

Abstract
Background
Genetic alterations affecting the MAPK/ERK pathway are common in lung adenocarcinoma (LAD). Early steps of the signaling pathway are most often affected with EGFR, KRAS and BRAF mutations encompassing over 70% of all alterations. Somatic mutations in MEK1, located downstream of BRAF, are rare and remain poorly defined as a distinct molecular subset.
Methods
Tumors harboring MEK1 mutations were identified through targeted screening of a large LAD cohort concurrently interrogated for recurrent mutations in MEK1, EGFR, KRAS, BRAF, ERBB2/HER2, NRAS, PIK3CA, and AKT. Additional cases were identified through a search of publically available cancer genomic datasets. Mutations were correlated with patient characteristics and treatment outcomes. Overall survival was compared to stage-matched patients with KRAS and EGFR mutant lung adenocarcinomas.
Results
We identified 36 MEK1 mutated cases among 6024 LAD (0.6%, 95% CI 0.42 to 0.85). The majority of patients were smokers (97%, n=35/36). There was no association with age, sex, race, or stage. The most common mutations were K57N (64%, 23/36) followed by Q56P (19%, 7/36), all mutually exclusive with other driver mutations in the targeted panel. Transversions G:C > T:A were predominant (89%, 31/35), in keeping with smoking-associated DNA damage. Additional less common somatic mutations were identified in the kinase domain, all of which are predicted to converge into a single interaction area based on in-silico 3D modeling.
Conclusion
MEK1 mutations define a distinct subset of lung cancers (∼1%) with potential sensitivity to MEK inhibitors. Mutations are predominantly transversions, in keeping with a strong association with smoking.
Keywords: MEK1, MAP2K1, NSCLC, Lung Cancer
Introduction
The MAPK/ERK pathway (RAS-RAF-MEK-ERK pathway) is one of the most extensively studied signal transduction pathways and regulates fundamental cell activities including proliferation, transcriptional regulation, differentiation and survival. Consistent with this critical role, defects in this pathway are implicated in the pathogenesis of several human malignancies, including lung cancer. Genetic alterations affecting the MAPK/ERK pathway are common in non-small cell lung carcinoma (NSCLC), particularly those affecting the upstream components of the signaling cascade. Mutations in EGFR and KRAS (1, 2), for instance, constitute over 70% of all reported driver mutations in this cancer, while those involving BRAF are less common but well described (3). Alterations in MAP2K1 (MEK1), a primary downstream effector of RAF kinases, are uncommon and consequently tumors harboring these mutations have remained poorly defined as a distinct molecular subset in lung cancer (4).
MEK1, also known as the mitogen activated protein kinase kinase 1 (MAP2K1), is a dual specificity kinase of the MAP2K/STE7 kinase family. This kinase family also includes five additional members but only MEK1 and MEK2 are known to participate in the MAPK/ERK cascade with pivotal roles in signal integration (5). These two kinases share a high degree of sequence homology and while they seem to perform similar functions, they remain distinct in the ways they contribute to regulated ERK activity (6). The functional activation of the MAPK/ERK pathway begins with the extracellular binding of ligand to a receptor tyrosine kinase, such as EGFR, and is followed by sequential activation of RAS, RAF and MEK. Once activated by RAF, MEK1/2 catalyzes the concomitant phosphorylation of threonine and tyrosine residues on ERK (MAPK) within a specific Thr-Glu-Tyr sequence at codons 202 to 204. This locks the kinase domain of MAPK into a catalytically competent conformation, ultimately leading to nuclear signals that regulate cellular growth and proliferation. To date, ERK (MAPK) remains the only known MEK catalytic substrate. This tight selectivity, along with the unique dual specificity kinase function, confers a strategic role in the integration of signals through the pathway to the MEK1/2 proteins.
Initial insight into the critical role of MEK in cell proliferation and tumorigenesis has come from engineered mutations used in in-vitro models. Deletions and mutations in the activation segment of MEK1 have been shown to constitutively activate the protein. Activated MEK1 mutants enhance cell proliferation, differentiation, and promote transformation (7-9). Conversely, dominant negative MEK1 mutants can prevent cellular proliferation in fibroblasts and transformed cell lines. Naturally occurring mutations in MEK1 and MEK2 have been described in association with cardio-facio-cutaneous (CFC) syndrome as a germline event (10). Additionally, somatic mutations have also been identified in small subsets of lung cancer (MEK1 only) (4, 11) and colorectal cancer (MEK1 and MEK2)(12, 13). More recently, MEK1 mutations have also been reported in melanomas in the setting of resistance to BRAF targeted therapy (14). Since MEK is a convergence point for multiple upstream kinases and intracellular signaling intermediaries, it constitutes a particularly attractive target for drug development and specific inhibitors are already available and are moving through various stages of clinical trials. Recently, trametinib, a selective inhibitor of MEK1 and MEK2, was approved by the FDA for use in BRAF mutant melanoma patients.
Despite the growing awareness of MEK1 mutations and their potential as drug targets, to date, no comprehensive report has focused on the characteristics of lung carcinoma patients harboring these mutations. In the current study, we report the largest collection of lung cancer tumors screened for recurrent mutations in this gene. We describe the clinical and pathologic characteristics of these tumors and make comparisons with other molecularly defined subsets in lung carcinoma.
Materials and Methods
Patient data
Clinical cases of lung adenocarcinoma (LAD) received for routine molecular profiling at two institutions (Memorial Sloan Kettering Cancer Center (MSKCC) and Vanderbilt-Ingram Cancer Center (VICC)) between January 2009 and June 2014 were identified for review. Clinical characteristics including smoking history, clinical stage, age, gender, race (as reported by patient) and treatment were recorded under IRB-approved waivers for specific subsets.
Tissue procurement and mutational profiling
Tumor specimens were obtained as standard of care for clinical management or with patients' consent under Institutional Review Board–approved protocols. Genomic DNA was extracted from tumor samples using standard procedures. Clinical testing for the detection of mutations in EGFR (exon 19 deletions and L858R) and KRAS (exon 2) was carried out by a combination of methods including fragment analysis, Sanger sequencing, mass spectrometry genotyping (Sequenom) or SNapShot assays as previously described (15-17). Extended mutation analysis for other recurrent point mutations in EGFR, KRAS, BRAF, ERBB2/HER2, NRAS, AKT, PIK3CA and MEK1 (MAP2K1) was performed by mass spectrometry or SNapShot assays. Specific MEK1 mutations interrogated in the extended panels included Q56P, K57N and D67N. To further assess other variants, mutation data for MEK1 was also extracted from publicly available sources, including TCGA, cBio Portal and the COSMIC data base.
In Vitro Studies
Functional assays
Human embryonic kidney 293H cells were cultured in DME-HG, 10% fetal calf serum, 2 mM glutamine, and 50 units/mL each of penicillin and streptomycin. Specific MAP2K1 mutations were generated from the MEK1-GFP plasmid (Addgene, 14746) using QuickChange Site-Directed Mutagenesis Kit (Stratagene). 293H cells were seeded for 70-90% confluency at the time of transfection, then transiently transfected with 10μg of wild-type or mutant MAP2K1 DNA using the Lipofectamine® 2000 Transfection Reagent. At 24 hours, cells were lysed in 1% NP-40 lysis buffer and processed for immunoblotting as previously described (18). Rabbit polyclonal antibodies recognizing phosphorylated ERK1/2 (Thr202/Tyr204), ERK1/2, and phosphorylated p90RSK (Thr359/Ser363) were obtained from Cell Signaling. Rabbit monoclonal antibodies recognizing RSK1/2/3 and GFP were obtained from Cell Signaling. After incubation with horseradish peroxidase-conjugated secondary antibodies, proteins were detected by chemiluminescence (SuperSignal® West Dura Chemiluminescent Substrate, Thermo Scientific) and visualized using a Fuji LAS-4000 (GE Life Sciences).
Drug treatments
Transfected 293H cells were grown under selection with Geneticin (Gibco). Cells were plated overnight, then treated with dimethyl sulfoxide (control) or 1 μM of AZD6244 for 0.5, 1, 2 or 6 hours.
Plasmid construction
To generate the MSCV-puro-MEK1 plasmid, MEK1 cDNA was excised from the pEGFP-N1-MEK1-GFP plasmid using XhoI/AgeI digestion, then ligated into the XhoI/AgeI-digested MSCV-puro vector (Gateway). MAP2K1 mutations were generated (specified above) from the MSCV-puro-MEK1 plasmid. All plasmids were verified by sequencing.
Viral production and infection of target cells
Retrovirus was generated by cotransfection of MSCV-puro vector, MSCV-puro-MEK1, or mutant MSCV-puro-MEK1 with psi-eco mouse packaging vector in 293T cells. NIH-3T3 cells were infected with virus-containing medium and 40 μg of polybrene (Santa Cruz Biotechnology) for 48 hours. Cells were then cultured in fresh medium and selected with 2 μg/mL of puromycin.
Soft agar colony formation assay
Log phase NIH-3T3, NIH-3T3-MSCV-puro, NIH-3T3-MSCV-puro-Mek1, and mutant NIH-3T3-MSCV-puro-Mek1 cells (1 × 104) were mixed with agar (0.33%) and plated over a bottom layer of 0.5% agar on 6-well plates.Treated cells were mixed with agar and 1 μM AZD6244, then plated. Cells were incubated at 37°C for 4 weeks. Wells were stained with crystal violet (Sigma-Aldrich) for 1 hour at 37°C. Colonies greater than 150 microns were imaged and counted using GelCount™ (Oxford Optronix). Two independent experiments were performed with each condition in duplicate wells per experiment.
Statistical analysis
Fisher's exact tests were applied to test associations among MEK1 mutations, smoking history, and race. Overall survival (OS) and progression-fee survival (PFS) were determined using the Kaplan-Meier method. For all MEK-mutated patients (stage I-IV), OS was calculated from the time of pathologic diagnosis of disease using Kaplan-Meier estimates. Comparisons of overall survival between MEK-mutant lung cancers and KRAS-mutant, EGFR-mutant, BRAF-mutant, ALK-rearranged, ROS1-rearranged, and RET-rearranged lung cancers were calculated from the time of diagnosis of metastatic disease.
Results
Mutation screening
Among 5330 LAD patients tested at two institutions (MSKCC and VICC), we identified 26 NSCLC patients harboring MEK1 mutations (MSKCC: 18, VICC: 8). The distribution of driver mutations in this cohort were as follows: EGFR 20%, ERRB2 1%, KRAS 32%, BRAF 2%, PIK3CA 3%, MEK1 0.6%, and NRAS 0.25%. 211 cases of squamous cell carcinoma were also tested at MSKCC with no MEK1 mutations identified.
In order to produce a more comprehensive view of lung adenocarcinoma-associated MEK1 alterations, we combined the above data sets with next generation sequencing data publically available through the cBio Portal (TCGA and Broad) (19, 20). Through this search we identified 9 additional MEK1 mutated cases among 421 LAD samples. One additional (non-overlapping) MEK1 positive case was identified in the COSMIC database (1/273). In total, this combined dataset included 36 MEK1 mutated cases among 6024 LAD (0.6%)
Spectrum of MEK1 mutations
Amongst the 26 cases of MEK1 mutated lung cancer identified at the two institutions, K57N was the most common mutation detected, encompassing 77% (20/26) of all variants while all remaining cases harbored the Q56P (6/26).No D67N mutations were identified. Both the K57N and the Q56P constitute transversions, G>T and A>C, respectively. In all cases, MEK1 mutations were mutually exclusive with all other alterations tested for in the panel including known activating mutations in EGFR, ERBB2, KRAS, NRAS and BRAF.
Amongst the 9 MEK1 mutated cases identified through publically available data sets, there were three novel mutations: M146I (1/9), G301X (1/9), S331R (1/9). Since the G301X mutation would be presumed to be inactivating and since the M146I and S331R mutations were reported in patients with concurrent KRAS mutations (G13C and G12V, respectively), these three MEK1 alterations were deemed as unlikely to be driver mutations. The six other cases harbored previously reported mutations in lung or other cancers including F53L (1/9), Q56P (1/9), K57N (2/9), E102_l103del (1/9), and C121S (1/9)(14, 19-21). No concurrent recurrent mutations were identified in these cases in EGFR, KRAS, BRAF, ERBB2/HER2, NRAS, AKT or PIK3CA genes. Finally, the single case indentified in the COSMIC database harbored a K57N mutation.
Among all 36 patients with MEK1 mutations, 92% (33/36) were G:C > T:A transversions. Most cases (86%, 31/36) were in exon 2.
The distribution of mutations is depicted on Figure 1. A detailed summary of positive cases is presented in Table 1.
Figure 1.

Top. Linear model of the MEK1 gene. Exons are represented by the numbered boxes. Red boxes indicate regions coding for the core kinase domain (residues 55–369) Bottom. Mapping of all mutations identified in this study and publically available data (cBio portal and Cosmic database). Each circle represents a single patient – Lung adenocarcinoma (green circle). Reported mutations in melanoma (light brown circle), colon carcinoma (brown circle), bladder (yellow), gastric carcinoma (blue), breast (light blue), head and neck (purple) are also mapped for comparison. D-Domain is the ERK docking site, Pro rich insert 262-307
Table 1. MAP2K1 (MEK1) mutated cases.
| AA Mutation | CDS | Frequency | Type of change |
|---|---|---|---|
| p.F53L | c.157T>C | 1 (3%) | Transition |
| p.Q56P | c.167A>C | 7 (19%) | Transversion |
| p.K57N | c.171G>T | 23 (64%) | Transversion |
| E102_I103del | c. 304_306delGAGATC | 1 (3%) | - |
| p.C121S | c.361T>A | 1 (3%) | Transversion |
| p.M146I | c.438G>C | 1 (3%) | Transversion |
| p.G301X | c. 901G>T | 1 (3%) | Transversion |
| p.S331R | c.991A>C | 1 (3%) | Transversion |
Characteristics of Patients with MEK1 mutations
The clinical characteristics of 36 patients with MEK1 mutations are summarized in Table 2a. The majority of patients were Caucasian (92%, 33/36), former or current smokers (97%, 35/36) with no gender predilection. Median pack-year history of smoking was 48. Patients most commonly presented with stage IV disease (39%, 14/36). The only never smoker in this series harbored a K57N mutation and was also the only patient of Asian ancestry.
Table 2a. Clinical characteristics of patients with MAP2K1 (MEK1) mutations.
| Smoking History | |
| Never | 3% (1/36) |
| Former <10 pack-years | 3% (1/36) |
| Former >10 pack-years | 94% (34/36) |
|
| |
| Pack Years | Median 48 pack-years (range 0-160) |
|
| |
| Age at diagnosis | Median 68 (range 48-89) |
|
| |
| Sex | M 50% (18/36) |
| F 50% (18/36) | |
|
| |
| Stage at Presentation | |
| IA | 19% (7/36) |
| IB | 11% (4/36) |
| IIA | 11% (4/36) |
| IIB | 11% (4/36) |
| IIIA | 3% (1/36) |
| IIIB | 6% (2/36) |
| IV | 39% (14/36) |
|
| |
| Race | |
| Caucasian | 91% (33/36) |
| Asian | 3% (1/36) |
| Unknown | 6% (2/36) |
Comparisons of MEK1 mutants versus EGFR and KRAS mutated subsets showed significant differences in the smoking history of MEK1 and EGFR-mutated cases. Compared to both EGFR and KRAS, MEK1 mutations were more common in males, although this difference only reached statistical significance in comparison to EGFR (Table 2b).
Table 2b. Comparison of clinical characteristics of MAP2K1 (MEK1) mutant patients versus other molecularly defined subsets.
| MAP2K1 (n=36) | KRAS (n=1254)** | EGFR (n=769)** | |
|---|---|---|---|
| Gender (female/male) | 18/18 | 788/466 (p=0.07) | 546/223* (p=0.006) |
| Median age (range) | 68 (48-89) | 68 (37-90) | 67 (30-92) |
| Smoking status (former or current/never smoker) | 35/1 | 1136/118 (p=0.36) | 347/422* (<0.00001) |
KRAS and EGFR groups are based on the MSKCC cohort
NOTE: p values in parentheses based on comparison with MEK1; only significant values are annotated.
Clinical outcomes and survival analysis
Early-Stage Disease (Stage IA-IIIA)
Of the 20 patients with stage IA-IIIA MEK1-mutated lung cancers, data on therapy was available for 15 patients. 14 of 15 patients underwent surgical resection of their lung cancers with curative intent. None of these patients (n=0/14) developed disease recurrence (median recurrence-free survival 17 months, range 1-66 months). At the time of last follow-up, 13 of 14 remained alive and disease-free, and 1 of 14 died of a non-malignant cause. Recurrent disease was only noted in the remaining patient who was not a surgical candidate. This patient was treated with sequential chemotherapy and radiation, developed distant recurrence 3.2 years after completion of therapy, and passed away secondary to cancer.
Advanced/Metastatic Disease (Stage IIIB- IV)
Thirteen patients presented with stage IV disease at diagnosis. The most common site of metastatic disease at diagnosis was bone (73% of patients with available data, n=8/11). Data on systemic therapy were available for 8 of 13 patients. The median number of lines of systemic therapy was 2 (range 1-4). Duration of disease control with systemic therapy was short, with a median PFS of 2.0 months [8 patients, 17 individual lines of therapy (n=5/17 platinum doublet or triplet, n=3/17 non-platinum doublet, n=5/17 single-agent cytotoxic, n=4/17 targeted therapy with erlotinib, nintedanib, erlotinib+MET inhibitor, PI3K/mTOR+MEK inhibitor)]. The patient who received targeted therapy with a combination of a MEK inhibitor and a PI3K/mTOR inhibitor on a phase I clinical trial developed progression of disease at first radiographic evaluation.
Two patients presented with locally advanced (stage IIIB) disease at diagnosis. Data on therapy was available for one of these patients who underwent chemoradiation with carboplatin and paclitaxel. During treatment, he was found to have metastatic disease to the adrenals. The median OS from date of pathologic diagnosis for all patients with stage IIIB/IV disease where survival data was available (n=12) was 1.2 years (15 months). All deaths (n=8/11 with known disease status at the time of last follow-up or death) were cancer-related.
Survival Comparison to Other Oncogene-Driven Lung Cancers
Median OS of all patients with MEK1-mutant lung cancers (stage IA-IV, n=27 with available data on survival) was 48.9 months (4 years). Median OS from the time of diagnosis of metastatic disease (n=11 patients with MEK-mutant lung cancers who developed or were diagnosed with metastatic disease, and with available survival data) was compared to other groups of oncogene-driven lung cancers (n=117 KRAS-mutant, n=102 EGFR-mutant, n=45 ALK-rearranged, n=18 RET-rearranged, n=10 ROS1-rearranged, n=10 BRAF-mutant).
No statistically significant differences in median OS from diagnosis of metastatic disease were found between MEK1-mutant and KRAS-mutant (14.9 vs 13.7 months, HR 0.89, p=0.75), or BRAF-mutant lung cancers (14.9 vs 20.9 months, HR 1.64, p=0.30). Median OS of MEK1-mutant lung cancers was inferior in comparison to EGFR-mutant (14.9 vs 29.7 months, HR 2.16, p=0.04), ALK-rearranged (14.9 vs 35.9 months, HR 2.64, p=0.01), ROS1-rearranged (14.9 vs 76 months, HR 4.00, p=0.02), and RET-rearranged lung cancers (14.9 months vs not reached, HR 3.25, p=0.03) (Figure 2).
Figure 2.

Kaplan–Meier curves for overall survival in patients with MEK1 mutations (time plotted in months). A. Overall survival for MSKCC and VICC patients. B. Overall survival for patients with advanced disease. C. Survival comparison to other oncogene-driven lung cancers -The overall survival of the MEK1-mutant cohort was similar to the KRAS and BRAF-mutant lung and significantly inferior to those patients with EGFR mutations and with rearrangements involving ALK, RET and ROS1
Functional characterization of MEK1 mutants
To determine how the two most common mutations (Q56P and K57N) identified in our cohort affect MAPK signaling, we assayed the ability of the mutants to induce ERK phosphorylation and its downstream effector ribosomal S6 kinase (p90RSK) in 293H cells. These were compared to other less common mutations including F53L, D67N, EI102del, C121S and E203K. Western blot analysis demonstrated increased kinase activity in all mutants, except for the D67N. ERK phosphorylation levels in the D67N mutant did not vary significantly from the wild-type (Figure 3A&B).
Figure 3.
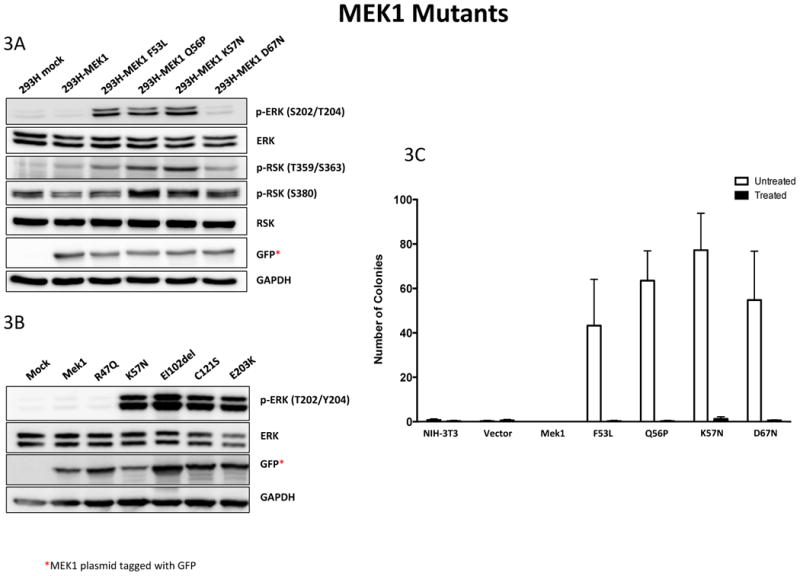
Functional characterization of MEK1 mutants. 293H cells were transfected with a wild-type or mutant GFP-tagged Mek1 plasmid. Cell lysates were collected after 24 hours and analyzed by Western blotting. 3A shows the F53L, Q56P and K57N mutants which display hyperactive downstream MAPK pathway signaling based upon phosphorylation of ERK and its downstream effector ribosomal S6 kinase (RSK). By contrast, the D67N mutant kinase activity is significantly lower than the other hyperactive mutants and not significantly different from the wild-type. 3B shows the EI102del, C121S and E203K mutants and display hyperactive downstream MAPK pathway signaling that was comparable to the K57N.
3C. NIH-3T3, NIH-3T3-vector, NIH-3T3-MEK1 and NIH-3T3-Mek1 mutant cells were cultured for 4 weeks in soft agar to assess anchorage-independent growth. Colonies greater than 150 microns in size formed in all four mutant cell lines but not in control and wild-type Mek1 cells. Treatment of mutant cell lines with 1 μM of Selumetinib (AZD6244) prevented colony formation in all the mutant cell lines.
Anchorage independent growth was also assessed for mutated cell lines harboring F53L, Q56P, K57N and D67N mutations. Colony formation after 4 week culture in soft agar showed colonies of greater than 150 microns in the 4 mutant cell lines tested but not in the control or wild-type MEK1 cells. Treatment with 1 μM Selumetinib inhibited colony formation in all the mutated cell lines (Figure 3C), Further assessment of ERK following treatment with Selumetinib at 0.5, 1, 2 or 6 hours showed significant ERK inhibition in all mutants after 30 minutes of treatment (Supplemental figure 1)
Discussion
Somatic mutations in MEK1 were first described in NSCLC at MSKCC by Marks et al (4) in 2008 and this led to the early inclusion of the known, recurrent MEK1 point mutations in our multiplex genotyping panel for lung cancer. Since then, only rare reports have been published describing 1 or 2 mutation positive patients (11, 22), making it difficult to define the molecular, clinical and pathologic characteristics of patients with these tumors. The current study represents the largest series of MEK1 mutant lung cancer to date. Here we describe the clinicopathologic characteristics associated with 36 MEK1 mutation-positive lung cancer patients identified among 6024 tested (0.6%), and we further explore the molecular pathology of these tumors. We demonstrate that mutations in MEK1 define a unique subset of lung cancer that is strongly associated with smoking and display a clinical behavior that most closely resembles KRAS mutated lung cancer. Although the frequency of MEK1 mutations in LAD is low, LAD is a common disease with at least 100,000 new cases in the US each year. Based on our estimated prevalence of 0.6% of LAD, this would represent over 600 lung cancer patients a year in the USA alone who could potentially benefit from specific targeted therapies directed at this driver oncogene.
Mutations in MEK1 were initially described in the germline setting in association with CFC syndrome (10). This observation was particularly noteworthy as it was the first description of naturally occurring mutations in this gene and offered additional insight into the biochemistry of MEK1 and MEK2 that could not be obtained from in-vitro models. Since then, additional mutations have been reported in CFC, and somatic variants have been found in a small proportion of solid tumors and in melanomas in the setting of resistance to BRAF inhibitors. More recently, MEK1 mutations have also been reported in a specific subset of hairy cell leukemias (23). Somatic mutations in MEK1 cluster in the same functional regions as the previously identified germline alterations. The first cluster lies in and around the MEK NRR (negative regulatory region), while the second cluster localizes to the region flanked between codons 121 and 130 at the center portion of the kinase domain. In somatic cancer mutations, there seems to be a third preferentially mutated region surrounding codon 203. It is important to note, that although these clusters are separated at the level of the amino acid sequence, when mapped into a 3D model, all preferential regions are brought together forming a single mutational hot-spot that is proximal to the NRR and the ATP-binding site and thus well positioned to influence catalytic activity (6). (Figure 4)
Figure 4. Hotspots mutations in MEK1 destabilize the alpha-helix.

A 3Dmodel of MEK1 showing the most common mutations identified in this study and selected mutations also identified in other solid tumors. Labeled residues identify the region of the mutation. Alpha helix – bright green (residues 43-67). Although in a linear model these mutations are separated into clusters in exons 2, 3 and 6, when mapped into a 3D model, mutations form a single hot spot.
In the current study, MEK1 testing was initially focused on point mutations in exon 2, namely Q56P, K57N and D67N, based on the variants identified in initial reports. To further characterize the spectrum of mutations affecting this gene, we integrated publicly available comprehensive mutation analysis results from lung adenocarcinoma studies (19, 20, 24) and non-overlapping data from the COSMIC database. In this combined dataset, 86% of all mutations were identified in exon 2. While this is influenced by the original screening design, comprehensive unbiased datasets also identify this as the preferentially mutated region in lung cancer.
At the time of its initial description in lung cancer, it was unclear how the reported mutations would affect MEK1 structure as the crystal structure of some regions had not been defined. Since then, the crystal structure has been solved for most of the protein, providing a structural rationale for the role of several residues and the impact of their alterations. In the study by Fischmann et al (25), the boundaries of the MEK1 consensus kinase domain were established to encompass the region of residues 55 to 369. Residues 43 to 61 fold into the alpha helix of the protein with significant packing interactions all along this segment. Residues F53, Q56 and K57 are located in the C terminal end of the alpha helix, the area which is most involved with contacts to the N terminal lobe of the protein and likely maintains helical integrity. Mutations in this region are likely to cause significant alterations in the helical structure that would deregulate kinase function due to their critical interface with the rest of the kinase domain. Of note, residue D67, mutated in colon cancer, lies outside the interface between the alpha helix and the rest of the N-terminal lobe. Its role in MEK1 constitutive activation is unclear based on the kinase structure. Interestingly, in our functional characterization, we find that the D67N mutant kinase activity is significantly lower than all other mutants tested (including F53L, Q56P, K57N, E102_I103del, C121S and E203K) and not significantly different from the wild-type in our hands (Fig 3). A previous study, however, shows that the D67N substitution does lead to constitutive activation of the MAPK pathway (26). In this study by Estep et al, the level of ERK phosphorylation of the D67N MEK1 was described to be less than that of MEK1 Y130C mutant used for comparison. Additional studies are required to better understand the role of this mutation in MEK1 constitutive activation and how the mutation compares to other hyperactive mutations.
Additional mutations identified in our search of public databases include 3 novel mutations: M146I, G301X, S331R and 2 previously described mutations, E102_I103del and C121S (14, 27). Our assessment of the later 2 shows these to be functional with hyperactive downstream MAPK pathway signaling that was comparable to the K57N (Figure 3B). The C121S mutation has also been associated with the development of resistance to both RAF and MEK inhibition in vitro (14). Of interest, the M146 residue is an important component within the binding pocket of the highly selective MEK inhibitor U0126 (23). The same binding pocket is shared by the MEK1 inhibitor PD-325901 which is currently in clinical trials for treatment of NSCLC and breast carcinoma (clinical trials.gov)(23). The potential implication of these mutations in the binding of the drug remains to be explored.
Morphologically, MEK1 mutated tumors in our study were all adenocarcinomas, although one case also had a squamous cell component. No mutations were identified among 211 morphological and immunophenotypically defined squamous cell carcinomas. The clinical characteristics of patients with MEK1 mutations were also examined. While we found no particular sex predilection, there was a strong association with smoking. Remarkably, all but 1 patient (35/36, 97%) were current or former smokers with a median smoking history of 48 pack years. In further support of the smoking association, we note that the vast majority of the identified mutations (89%) were G:C>T:A transversions, a pattern known to occur in association with direct exposure to tobacco carcinogens. A similar relationship has also been observed for KRAS (28-30) and TP53 mutations in lung cancers from smokers (31). In contrast, MEK1 mutations in melanoma, reported in up to 6% cases (32), predominantly involve C>T (majority) and G>A transitions, changes frequently associated with exposure to UV radiation (32, 33). Similar to melanoma, although based on a fewer number of reported cases, mutations in colon carcinomas are also transition predominant. Of note, and also in contrast to the data available in melanoma, where MEK1 mutations are often found to co-exist with BRAF or NRAS mutations (32-34), in lung cancers we find most MEK1 mutations to be mutually exclusive with all other established driver mutations including EGFR, KRAS, NRAS, BRAF, HER2, PIK3CA and fusions involving ALK. Only 2 novel, non-recurrent MEK1 mutations had concurrent KRAS alterations (both G>T transversions).These two cases could represent passenger mutations in MEK1 in smoking-associated lung cancers with a high mutation load.
In this series, median overall survival from the diagnosis of metastatic disease was not significantly different between patients harboring MEK1-mutant lung cancers and other lung cancers that have been associated with smoking including those harboring KRAS- and BRAF-mutations. In contrast, the survival of patients with MEK1-mutant lung cancers was significantly inferior to that of patients whose lung cancers have been associated with a never or former light smoking history (EGFR-mutant, ALK-rearranged, ROS1-rearranged, and RET-rearranged lung cancers).
Overall, our data suggest that mutations in MEK1, although uncommon, define a molecular subset of LAD that is distinct and potentially “druggable”. While MEK inhibitors are currently in clinical testing and are approved either as a single-agent or in combination with a second targeted agent in BRAF-mutant melanomas, to the best of our knowledge, no ongoing or completed phase 1 or phase 2 trial of a MEK-selective inhibitor has yet enriched for MEK-mutant non-small cell lung cancer patients and trials such as this remains an unmet need. In the past, testing for these rare mutations has been a bottleneck in this setting but more comprehensive tumor genotyping approaches should increasingly make it feasible to stratify patients on the basis of MEK1 tumor genotype and this would have secondary benefits in furthering the understanding of the biology of these tumors and the clinical development of MEK1 inhibitors.
Supplementary Material
Statement of Translational Relevance.
Although genetic alterations activating the MAPK signaling pathway are common in lung adenocarcinoma, somatic mutations in MEK1, located downstream of BRAF, are relatively rare and have remained poorly defined as a distinct molecular subset.
The present study of 36 MEK1 mutation-positive lung cancer patients represents the first systematic analysis of this molecular subset of lung adenocarcinoma. We show that mutations in MEK1 define a distinct subset of lung cancer that is strongly associated with smoking and that the mutations are recurrent, clustered, activating, and potentially “druggable”. Though rare, based on estimated lung adenocarcinoma incidence, mutations in MEK1 may account for over 600 lung cancer patients a year in the USA alone who could benefit from specific targeted therapies directed at this driver oncogene.
Acknowledgments
Financial Support: NIH P01 CA129243 (to M. Ladanyi, M.G. Kris)
Bibliography
- 1.Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2008;21(Suppl 2):S16–22. doi: 10.1038/modpathol.3801018. [DOI] [PubMed] [Google Scholar]
- 2.Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:5731–4. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:2046–51. doi: 10.1200/JCO.2010.33.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer research. 2008;68:5524–8. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–5. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 6.Bromberg-White JL, Andersen NJ, Duesbery NS. MEK genomics in development and disease. Briefings in functional genomics. 2012;11:300–10. doi: 10.1093/bfgp/els022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–52. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 8.Mansour SJ, Candia JM, Gloor KK, Ahn NG. Constitutively active mitogen-activated protein kinase kinase 1 (MAPKK1) and MAPKK2 mediate similar transcriptional and morphological responses. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research. 1996;7:243–50. [PubMed] [Google Scholar]
- 9.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–70. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki H, Hikosaka Y, Kawano O, Moriyama S, Yano M, Fujii Y. MEK1 and AKT2 mutations in Japanese lung cancer. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2010;5:597–600. doi: 10.1097/JTO.0b013e3181d35236. [DOI] [PubMed] [Google Scholar]
- 12.Choi YL, Soda M, Ueno T, Hamada T, Haruta H, Yamato A, et al. Oncogenic MAP2K1 mutations in human epithelial tumors. Carcinogenesis. 2012;33:956–61. doi: 10.1093/carcin/bgs099. [DOI] [PubMed] [Google Scholar]
- 13.Murugan AK, Dong J, Xie J, Xing M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell cycle. 2009;8:2122–4. doi: 10.4161/cc.8.13.8710. [DOI] [PubMed] [Google Scholar]
- 14.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arcila M, Lau C, Nafa K, Ladanyi M. Detection of KRAS and BRAF mutations in colorectal carcinoma roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. The Journal of molecular diagnostics: JMD. 2011;13:64–73. doi: 10.1016/j.jmoldx.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO molecular medicine. 2010;2:146–58. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Z, Dias-Santagata D, Duke M, Hutchinson K, Lin YL, Borger DR, et al. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. The Journal of molecular diagnostics: JMD. 2011;13:74–84. doi: 10.1016/j.jmoldx.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer research. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou JX, Yang H, Deng Q, Gu X, He P, Lin Y, et al. Oncogenic driver mutations in patients with non-small-cell lung cancer at various clinical stages. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2013;24:1319–25. doi: 10.1093/annonc/mds626. [DOI] [PubMed] [Google Scholar]
- 23.Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nature genetics. 2014;46:8–10. doi: 10.1038/ng.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischmann TO, Smith CK, Mayhood TW, Myers JE, Reichert P, Mannarino A, et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry. 2009;48:2661–74. doi: 10.1021/bi801898e. [DOI] [PubMed] [Google Scholar]
- 26.Estep AL, Palmer C, McCormick F, Rauen KA. Mutation analysis of BRAF, MEK1 and MEK2 in 15 ovarian cancer cell lines: implications for therapy. PloS one. 2007;2:e1279. doi: 10.1371/journal.pone.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahrendt SA, Decker PA, Alawi EA, Zhu Yr YR, Sanchez-Cespedes M, Yang SC, et al. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–30. doi: 10.1002/1097-0142(20010915)92:6<1525::aid-cncr1478>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 29.Dogan S, Shen R, Ang DC, Johnson ML, D'Angelo SP, Paik PK, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6169–77. doi: 10.1158/1078-0432.CCR-11-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hecht SS. Tobacco smoke carcinogens and lung cancer. Journal of the National Cancer Institute. 1999;91:1194–210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 31.Hainaut P, Pfeifer GP. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–74. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 32.Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nature genetics. 2012;44:133–9. doi: 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- 33.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi H, Moriceau G, Kong X, Koya RC, Nazarian R, Pupo GM, et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer discovery. 2012;2:414–24. doi: 10.1158/2159-8290.CD-12-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.