Lys4 | Rabbit | Millipore | 07-030 | 1:1000 |
| Hsp90* | Total | Rabbit | SCBT | sc-7947 | 1:2000 |
| IκBα | Total | Mouse | CST | 4814 | 1:2000 |
| KLF4 | Total | Rabbit | SCBT | sc-20691 | 1:1000 |
| KRT5 | Total | Chicken | Covance | SIG-3475 | 1:2000 |
| Lamin A/C | Total | Mouse | SCBT | sc-7292 | 1:1000 |
| MCL1 | Total | Rabbit | SCBT | sc-819 | 1:1000 |
| p38* | Total | Rabbit | SCBT | sc-535 | 1:5000 |
| PDI | Total | Mouse | Thermo | MA3-019 | 1:2000 |
| Smad2 | Total | Mouse | CST | 3103 | 1:2000 |
| p-Ser245/250/255 | Rabbit | CST | 3104 | 1:1000 | |
| Tubulin* | Total | Chicken | Abcam | ab89984 | 1:20,000 |
| Total | Rabbit | CST | 2148 | 1:2000 | |
| Vinculin* | Total | Mouse | Millipore | 05-386 | 1:10,000 |
Used as loading controls; BD, BD Biosciences; CST, Cell Signaling Technology; SCBT, Santa Cruz Biotechnology; Thermo, Thermo Fisher Scientific
Stripping, reprobing, and the total-protein control
Aside from loading controls for total cellular content, immunoblots that quantify protein modification states should contain an additional control: an immunoblot for the total protein. This control serves to gauge how much of the observed change in protein modification can be explained by differences in target abundance. For rapid experiments that are expected to avoid protein turnover and synthesis (such as the one shown in Fig. 6), the total-protein control can additionally contribute to a panel of loading controls if changes in band intensity are clearly co-fluctuating with other loading controls. However, for comparisons on long time scales, across different cell types, or with rapid protein turnover, the total-protein control is specifically important on its own and may not be a reliable indicator of loading.
A common way to estimate target protein abundance from a modification-specific immunoblot is to strip the membrane of antibody and then reprobe with an antibody that recognizes the total protein (or at least the unmodified form). Essential to this approach is confirming that the stripping conditions have fully removed the original modification-specific antibody from the membrane of choice (PVDF or nitrocellulose). If not, the reprobed blot will show artifacts from the residual first antibody. As an example, I acutely stimulated AC16 cells with epidermal growth factor (EGF) for 5 minutes and immunoblotted for phosphorylated extracellular signal-regulated kinases 1 and 2 (p-ERK1/2) with an antibody that is difficult to remove. Technical replicates of the same two lysates showed good reproducibility in the observed EGF-stimulated induction of p-ERK1/2, and loading controls were consistent across the membrane (Fig. 7A). After cutting the membrane into thirds, I tested three stripping conditions: (i) a gentle low pH glycine buffer solution (1.5% glycine [pH 2.2], 0.1% SDS, 1% Tween), (ii) a more stringent guanidinium solution (6 M guanidine-HCl) (30), and (iii) a high stringency SDS plus β-mercaptoethanol solution with heat (62.5 mM Tris [pH 6.8], 2% SDS, 100 mM β-mercaptoethanol at 50°C) (31). The stripped membranes were blocked identically and reprobed for total ERK1/2 along with three additional loading controls. Reprobed membranes were imaged and displayed identically to emphasize differences in the immunoblot signal detected (Fig. 7B).
Fig. 7.
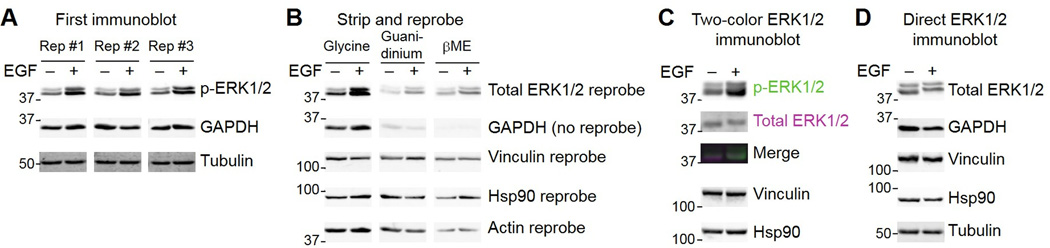
Membrane stripping and reprobing is a quantitative tradeoff between antibody removal and total protein loss. (A) Replicate immunoblots for phosphorylated ERK1/2 phosphorylated on Thr202 and Tyr204 of ERK1 or Thr185 and Tyr187 of ERK2 (p-ERK1/2) in AC16 cells stimulated with 100 ng/ml EGF for 5 minutes. GAPDH and tubulin were used as loading controls in the first immunoblot. (B) Reprobe of the membrane in A for total ERK1/2 after stripping with glycine buffer, guanidinium, or β-mercaptoethanol (βME) stripping buffer. Vinculin, Hsp90, and actin were used as loading controls for the reprobed blots. (C) Two-color fluorescence immunoblot for p-ERK1/2 (green) and total ERK1/2 (magenta) of the same lysates as in A. Vinculin and Hsp90 were used as loading controls. (D) Direct immunoblot for total ERK1/2 of the same lysates as in A. A lower percentage polyacrylamide gel was used in C and D to emphasize the total ERK1/2 upshift after stimulation with EGF. GAPDH, vinculin, Hsp90, and tubulin were used as loading controls. Data are representative of two experiments.
The experiment revealed that the low pH glycine strip was ineffective at removing the antibodies. Indeed, “total” ERK1/2 looked identical to p-ERK1/2, and there was even a clear artifact of residual GAPDH antibody staining. By contrast, the guanidinium strip removed most of the GAPDH antibody but left a clear p-ERK1/2 artifact in the total ERK1/2 reprobe, with increased staining in the +EGF lane. The SDS plus β-mercaptoethanol strip completely removed the GAPDH antibody and yielded the closest approximation of total ERK1/2 abundance, although with substantial loss of total protein from the membrane based on the immunoreactivity of the three additional loading controls (compare the glycine-stripped membrane to the membrane stripped with SDS plus β-mercaptoethanol). Moreover, after SDS plus β-mercaptoethanol stripping, the total ERK1/2 bands still showed an artifactual increase in the EGF-treated sample. To avoid the need to strip and reprobe the same blot, an alternative is to use two-color fluorescence detection with phosphorylation-specific and total antibodies that are raised in different hosts and against different epitopes (Fig. 7C). Another option is to measure total target protein by immunoblotting a replicate set of samples and confirming that the matched loading controls are comparable to those in the modification-specific immunoblot (Fig. 7D).
Advanced quantitative immunoblotting: absolute quantification
For some computational models of biochemical networks, the absolute abundances of specific cellular proteins are needed (35, 36). Such applications require explicit calibration using proper absolute standards on the same immunoblot. For the cellular proteins of interest, appropriate standards are purified recombinant proteins, which have been calibrated against a purified protein of known mass.
To illustrate this process, I quantified the absolute masses of ERK2 and p38 (per 25 µg cellular extract) in HT-29 cells and AC16 cells (file S4). Absolute quantification enabled ERK2-p38 comparisons within each cell line as well as between cell lines.
Recombinant ERK2 and p38 proteins were cloned, purified, and quantified by protein assay. Using a known mass of bovine serum albumin (obtained as a calibrated standard from a protein assay kit), a standard curve was constructed by running serially diluted bovine serum albumin on a polyacrylamide gel alongside recombinant glutathione-S-transferase-tagged purified ERK2 and p38 (22) (Fig. 8A). The gel was then stained with Coomassie blue and scanned for its near-infrared fluorescence (37), yielding a digital image for densitometry in ImageJ (see Materials and Methods). To accommodate some degree of band saturation and improve the dynamic range of detection, I fit the protein band intensities to a simple hyperbolic curve:
with two free parameters (a, b) that were estimated by least-squares regression. The modeled fit captured all of the albumin standards and enabled mapping the measured GST-ERK2 and GST-p38 band densities to total protein amount (Fig. 8B). Dividing by the volume of sample loaded into the gel resulted in an estimated concentration of full-length protein in the recombinant preparation.
Fig. 8.
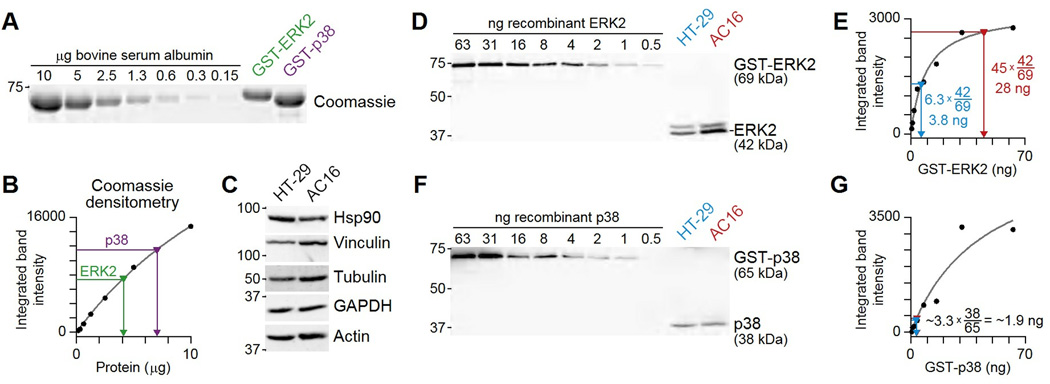
Workflow for absolute protein quantification. (A) Serial dilution of an albumin standard to calibrate recombinant purifications of GST-ERK2 and GST-p38 by Coomassie staining. (B) Albumin band intensity (black) plotted as a function of protein and fit to a hyperbolic model (gray) that infers the amounts of GST-ERK2 (green) and GST-p38 (purple) protein. (C) HT-29 and AC16 cells have roughly equal protein constituents by mass based on the amount of Hsp90, vinculin, tubulin, GAPDH, and actin detected in 25 µg of each sample. (D) Serial dilution of the GST-ERK2 standard to calibrate endogenous abundances of ERK2 in HT-29 and AC16 cells. (E) GST-ERK2 band intensity (black) plotted as a function of protein input and fit to a hyperbolic model (gray) that infers the amount of ERK2 in HT-29 cells (blue) and AC16 cells (red). (F) Serial dilution of the GST-p38 standard to calibrate endogenous abundances of p38 in HT-29 and AC16 cells. (E) GST-p38 band intensity (black) plotted as a function of protein input and fit to a hyperbolic model (gray). The model was used to infer the amount of ERK2 in HT-29 cells (blue) and AC16 cells (red). Data are representative of two experiments. See file S4 for raw images and calculations.
Size separation of the recombinant protein before quantification is critically important, because purified preparations often contain cleavage products that add to total protein but are not immunoreactive. Commercial vendors of recombinant protein may simply quantify total protein (full length plus fragments that are not useful for calibration), ultimately resulting in an overestimation of cellular protein. For calibration, one should also remember that unknown samples must fall between calibration samples that are well fit by the linear or hyperbolic curve. Samples that fall outside the calibration range will cause the linear or hyperbolic equation to make extrapolations that could be highly inaccurate.
Before making comparisons between cell lines, I verified that loading the same total mass of HT-29 and AC16 extract gave approximately equivalent band intensities for several loading controls (Fig. 8C). Hsp90 abundance was higher (per total mass) in HT-29 cells, whereas vinculin and tubulin were higher in AC16 cells, and GAPDH and actin were approximately equal. The uncorrelated abundance of these “housekeeping proteins” suggests that the cell extracts can be fairly compared on a total-protein basis. The results further reinforced the importance of using multiple loading controls.
To quantify the protein of interest in cellular extracts, I ran the calibrated standards and the extracts alongside one another and blotted with an antibody that detects both the recombinant protein and the protein of interest (Fig. 8D to G). Note that the ERK2 calibration cannot be used to quantify the ERK1 band at 44 kDa because of differences in the immunoreactive epitope. In addition, a mass correction is needed to account for the size difference between the GST-tagged recombinant protein and the endogenous protein, because immunoblotting quantifies antigen independently of its mass (the correction is 42/69 for ERK2 and 38/65 for p38; Fig. 8E and G). This analysis showed that AC16 cells had >7-fold more ERK2 protein compared to HT-29 cells, despite only ~2-fold difference in band intensity (Fig. 8E). The limited different in band intensity was due to the saturation of the ERK2 immunoblot at high protein concentrations, which was captured by the calibration curve. ERK2 abundance in AC16 cells was also high compared to the abundance of p38, which had a concentration that was an order of magnitude lower (Fig. 8G). Although laborious, this type of absolute quantification can be done systematically for multiple cellular proteins and provide new insight into signaling function (38).
Discussion
Just because we can put numbers on an image does not imply that we should—a quantified biomolecule should relate directly to the true quantity of that biomolecule if it is to be meaningful (8, 39–43). What are the implications of pseudoquantitative immunoblotting? When chemiluminescence is used cavalierly, there is a danger of wildly exaggerated claims. For example, a twofold change in Hsp90 abundance might appear as a ~fivefold change in the intensity of the band on an immunoblot (compare the ECL values for ~6 µg and ~3 µg in Fig. 5B). A twofold change in GAPDH abundance could be “quantified” as a >20-fold change on the basis of band intensity (compare the ECL values for 25 µg and ~13 µg in Fig. 5C). This makes numerical results look very impressive, but the truth is still a twofold change in abundance.
With fluorescence detection, there is not the danger of a runaway reaction, but a persistent concern is saturation (Fig. 4). Saturated immunoblots do not overestimate a change in protein; instead, they can substantially underestimate it. To demonstrate, I plotted dilutions of an unstimulated and stimulated extract for a theoretical immunoblot band that is hyperbolically saturated (Fig. 9). There is a clear threefold change in abundance when the target is immunoblotted under conditions where the linear approximation is accurate (<10 µg extract in this example). However, overloading the gel with 50 µg of extract reduces the difference to 1.4 fold. This type of dampening has led some to conclude that fluorescence detection is not sensitive compared chemiluminescence (especially on film). However, the comparison is not fair if the 1.4-fold difference in antibody binding has been exaggerated by a nonlinear chemiluminescent reaction (Fig. 5B and C).
Fig. 9.

Quantifying partially saturated immunoblots can dramatically underestimate differences between samples. In this theoretical example, a serial dilution is performed with unstimulated and stimulated extracts. The relative change in the linear range of the immunoblot is 99 (blue) /33 (red) ~ threefold, whereas the relative change at fivefold higher loading is only 1.4 fold (36%).
The analysis here also illustrated that quantifying phosphorylation-specific immunoblots as a “phosphorylated-to-total ratio” is fraught with both numerical complications (Fig. 6) and potential experimental artifacts (Fig. 7). The ratio is further prone to be misinterpreted as a phosphorylation stoichiometry, which cannot be calculated in experiments that use different antibodies to detect the phosphorylated and total protein because of differences in antibody affinity. Calculating phosphorylation stoichiometry by immunoblotting requires electrophoretic conditions that separate the phosphorylated and total forms by mobility (32). Modification stoichiometry can be critical under certain conditions (33), but there are also examples where increases in total protein have impacted signal flow (34). Considering these caveats, my conclusion is that phosphorylated to total ratios based on stripped and reprobed membranes should be avoided with immunoblot data.
A major challenge in evaluating the immunoblot data of others is that publications and manuscripts will often omit details on acquisition that are considered “routine”. Those details matter, because the evaluation criteria of an immunoblot detected by film is different than one detected by fluorescence. When such information is missing, readers and reviewers can distinguish a film exposure by the hazy gray of the background and the blurred borders of bands resulting from the flatbed optical scan of a film at arbitrarily high resolution (44). Digitally acquired immunoblots will often have a whiter background with crisper bands that may appear pixelated due to binning on a CCD camera or the step size of a fluorescence scanner. Although some may find them less aesthetically appealing, digitally acquired images provide the more-accurate representation of band intensity as it relates to sample abundance.
The message of this Research Resource is not that chemiluminescence cannot be quantitative or that film exposures are always inappropriate. Rather, I want to convey that with numbers comes great responsibility. There are straightforward ways to diagnose immunoblot accuracy (Figs. 4, 5, and 8) and precision (Fig. 6). We should all be encouraged to complete these diagnostics on our own targets and immunoblot setups before diving in to generate “real data”. The stakes are simply too high to do otherwise.
Materials and Methods
Cell lines, stimulation, and lysis
HT-29 cells were obtained from the American Type Culture Collection and maintained as recommended. The 5E clone of MCF10A cells was isolated and maintained as previously described (8). AC16 cells (15) were purchased from Dr. Mercy Davidson (Columbia University) and maintained in DMEM/F-12 medium (Life Technologies) plus 12.5% tetracycline-free fetal bovine serum (Clontech) and penicillin-streptomycin (Gibco).
Cells were stimulated with the indicated concentrations of anti-APO-1-3 crosslinking antibody (Axxora), TGFβ (Peprotech), or EGF (Peprotech) for the indicated times, washed with ice-cold PBS, and then lysed in RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 5 mM EDTA supplemented with 10 µg/ml aprotinin, 10 µg/ml leupeptin, 1 µg/ml pepstatin, 1 mM PMSF, 1 µg/ml microcystin-LR, and 200 µM sodium orthovanadate), NP-40 buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% NP-40 substitute, 5 mM EDTA supplemented with 10 µg/ml aprotinin, 10 µg/ml leupeptin, 1 µg/ml pepstatin, 1 mM PMSF, 1 µg/ml microcystin-LR, and 200 µM sodium orthovanadate), or dithiothreitol-containing Laemmli sample buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 100 mM dithiothreitol, and 0.01% bromophenol blue). Laemmli sample buffer lyses were performed at room temperature to avoid precipitating the SDS, and viscosity of these lysates was reduced by passing them vigorously through a 25-gauge needle 5–10 times. For RIPA and NP-40 lysates, protein concentrations were determined with the bicinchoninic acid assay (Thermo Fisher Scientific).
For solubilization comparisons (Fig. 1), HT-29 cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors as described above. After incubation on ice and centrifugation, the RIPA-insoluble pellet was boiled for 5 minutes in an equivalent volume of dithiothreitol-containing Laemmli sample buffer.
Immunoblotting: polyacrylamide gel electrophoresis
Immunoblotting was performed as described (12, 45, 46), but the details of the implementation will be elaborated upon here. Samples were prepared in dithiothreitol-containing Laemmli sample buffer to a total volume of 20 µl or 40 µl. 8, 10, 12, or 15% polyacrylamide gels of 1.5-mm thickness were cast according to (1), and samples were electrophoresed in Tris-glycine running buffer (25 mM Tris base, 250 mM glycine, 0.1% SDS) at 130V until the dye front reached the end of the gel.
Immunoblotting: electrophoretic transfer
Proteins from the polyacrylamide gel were transferred to a polyvinyldifluoride membrane (Millipore; Immobilon-FL, 0.45-µm thickness) in a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad) under modified Towbin conditions (4) (25 mM Tris, 192 mM glycine, 0.0375% SDS, 10% methanol unless otherwise indicated). Transfers were electrophoresed at 100V for 1 hour under ambient conditions with an ice block in the transfer tank and the transfer tank surrounded by ice.
Immunoblotting: membrane blocking
After transfer, the molecular weight markers on the membrane were overwritten with a lead pencil (to provide 700-channel fluorescence), and the membrane was blocked with 0.5× blocking buffer: Odyssey blocking buffer [LI-COR; #927-40000] diluted in an equal volume of PBS. Although not observed for the immunoblots here, some phosphorylation site-specific antibodies can be competed away from their target epitopes with PBS buffers. In this circumstance, the 0.5× blocking buffer should be prepared with Odyssey blocking buffer [LI-COR; # 927-50000] and diluted in an equal volume of TBS throughout the procedure. TBS buffers were also used for the fluorescence-chemiluminescence comparison (Fig. 5). All blocking steps used a surface-to-volume ratio of 5 ml 0.5× blocking solution per 2 ¾-inch × 3 ¼-inch membrane from a 15-well minigel. The membrane and 0.5× blocking solution were sealed in a plastic bag and incubated on a rotating platform for 1 hour at room temperature.
Immunoblotting: antibody probing
After blocking, membranes were incubated with 0.5× blocking solution + 0.1% Tween-20 containing primary antibodies recognizing the proteins or epitopes listed in Table 1. All primary antibody steps used a surface-to-volume ratio of 5 ml primary antibody solution per 2 ¾-inch × 3 ¼-inch membrane from a 15-well minigel. The membrane and primary antibody solution were sealed in a plastic bag and incubated on a rotating platform overnight at 4°C.
Antibody pairs raised in different species were routinely multiplexed when using two-color fluorescence detection. In addition, primary antibodies with negligible off-target bands could be multiplexed in the same detection channel if the molecular weights of their protein targets could be clearly resolved from one another. This single-color multiplexing enabled concurrent detection of multiple loading controls. Common primary single-channel combinations included antibodies recognizing tubulin (50 kD) + GAPDH (36 kD), Hsp90 (90 kD) + p38 (38 kD), vinculin (120 kD) + actin (42 kD), and vinculin (120 kD) + tubulin (50 kD) + GAPDH (36 kD).
Immunoblotting: fluorescence detection
Membranes were removed from primary antibody solution and washed on a rocking platform for 4 × 5 minutes in ~25 ml of PBS + 0.1% Tween-20 (PBS-T). After washing, membranes were incubated with 0.5× blocking solution + 0.1% Tween-20 + 0.01% SDS containing one or more of the following secondary antibodies: IRDye800CW-conjugated goat anti-rabbit (LI-COR; #926-32211, 1:20,000), IRDye800CW-conjugated goat anti-mouse (LI-COR; #926-32210, 1:20,000), IRDye680-conjugated goat anti-rabbit (LI-COR; #926-32221, 1:20,000), IRDye680LT-conjugated goat anti-mouse (LI-COR; #926-68020, 1:20,000), IRDye680LT-conjugated donkey anti-chicken (LI-COR; #926-68028, 1:20,000). All secondary antibody steps used a surface-to-volume ratio of 5 ml primary antibody solution per 2 ¾-inch × 3 ¼-inch membrane from a 15-well minigel. The membrane and secondary antibody solution were sealed in a plastic bag and incubated on a rotating platform for 1 hour at room temperature.
Membranes were removed from secondary antibody solution and washed on a rocking platform for 4 × 5 minutes in ~25 ml of PBS-T. To remove residual Tween-20, which is highly autofluorescent in the 700 fluorescence channel, the membrane was washed for 5 minutes in ~25 ml of PBS before scanning. Fluorescence images were obtained on an Odyssey infrared scanner (LI-COR) at 169 µm resolution and 0 mm focus offset. Fluorescence channel intensities ranged from 5.0–8.5 depending on the immunoblot.
Immunoblotting: chemiluminescence detection
Membranes were removed from primary antibody solution and washed on a rocking platform for 4 × 5 minutes in ~25 ml of TBS + 0.1% Tween-20 (TBS-T). After washing, membranes were incubated with 0.5× blocking solution + 0.1% Tween-20 containing horseradish peroxidase-conjugated goat anti-rabbit (Jackson ImmunoResearch; #111-035-144, 1:10,000) or anti-mouse (Jackson ImmunoResearch; #115-035-146, 1:10,000). All secondary antibody steps used a surface-to-volume ratio of 5 ml primary antibody solution per 2 ¾-inch × 3 ¼-inch membrane from a 15-well minigel. The membrane and secondary antibody solution were sealed in a plastic bag and incubated on a rotating platform for 1 hour at room temperature.
Membranes were removed from secondary antibody solution and washed on a rocking platform for 4 × 5 minutes in ~25 ml of TBS-T. To enable a fair comparison with fluorescence detection, the membrane was washed for 5 minutes in ~25 ml of TBS before exposing. Membranes were covered with an enhanced chemiluminescence solution comprised of 1.25 mM luminol, 2 mM 4-iodophenylboronic acid, and 0.0162% H2O2 (16). Alternatively, membranes were covered with SuperSignal West Femto reagent according the manufacturer’s instructions (Thermo Fisher Scientific). Chemiluminescent exposures were captured on a ChemiDoc MP gel imager (Bio-Rad) with “Chemi Hi Resolution” settings (2×2 camera binning). Exposure times were set manually to fill the bit depth of the CCD camera without saturating any binned pixels.
Immunoblotting: stripping and reprobing
For the low pH glycine strip, membranes were incubated with low pH glycine buffer solution (1.5% glycine [pH 2.2], 0.1% SDS, 1% Tween) for 2 × 10 minutes at room temperature on a rocking platform. The stripped membranes were washed 2 × 10 minutes in ~25 ml of PBS before blocking and immunodetection as described above. For the guanidinium strip, membranes were incubated with 6 M guanidine-HCl for 10 minutes at room temperature on a rocking platform, followed by a 5 minutes wash with PBS before blocking and immunodetection as described above. For the SDS plus β-mercaptoethanol strip, membranes were incubated with SDS plus β-mercaptoethanol solution (62.5 mM Tris [pH 6.8], 2% SDS, 100 mM β-mercaptoethanol) and incubated in a dry-air oven at 50°C for 30 minutes with occasional agitation by hand. Stripped membranes were washed 3 × 5 minutes in ~25 ml of PBS before blocking and immunodetection as described above.
Total-protein staining after electrophoretic transfer
PVDF membranes were stained for total protein with Memcode Reversible Protein Stain (Thermo Scientific) according to the manufacturer’s recommendations. Erasure of the Memcode stain was performed with the Eraser/Methanol solution for 20 minutes. For total-protein staining with Ponceau S, membranes were incubated with 0.1% (w/v) Ponceau S in 5% acetic acid for 5 minutes and washed 2 × 5 minutes in 10% acetic acid, followed by washes of 5 minutes in 100% methanol and 5 minutes in a 70/30/4 volume ratio of methanol, acetic acid, and PEG-400. For erasing, Ponceau S-stained membranes were treated with 0.1 N NaOH for 30 seconds and washed with running deionized water for two minutes.
For both total-protein stains, digital images were captured on a ChemiDoc MP gel imager (Bio-Rad) with “Colorimetric” settings (2×2 camera binning). Fluorescence images were obtained on an Odyssey infrared scanner (LI-COR) at 169 µm resolution and 0 mm focus offset, with a 700-channel intensity of 5.0 and an 800-channel intensity of 8.0.
Coomassie staining and digital image acquisition
Polyacrylamide gels were stained with 0.1% w/v Coomassie blue R250 in 40% methanol and 10% glacial acetic acid on a rocking platform for 1 hour at room temperature and then destained for several hours with 30% methanol + 10% glacial acetic acid until the background was acceptable. The stained gel was scanned on an Odyssey infrared scanner (LI-COR) at 169 µm resolution and 0.5 mm focus offset in the 700 fluorescence channel.
Image densitometry
Raw 16-bit TIFs were opened in ImageJ (5) and rotated to align immunoblot bands horizontally in the window. The rectangle tool was then used to select lanes containing the band of interest. The width of the lane rectangle was drawn as wide as possible without causing overlap with bands from adjacent lanes. The height of the lane rectangle was drawn long enough to get a sample of the local background surrounding the band of interest. Lane profiles were plotted with the gel analysis plugin, and background was subtracted by connecting the background intensity profiles to the left (top) and the right (bottom) of the band of interest by using the line tool. Last, the magic-wand tool was used to calculate the integrated area within the band profile of interest and obtain the final raw densitometry value.
Recombinant protein purification
Recombinant GST-ERK2 and GST-p38 were prepared by glutathione affinity chromatography in RIPL cells (Stratagene) as described (22).
Statistical analysis
Serial dilutions were fit to linear or hyperbolic models by least-squares regression in Igor Pro (WaveMetrics). The χ2 statistic from each model fit was used together with the number of data points and the number of fitted parameters (one for the linear model, two for the hypergeometric model) to calculate an F statistic that compares goodness-of-fit between models. To correct for multiple-hypothesis testing, false-discovery rates were calculated according to Benjamini and Hochberg (47).
Supplementary Material
Acknowledgments
I thank Dan Gioeli, Mike Weber, and David Brautigan for comments on this manuscript, Zeinab Chitforoushzadeh and Sameer Bajikar for lysate preparations, Millie Shah for recombinant proteins, Timothy Allen and Christian Smolko for Memcode Reversible Protein Stain, Suzanne Gaudet for immunoblotting anecdotes, Mercy Davidson for providing access to AC16 cells, and Cheryl Borgman for copy-editing the manuscript.
Funding: This work was supported by the National Institutes of Health (#1-R21-AI105970), the American Cancer Society (#120668-RSG-11-047-01-DMC), the Pew Charitable Trusts (#2008-000410-006), and the David and Lucile Packard Foundation (#2009-34710).
Footnotes
Supplementary Materials
Fig. S1. Challenges with using total-protein stains for normalization of quantitative immunoblots.
File S1. Raw 16-bit images and densitometry calculations from Fig. 4.
File S2. Raw 16-bit images and densitometry calculations from Fig. 5.
File S3. Raw 16-bit images and densitometry calculations from Fig. 6.
File S4. Raw 16-bit images and densitometry calculations from Fig. 8.
Author contributions: K.A.J. conceived of the work, performed all immunoblots and quantitative analyses, and wrote the paper.
Competing interests: The author declares no competing financial interests.
References and Notes
- 1.Green MR, Sambrook J. Molecular cloning: a laboratory manual. ed. 4th. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2012. p. 1890. [Google Scholar]
- 2.Taylor SC, Posch A. The design of a quantitative western blot experiment. Biomed Res Int. 2014;2014:361590. doi: 10.1155/2014/361590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor SC, Berkelman T, Yadav G, Hammond M. A defined methodology for reliable quantification of Western blot data. Mol Biotechnol. 2013;55:217–226. doi: 10.1007/s12033-013-9672-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngoka LC. Sample prep for proteomics of breast cancer: proteomics and gene ontology reveal dramatic differences in protein solubilization preferences of radioimmunoprecipitation assay and urea lysis buffers. Proteome Sci. 2008;6:30. doi: 10.1186/1477-5956-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 8.Janes KA, Wang CC, Holmberg KJ, Cabral K, Brugge JS. Identifying single-cell molecular programs by stochastic profiling. Nat Methods. 2010;7:311–317. doi: 10.1038/nmeth.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 10.Gaudet S, Janes KA, Albeck JG, Pace EA, Lauffenburger DA, Sorger PK. A Compendium of Signals and Responses Triggered by Prodeath and Prosurvival Cytokines. Mol Cell Proteomics. 2005;4:1569–1590. doi: 10.1074/mcp.M500158-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Janes KA, Gaudet S, Albeck JG, Nielsen UB, Lauffenburger DA, Sorger PK. The Response of Human Epithelial Cells to TNF Involves an Inducible Autocrine Cascade. Cell. 2006;124:1225–1239. doi: 10.1016/j.cell.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 12.Wang CC, Bajikar SS, Jamal L, Atkins KA, Janes KA. A time- and matrix-dependent TGFBR3-JUND-KRT5 regulatory circuit in single breast epithelial cells and basal-like premalignancies. Nat Cell Biol. 2014;16:345–356. doi: 10.1038/ncb2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacKintosh C, Beattie KA, Klumpp S, Cohen P, Codd GA. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 1990;264:187–192. doi: 10.1016/0014-5793(90)80245-e. [DOI] [PubMed] [Google Scholar]
- 14.Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 15.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Haan C, Behrmann I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J Immunol Methods. 2007;318:11–19. doi: 10.1016/j.jim.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 17.Marcus E. Credibility and reproducibility. Cell. 2014;159:965–966. doi: 10.1016/j.cell.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 18.Olson BJ, Markwell J. Assays for determination of protein concentration. Chapter 3. Curr Protoc Protein Sci. 2007;(Unit 3):4. doi: 10.1002/0471140864.ps0304s48. [DOI] [PubMed] [Google Scholar]
- 19.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 20.Jensen KJ, Garmaroudi FS, Zhang J, Lin J, Boroomand S, Zhang M, Luo Z, Yang D, Luo H, McManus BM, Janes KA. An ERK-p38 subnetwork coordinates host cell apoptosis and necrosis during coxsackievirus B3 infection. Cell Host Microbe. 2013;13:67–76. doi: 10.1016/j.chom.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang BH, Jensen KJ, Hatch JA, Janes KA. Simultaneous profiling of 194 distinct receptor transcripts in human cells. Sci Signal. 2013;6:rs13. doi: 10.1126/scisignal.2003624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bose AK, Janes KA. A high-throughput assay for phosphoprotein-specific phosphatase activity in cellular extracts. Mol Cell Proteomics. 2013;12:797–806. doi: 10.1074/mcp.O112.024059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brody JP, Williams BA, Wold BJ, Quake SR. Significance and statistical errors in the analysis of DNA microarray data. Proc Natl Acad Sci U S A. 2002;99:12975–12978. doi: 10.1073/pnas.162468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tricarico C, Pinzani P, Bianchi S, Paglierani M, Distante V, Pazzagli M, Bustin SA, Orlando C. Quantitative real-time reverse transcription polymerase chain reaction: normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal Biochem. 2002;309:293–300. doi: 10.1016/s0003-2697(02)00311-1. [DOI] [PubMed] [Google Scholar]
- 25.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, Sapkota G, Pan D, Massague J. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shapiro GI. Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin Cancer Res. 2004;10:4270s–4275s. doi: 10.1158/1078-0432.CCR-040020. [DOI] [PubMed] [Google Scholar]
- 28.Salinovich O, Montelaro RC. Reversible staining and peptide mapping of proteins transferred to nitrocellulose after separation by sodium dodecylsulfate-polyacrylamide gel electrophoresis. Anal Biochem. 1986;156:341–347. doi: 10.1016/0003-2697(86)90263-0. [DOI] [PubMed] [Google Scholar]
- 29.Taborsky G. Phosphoproteins. Adv Protein Chem. 1974;28:1–210. doi: 10.1016/s0065-3233(08)60230-2. [DOI] [PubMed] [Google Scholar]
- 30.Gallagher S, Winston SE, Fuller SA, Hurrell JG. Immunoblotting and immunodetection. Chapter 8. Curr Protoc Immunol. 2001;(Unit 8):10. doi: 10.1002/0471142727.mb1008s83. [DOI] [PubMed] [Google Scholar]
- 31.Kaufmann SH, Ewing CM, Shaper JH. The erasable Western blot. Anal Biochem. 1987;161:89–95. doi: 10.1016/0003-2697(87)90656-7. [DOI] [PubMed] [Google Scholar]
- 32.English JG, Shellhammer JP, Malahe M, McCarter PC, Elston TC, Dohlman HG. MAPK feedback encodes a switch and timer for tunable stress adaptation in yeast. Sci Signal. 2015;8:ra5. doi: 10.1126/scisignal.2005774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stemmann O, Zou H, Gerber SA, Gygi SP, Kirschner MW. Dual inhibition of sister chromatid separation at metaphase. Cell. 2001;107:715–726. doi: 10.1016/s0092-8674(01)00603-1. [DOI] [PubMed] [Google Scholar]
- 34.Hsu YH, Yao J, Chan LC, Wu TJ, Hsu JL, Fang YF, Wei Y, Wu Y, Huang WC, Liu CL, Chang YC, Wang MY, Li CW, Shen J, Chen MK, Sahin AA, Sood A, Mills GB, Yu D, Hortobagyi GN, Hung MC. Definition of PKC-alpha, CDK6, and MET as Therapeutic Targets in Triple-Negative Breast Cancer. Cancer Res. 2014;74:4822–4835. doi: 10.1158/0008-5472.CAN-14-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sturm OE, Orton R, Grindlay J, Birtwistle M, Vyshemirsky V, Gilbert D, Calder M, Pitt A, Kholodenko B, Kolch W. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci Signal. 2010;3:ra90. doi: 10.1126/scisignal.2001212. [DOI] [PubMed] [Google Scholar]
- 36.Schoeberl B, Eichler-Jonsson C, Gilles ED, Muller G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat Biotechnol. 2002;20:370–375. doi: 10.1038/nbt0402-370. [DOI] [PubMed] [Google Scholar]
- 37.Luo S, Wehr NB, Levine RL. Quantitation of protein on gels and blots by infrared fluorescence of Coomassie blue and Fast Green. Anal Biochem. 2006;350:233–238. doi: 10.1016/j.ab.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 38.Kiel C, Verschueren E, Yang JS, Serrano L. Integration of protein abundance and structure data reveals competition in the ErbB signaling network. Sci Signal. 2013;6:ra109. doi: 10.1126/scisignal.2004560. [DOI] [PubMed] [Google Scholar]
- 39.Janes KA, Lauffenburger DA. A biological approach to computational models of proteomic networks. Curr Opin Chem Biol. 2006;10:73–80. doi: 10.1016/j.cbpa.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Janes KA, Lauffenburger DA. Models of signalling networks - what cell biologists can gain from them and give to them. J Cell Sci. 2013;126:1913–1921. doi: 10.1242/jcs.112045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janes KA, Albeck JG, Peng LX, Sorger PK, Lauffenburger DA, Yaffe MB. A high-throughput quantitative multiplex kinase assay for monitoring information flow in signaling networks: application to sepsis-apoptosis. Mol Cell Proteomics. 2003;2:463–473. doi: 10.1074/mcp.M300045-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Shults MD, Janes KA, Lauffenburger DA, Imperiali B. A multiplexed homogeneous fluorescence-based assay for protein kinase activity in cell lysates. Nat Methods. 2005;2:277–284. doi: 10.1038/nmeth747. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Janes KA. Stochastic profiling of transcriptional regulatory heterogeneities in tissues, tumors and cultured cells. Nat Protoc. 2013;8:282–301. doi: 10.1038/nprot.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gassmann M, Grenacher B, Rohde B, Vogel J. Quantifying Western blots: pitfalls of densitometry. Electrophoresis. 2009;30:1845–1855. doi: 10.1002/elps.200800720. [DOI] [PubMed] [Google Scholar]
- 45.Bajikar SS, Fuchs C, Roller A, Theis FJ, Janes KA. Parameterizing cell-to-cell regulatory heterogeneities via stochastic transcriptional profiles. Proc Natl Acad Sci U S A. 2014;111:E626–E635. doi: 10.1073/pnas.1311647111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Brugge JS, Janes KA. Intersection of FOXO- and RUNX1-mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proc Natl Acad Sci U S A. 2011;108:E803–E812. doi: 10.1073/pnas.1103423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- 48.Trauth BC, Klas C, Peters AM, Matzku S, Moller P, Falk W, Debatin KM, Krammer PH. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245:301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.