

Abstract
Non-ribosomal peptide synthetases (NRPSs) are giant enzymes comprised of modules that house repeated sets of functional domains, which select, activate and couple amino acids drawn from a pool of nearly 500 potential building blocks.1 The structurally and stereochemically diverse peptides generated in this manner underlie the biosynthesis of a large sector of natural products. Many of their derived metabolites are bioactive such as the antibiotics vancomycin, bacitracin, daptomycin and the β-lactam-containing penicillins, cephalosporins and nocardicins. Although penicillins and cephalosporins are synthesised from a classically derived NRPS tripeptide (from ACVS, δ-(L-α-aminoadipyl)–L-cysteinyl–D-valine synthetase)2, we now report an unprecedented NRPS activity to both assemble a serine-containing peptide and mediate its cyclisation to the critical β-lactam ring of the nocardicin family of antibiotics. A histidine-rich condensation (C) domain, which typically carries out peptide bond formation during product assembly, was found to also synthesise the embedded 4-membered ring. Here, a mechanism is proposed and supporting experiments are described, which is distinct from the pathways that have evolved to the three other β-lactam antibiotic families: penicillin/cephalosporins, clavams and carbapenems. These findings raise the possibility that β-lactam rings can be regio- and stereospecifically integrated into engineered peptides for application as, for example, targeted protease inactivators.3,4
Despite their widespread use for more than half a century, the β-lactam antibiotics, represented most familiarly by the semisynthetic penicillins and cephalosporins, remain the most frequently prescribed anti-infectives in human medicine.5,6 Four structurally distinct clans occur naturally, and the more recently discovered of these and their synthetic variants are of increasing importance to combat the rising specter of antibiotic resistant infectious diseases.7,8 Members of this group of antibiotics contain monocyclic and fused bicyclic β-lactams whose high energy, strained-ring skeletons are essential to their antimicrobial activities. Markedly different but chemically efficient biosynthetic pathways have evolved to each of the penicillin and cephalosporin (e.g. isopenicillin N and cephalosporin C),9 clavulanic acid10 and the carbapenem (e.g. thienamycin)11 groups (Fig. 1a and b). Ironically, the fourth and structurally simplest clan of monocyclic β-lactams, exemplified by nocardicin G (Fig. 2b), has remained an unsolved problem.12,13
Figure 1. Representative members of the family of β-lactam antibiotics.
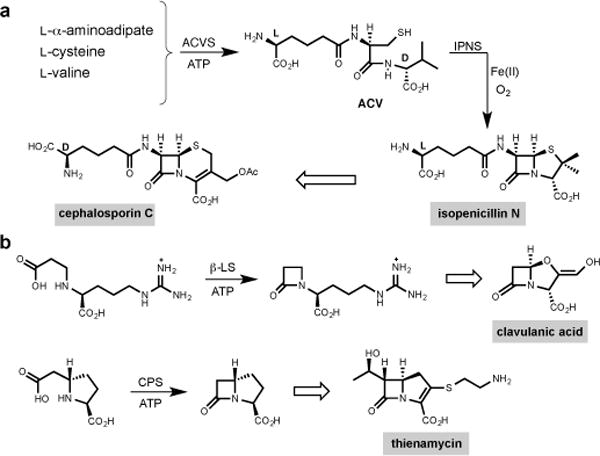
a, ACV [δ-(L-α-aminoadipic acid)–L-cysteine–D-valine] is an NRPS-derived tripeptide from ACV synthetase (ACVS). Isopenicillin N synthase (IPNS) catalyses oxidative β-lactam formation and bicyclisation of ACV to form isopenicillin N with a single molecule of dioxygen and release of two molecules of water. Cephalosporin C is derived after isopenicillin N is epimerised to penicillin N and oxidative ring expansion occurs. b, The clavams and carbapenems are exemplified by clavulanic acid and thienamycin, respectively. Formation of the β-lactam ring that ultimately appears in clavulanic acid and thienamycin is catalyzed by β-lactam synthetase (β-LS) and carbapenam synthetase (CPS), respectively, where transiently formed acyl adenylates are cyclized to β-lactam containing pathway intermediates, AMP and inorganic diphosphate.
Figure 2. Biosynthesis of nocardicin A.
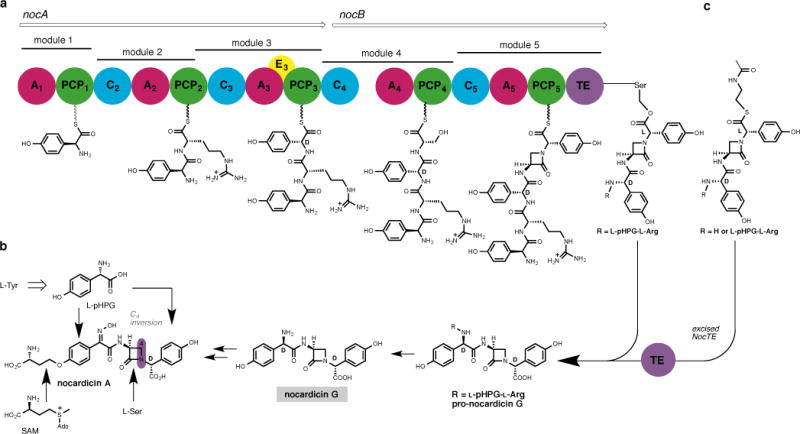
a, The nocardicin NRPS contains five modules, encoded by nocA and nocB, which together activate and condense in order L-pHPG, L-Arg, D-pHPG, L-Ser and L-pHPG. The TE domain catalyses C-terminal pHPG epimerisation and hydrolysis yielding pro-nocardicin G. b, Whole-cell experiments showed nocardicin A is derived from 2 units of L-pHPG, L-Ser and S-adenosyl-L-methionine (SAM), and from nocardicin G. c, Previous experiments with excised TE demonstrate that tri- and pentapeptide thioesters containing a preformed β-lactam ring are converted to nocardicin G (R = H) and pro-nocardicin G (R = L-pHPG–L-Arg), respectively.
The nocardicin NRPS encompasses two megaenzymes, NocA and NocB, which together comprise five modules (Fig. 2a). Each module contains an adenylation (A) domain that binds ATP, selects its cognate building block and carries out substrate acyl adenylation. The activated amino acid is then translocated as its aminoacyl thioester to the 4′-phosphopantetheine “arm” of the downstream post-translationally modified peptidyl carrier protein (PCP). Condensation (C) domains mediate substrate inter-module amide bond formation to yield peptides of length and sequence defined by the NRPS(s). An epimerisation (E) domain is embedded in module 3, which converts its associated amino acid residue from the L- to the D- configuration. Finally, catalytic turnover of the NRPS is achieved through disconnection of the final peptide product by the C-terminal thioesterase (TE) domain.
Although the roles of NocA and NocB have remained enigmatic, it was established early on that the O-homoseryl terminus of nocardicin A is derived in an unusual transfer reaction from S-adenosyl-L-methionine (SAM)14 and the β-lactam carbons arise from L-serine (Fig. 2b).15 The two modified p-(hydroxyphenyl)glycine (pHPG) units originate as catabolic products of L-tyrosine.16 Nocardicin G (Fig. 2b), the simplest of the nocardicins, is a key pathway intermediate to nocardicin A.17 As a consequence, it was initially thought that modules 1 and 2 of NocA were inactive to account for an apparent tripeptide NRPS precursor to nocardicin G. Subsequent experimentation, however, demonstrated that all five modules of NocA and NocB are essential to antibiotic production,12 and careful analysis of each dissected A domain gave the predicted product as L-pHPG–L-Arg–D-pHPG–L-Ser–L-pHPG, a pentapeptide.13 The role of the N-terminal L-pHPG–L-Arg in the biosynthesis was unclear as was the means by which the C-terminal pHPG epimerised from the L- to D-configuration present in nocardicin G. Recent experiments with the nocardicin TE domain (NocTE) shed light on these questions and defined the central problem of β-lactam formation. A series of predicted tri- and pentapeptide and potential seryl O-activated peptide thioesters all failed to undergo hydrolysis at rates greater than controls. On the other hand, the corresponding tri- and pentapeptide thioesters now bearing a preformed β-lactam ring from cyclisation of the seryl residue were not only rapidly hydrolysed but also completely epimerised to the C-terminal D-stereochemistry (Fig. 2c).18 NRPS epimerase activity by a TE domain was unprecedented, but in this specific instance owes to the anomalously high acidity of a pHPG α-hydrogen relative to other α-amino acids.18 Competition experiments established that the L,L,D,L,L-pentapeptide β-lactam thioester is the preferred NocTE substrate,18 a finding fully in accord with the requirement that all five modules of NocA/B are necessary for nocardicin biosynthesis.
Although NocTE catalyses C-terminal epimerisation and hydrolytic product release, it was not observed to mediate β-lactam synthesis.18 Azetidinone formation, therefore, must logically occur upstream on the NRPS after introduction of the last pHPG unit in module 5 from which the β-lactam ring nitrogen arises. In principle, formation of the embedded β-lactam ring could take place either in cis in this module or occur in trans. The latter alternative invokes the action of auxiliary enzyme(s), which are increasingly precedented in NRPS biochemistry.19 Among the mechanisms that can be visualized are in trans activation of the seryl hydroxyl group by, for example, phosphorylation or acylation, and intramolecular nucleophilic substitution (SNi) by the adjacent amide to form the critical C4–N bond, a process well supported by chemical precedent20 and consistent with the observation of stereochemical inversion at the seryl β-carbon.16 Bioinformatic analysis and biochemical experiments, however, did not point to candidate auxiliary enzyme(s) encoded by the nocardicin biosynthetic gene cluster.21 As a consequence, experiments were first undertaken to probe the in cis strategy with unexpected results.
The termination module of NocB, module 5, is composed of four domains: C5, A5, PCP5 and TE. This 144 kDa protein was heterologously expressed in Escherichia coli with a His6 tag and purified by affinity chromatography. Complete conversion to its corresponding holo form was ensured by Sfp-mediated 4′-phosphopantetheinyl transfer from coenzyme A (CoASH).22,23 The final chemical transformations catalysed by the termination module were successfully reconstituted in vitro through incubation of the predicted tetrapeptide-modified PCP domain from module 4 (PCP4) with holo-module 5. Bearing in mind that all five modules of NocA/B are required for production of nocardicin A in N. uniformis, and that the β-lactam-containing pentapeptide is preferentially processed by NocTE over the corresponding tripeptide,18 L-pHPG–L-Arg–D-pHPG–L-Ser-CoA (1, Fig 3a) was prepared (Supplementary Information) and linked to apo-PCP4 in an Sfp-mediated transfer to create L-pHPG–L-Arg–D-pHPG–L-Ser-S-PCP4 (2, Fig. 3b and Extended Data Fig. 1). It was anticipated that module 5 would activate L-pHPG in the presence of ATP and present this amino acid on PCP5 for reaction with the tetrapeptide delivered to module 5 by PCP4. Indeed, when holo-module 5, 10 equivalents of tetrapeptidyl-S-PCP4 2, L-pHPG and ATP were combined, smooth conversion to the pentapeptide β-lactam (pro-nocardicin G) was observed (Fig. 3c and Extended Data Fig. 2). Monitoring product formation by HPLC in a time-course experiment and simultaneous consumption of tetrapeptidyl-S-PCP4 2 by ESI-MS revealed a 1:1 correlation in accord with full catalytic turnover (Fig. 3d). Control experiments lacking L-pHPG or L-pHPG and ATP showed no product formation.
Figure 3. Analysis of the reactions catalysed by module 5.
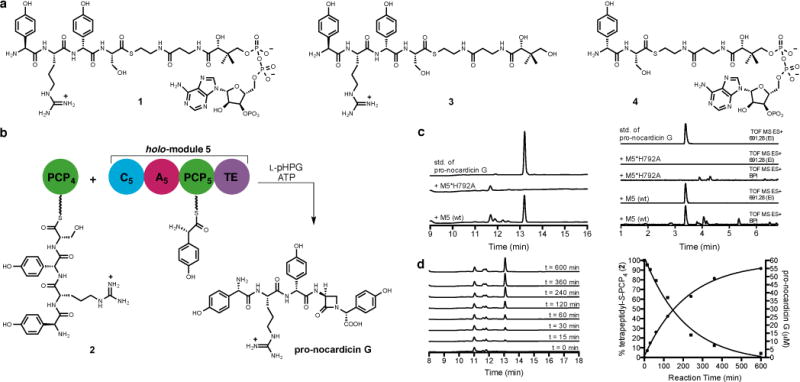
a, Substrates used in this study. b, Incubation of tetrapeptidyl-S-PCP4 2 with holo-module 5, ATP and L-pHPG produced β-lactam containing pro-nocardicin G. c, Left: HPLC traces of products obtained after incubation of tetrapeptidyl-S-PCP4 2 and indicated construct, ATP and L-pHPG. Pro-nocardicin G was observed in the wild-type reaction [+M5(wt)] but not in the mutant (+M5*H792A), verified by comparison to synthetic standard (top trace). Right: LC-MS traces of products obtained after incubation of tetrapeptidyl-S-PCP4 2 and holo-module 5, ATP and L-pHPG. Pro-nocardicin G was observed in the wild-type reaction [+M5(wt)] but not in the mutant (+M5*H792A), verified by comparison to synthetic standard (top trace). BPI = base peak ion, TOF = time of flight, ES = electrospray ionization, EI = extracted ion. Calculated exact mass of pro-nocardicin G = 691.2835 [M+H]+. d, Left: Time-course study of tetrapeptidyl-S-PCP4 2 and holo-module 5 supplemented with L-pHPG and ATP. Appearance of pro-nocardicin G was analyzed by HPLC. Right: Plots of pro-nocardicin G production (HPLC) and the corresponding conversion of 2 to the unloaded holo-PCP4, (ESI-MS). Less than 10% hydrolysis of 2 was observed during the 10-h experiment.
In a negative control experiment, the in vitro reconstitution experiment was repeated with a point mutant of C5 where the second histidine residue of the conserved active site HHxxxDG sequence, known to be essential for amide bond formation,24 was replaced by alanine (H792A). No new products were detected (Fig. 3c). To further define acceptable substrates for C5, the L-pHPG–L-Arg–D-pHPG–L-Ser-S-pantetheine (3, Fig 3a) substrate mimic was prepared (Supplementary Information) but did not yield pro-nocardicin G when incubated with holo-module 5, ATP and L-pHPG (Extended Data Fig. 3a and b), a result that underscores the critical importance PCP4•C5 domain•domain interaction plays to β-lactam formation. Next the dipeptide D-pHPG–L-Ser-CoA (4, Fig 3a) was prepared (Supplementary Information) and loaded onto apo-PCP4 as before to afford D-pHPG–L-Ser-S-PCP4 (5, Extended Data Fig. 4). When this construct was generated in the presence of holo-module 5, L-pHPG and ATP, nocardicin G was not detected (Extended Data Fig. 3c and d). These data suggest that the L-pHPG–L-Arg “leader” present in the tetrapeptidyl-S-PCP4 2 plays a vital role in the binding and/or recognition of the upstream tetrapeptidyl intermediate in C5, enabling peptide extension and β-lactam formation to occur.
Examination of the primary sequence of C5 showed no unusual insertions or deletions except that, in addition to the conserved HHxxxDG catalytic motif emblematic of condensation domains, a third His residue (H790) lies directly upstream of the His dyad (Extended Data Fig. 5). Analysis also revealed features of a DCL domain despite receiving an L-seryl tetrapeptide from PCP4 (Extended Data Table 1). We propose a mechanism in which His790 catalyses β-elimination of hydroxide (water) from the seryl residue of the PCP4-bound tetrapeptidyl-thioester and PCP5-tethered L-pHPG carries out β-addition with overall inversion of configuration at the seryl(dehydroalanyl) β-carbon dictated by earlier stereochemical experiments (Fig. 4a).25 The transient loss of the L-seryl stereocenter during the β-elimination/addition may account for the DCL characteristics of C5. The resulting β-aminothioester 6 is then proposed to undergo unconventional amide bond cyclisation (allowed 4-exo-trig), thermodynamically driven by amide bond formation from the active PCP4 thioester. The PCP5-bound pentapeptide β-lactam (pro-epi-nocardicin G) is poised for delivery to NocTE for C-terminal epimerisation and hydrolytic product release.
Figure 4. Proposed β-lactam formation mechanism.
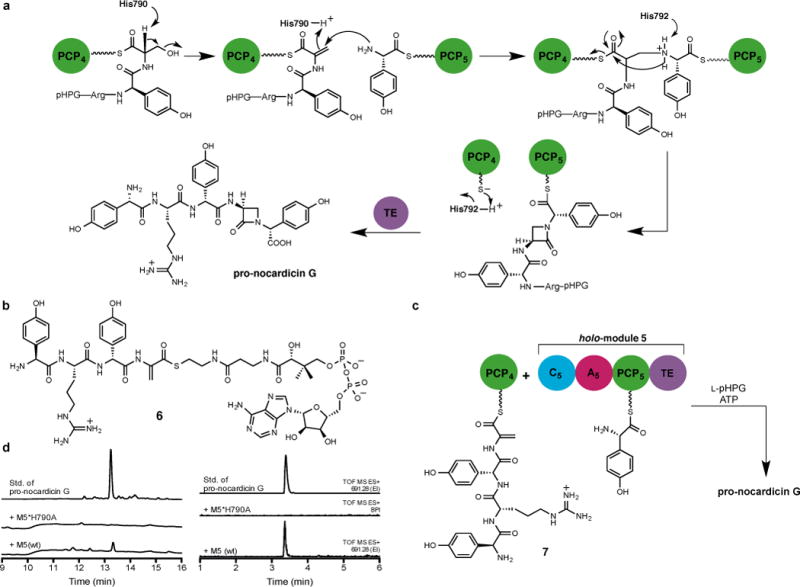
a, Proposed mechanism of β-lactam formation in C5. Tentative catalytic roles of histidine residues are indicated. b, Substrate used in this study. c, Incubation of dehydroalanyl tetrapeptidyl-S-PCP4 7 with holo-module 5, ATP and L-pHPG gave pro-nocardicin G. d, Left: HPLC traces of products obtained after incubation of dehydroalanyl tetrapeptidyl-S-PCP4 7 and indicated holo-module 5, ATP and L-pHPG. Pro-nocardicin G was observed in the wild-type reaction [+M5(wt)] but not in the mutant (+M5*H790A), verified by comparison to synthetic standard (top trace). Right: LC-MS traces of products obtained after incubation of dehydroalanyl tetrapeptidyl-S-PCP4 7 and indicated holo-module 5 construct, ATP and L-pHPG. Pro-nocardicin G was observed in the wild-type reaction [+M5(wt)] but not in the mutant (+M5*H790A), verified by comparison to synthetic standard (top trace). TOF = time of flight, ES = electrospray ionization, EI = extracted ion.
To support this mechanistic hypothesis, we prepared a mutant of module 5 in which the tentative catalytic His was replaced by alanine (M5*H790A). Repeating the experiments with PCP4-bound tetrapeptide 2 and mutant M5*H790A, L-pHPG and ATP yielded no product (Extended Data Fig. 6). Similarly site-specific mutation of the His residue typically involved in peptide bond formation (M5*H792A) also gave no reaction, as anticipated. In a further test of the proposed mechanism, the reactive dehydroalanyl tetrapeptide intermediate (6, Fig. 4b) was synthesised (Supplementary Information) and used in an Sfp-catalysed reaction to afford the corresponding L-pHPG–L-Arg–D-pHPG–dehydroalanyl-S-PCP4 substrate (7, Fig. 4c and Extended Data Fig. 7). In the course of preparing this sensitive material, it was discovered that the addition of sulfur, phosphorus and nitrogen nucleophiles occurred preferentially 1,4 rather than 1,2 in keeping with the hypothetical reactivity posed in Fig. 4a. When PCP4-bound dehydroalanyl tetrapeptide 7 was incubated with wild-type holo-module 5, L-pHPG and ATP, β-lactam formation was once again observed (Fig. 4d and Extended Data Fig. 8 and 9). Further insight into this process was afforded by the M5*H790A mutant, which did not support complete reaction of the dehydroalanyl substrate to the β-lactam product (Fig. 4d). The proposed catalytic residue H790 must not only act as a base to promote β-elimination but also serve as the acid to consummate amine (L-pHPG-S-PCP5) β-addition. Although interfering with the proper cycling of the protonation state of the enzyme can be partially compensated in the wild-type protein, it cannot in the M5*H790A mutant.
NRPS C domains are pseudodimeric proteins whose N- and C-terminal subdomains are joined to form an extended V-shaped substrate channel that accommodates the donor and acceptor aminoacyl reactants, each delivered by extended pantetheinyl “arms” from proximal PCP domains.26 The centrally located HHxxxDG motif promotes peptide bond formation and transfer of the growing peptide chain to the downstream PCP domain. The unprecedented β-lactam formation catalysed by C5 is distinct from the iron-mediated oxidative cyclisation to penicillin (Fig. 1a) and the ATP-driven β-amino acid closures that lead ultimately to clavulanic acid and all of the carbapenems (Fig. 1b),27 and it does not correlate to the heterocyclisation of serine residues to oxazolidine rings.28 There is no stereoelectronic imperative that β-elimination/addition reactions must occur with overall retention of stereochemistry.29 It can be readily appreciated that departure of the seryl OH and conjugate addition of the pHPG amine can take place on opposite faces of the dehydroalanyl intermediate 7 to achieve configurational inversion. This facile β-elimination from a thioester-bound seryl residue stands in contrast to the generation of dehydroalanyl components and lanthionine bridges in ribosomally synthesised and post-translationally modified natural products (RiPPs)30 where a prior O-phosphorylation intervenes to facilitate this elimination step. Impressive synthetic efficiency is achieved by the NocB termination module where previously unknown NRPS catalytic capabilities are captured in a non-oxidative route to β-lactams. Parsing a universe of >25,000 NRPS sequences to those that contain both a C domain bearing a HHHxxxDG motif and an immediately upstream A domain that is confidently predicted to activate Ser (or Thr) yielded only four Ser hits, two of which are known nocardicin producers13, and, interestingly, five Thr hits (Extended Data Table 1). The products of the latter seven, and whether β-lactam containing or not, are not known. The exceeding rarity of even potential β-lactam synthesis by an NRPS is underscored by these findings, but its discovery at once broadens the engineering and synthesis goals that can now be contemplated for this versatile class of giant modular enzymes.
Methods
Synthesis of all compounds used in this study can be found in the associated Supplementary Information.
General Methods
Analytical HPLC analyses of enzymatic reactions were performed on an Agilent model 1200 HPLC equipped with a multi-wavelength UV-vis detector in conjunction with a reverse-phase Phenomenex Luna 5u phenyl/hexyl analytical column (250 × 4.60 mm ID). Water + ACN + 0.1% TFA: 0–5 min isocratic 93% water + 7% ACN + 0.1% TFA, 5–22 min gradient 7% to 50% ACN + 0.1% TFA, 22–25 min gradient 50% to 7% ACN + 0.1% TFA, 25–35 min isocratic 93% water + 7% ACN + 0.1% TFA. Flow rate = 1.0 mL/min.
UPLC-HRMS samples were analyzed on a Waters Acquity H-Class UPLC system equipped with a multi-wavelength UV-vis diode array detector in conjunction with a Waters Acquity BEH UPLC column packed with an ethylene bridged hybrid C-18 stationary phase (2.1 mm × 50 mm, 1.7 um) in tandem with HRMS analysis by a Waters Xevo-G2 Q-ToF ESI mass spectrometer. Mobile phase: 100% water + 0.1% formic acid 0–1 min, 1 – 7.5 min 80% ACN + 0.1% formic acid, 7.5 min – 8.4 min isocratic 80% ACN + 0.1 % formic acid, 8.4 – 10 min 100% water + 0.1 % formic acid. Flow rate = 0.3 mL/min.
Cloning, expression and purification of His6-module 5
The module 5 gene containing C-A-PCP5-TE of the termination module in NocB was PCR amplified from the pMG0531 cosmid21 containing nocA and nocB genes using the M5-forward and M5-reverse primers (Supplementary Table 1) and Herculase-HF DNA polymerase (Agilent Technologies). The resulting PCR product was incorporated into a pCRBlunt-TOPO subcloning vector (Invitrogen) and sequence verified (JHU Core Sequencing Facility). The pCRBlunt-M5 construct was digested with NdeI and HindIII (NEB) and ligated with T4 DNA ligase (NEB) into a similarly digested pET28b (Novagen) vector to create the corresponding N-terminal 6x-His fusion construct.
Apo-Module 5 was expressed using the pET28b-M5 vector in Rosetta 2(DE3)/pLysS E. coli cells (Novagen) and cultured at 37 °C in 1 litre of 2xYT broth supplemented with 50 μg/mL kanamycin and 50 μg/mL chloramphenicol. Upon reaching an absorbance at 600 nm of 0.7, the temperature of the culture was reduced to 4 °C for 1 h. The temperature of the culture was raised to 18 °C and expression was induced with1 mM isopropyl α-D-thiogalactopyranoside (IPTG) and grown at 18 °C for 18 h.
The cells were harvested by centrifugation (5,000 × g, 15 min, 4 °C) and stored at −80 °C. Cells were thawed in lysis buffer (50 mM phosphate, 300 mM NaCl, pH 8.0) and disrupted by sonication (60% amplitude, 9 sec on/off, 3 min) on ice. Cell debris was removed by centrifugation (25,000 × g, 30 min, 4 °C) and the clarified cell lysate was incubated with 2 mL of 50% suspension/L of cell culture of TALON® metal affinity resin (Clontech) for 1–2 h at 4 °C in a batch-binding format. The suspension was loaded onto a gravity column and washed with 2 volumes of lysis buffer. The desired protein was eluted with a stepwise gradient of imidazole (20–300 mM) in lysis buffer. Fractions containing the purified protein, as determined by SDS-PAGE with Coomassie staining, were pooled and dialyzed against 3 litres of assay buffer twice containing assay buffer (50 mM HEPES, 25 mM NaCl, pH 7.5). Protein concentrations were quantified by Bradford assay.
Construction, expression and purification of His6-module 5 point mutants (M5*H790A and M5*H792A)
Site-directed mutagenesis designed to alter His790 (H790A) and His792 (H792A) was carried out using the splicing by overlap extension method, from the pET28b-M5 vector, using the appropriate DNA primers for the desired mutant (Supplementary Table 2). The reverse primers used in these PCR reactions employed a native PstI restriction site in the module 5 nucleotide sequence. The resulting extension-overlap PCR product was incorporated into a pCRBlunt-TOPO subcloning vector and sequence verified. The pCRBlunt-M5* mutant construct was digested with NdeI and PstI and this extension-overlap product was ligated into a similarly digested pET28b-M5 to provide the desired C domain mutant of full-length M5. Expression and purification of mutant constructs was achieved similarly through procedures described for the wild-type protein.
Construction, expression and purification of His6-PCP4
Gene pcp4 from nocB was PCR amplified from the pMG0531 cosmid using the PCP4-forward and PCP4-reverse primers (Supplementary Table 1) and Herculase-HF DNA polymerase. The resulting PCR product was incorporated into a pCRBlunt-TOPO subcloning vector and sequence verified. The pCRBlunt-PCP4 construct was digested with NdeI and NotI and ligated with T4 DNA ligase into a similarly digested pET28b vector to create the corresponding N-terminal 6×-His fusion construct. Expression and purification of the PCP4 monodomain was achieved similarly through procedures described for the module 5 constructs.
In-vitro reconstitution of module 5 activity
Loading of peptidyl-S-CoA onto apo-PCP4
The apo-PCP4 constructs were converted to their holo forms by an Sfp-mediated transfer of the desired peptidyl-S-CoA substrate with the apo-PCP4 construct. 200 μM of apo-PCP4 was incubated with 250 μM of desired peptidyl-S-CoA substrate in assay buffer supplemented with 10 mM MgCl2. 4′-Phosphopantetheine transfer reactions were initiated by the addition of 2 μM of Sfp and the enzymatic mixture was incubated for 45 min at room temperature. Excess peptidyl-S-CoA reagent was removed through serial dilutions of the reaction mixture. This was achieved by adding 3 volumes of assay buffer to the reaction mixture and concentrating the mixture back down to the initial volume using a 3k MWCO Amicon Ultra centrifugal filter (Milllipore). This dilution procedure was repeated 3 times.
Generation of holo-module 5 construct
To a separate 1.5 mL tube, 20 μM of apo-module 5 construct (either wild-type or mutant) was incubated with 40 μM coenzyme A in assay buffer supplemented with 10 mM MgCl2. 4′-Phosphopantetheine transfer was initiated by the addition of 2 μM of Sfp and the reaction was left to stand for 45 min at room temperature. Excess CoA reagent was removed through serial dilutions of the enzymatic mixture as before.
Module 5 catalysed β-lactam formation
Holo-module 5 constructs were supplemented with 5 mM ATP and 2 mM L-pHPG and left to stand for 5 min in assay buffer. Condensation reactions were initiated by adding equal volumes of peptidyl-S-PCP4 construct with holo-module 5 and left to stand for 2 h. The reaction contained 100 μM peptidyl-S-PCP4, 10 μM M5 2.5 mM ATP and 1 mM L-pHPG in assay buffer. Proteins were removed by centrifugation through a 3k MWCO Amicon Ultra centrifugal filter. The filtrate was directly analyzed by HPLC and products of interest were collected over multiple injections and concentrated by lyophilization. The concentrated samples were re-suspended in 70 μL of 95: 5 water: ACN + 0.1% formic acid and directly analyzed by LC-MS.
Incubation of tetrapeptidyl-S-pantetheine 3 with holo-module 5
Reactions in which pantetheinyl substrate 3 was substituted for the holo-PCP4 construct contained 1 mM 3, 10 μM holo-module 5 construct, 1 mM L-pHPG and 2.5 mM ATP and left to stand for 2 h at room temperature in assay buffer. Reactions were quenched and analyzed as described above.
Extended Data
Extended Data Figure 1. Mass spectrometric verification of apo to holo conversion of PCP4 with Sfp and L-pHPG-L-Arg-D-pHPG-L-Ser-S-coenzyme A (1), forming L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2).
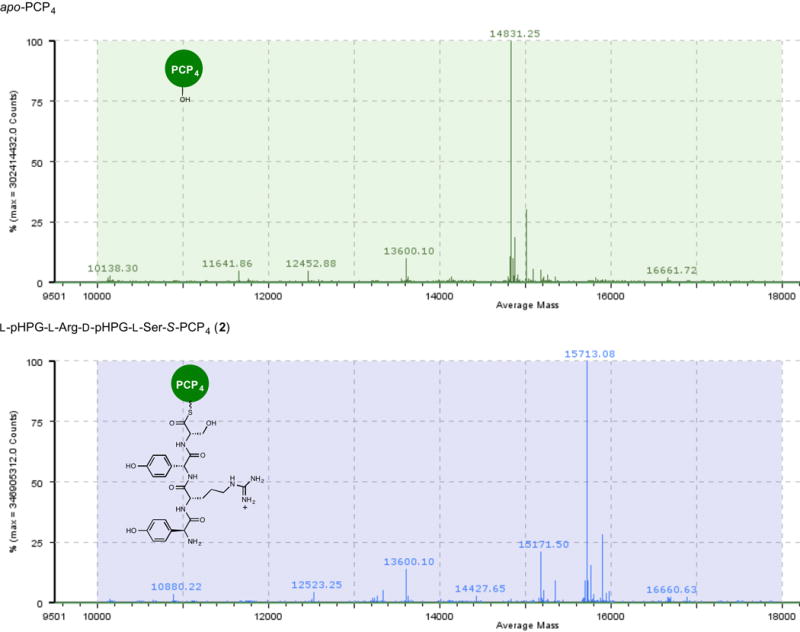
Top: mass spectrum (ESI+) of apo-PCP4. Bottom: mass spectrum (ESI+) of L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2) derived from treatment of apo-PCP4 with Sfp and corresponding synthetic tetrapeptidyl-CoA substrate 1.
Extended Data Figure 2. Mass spectrum of major product generated from reaction catalysed by holo-module 5 supplemented with ATP, L-pHPG and L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2).
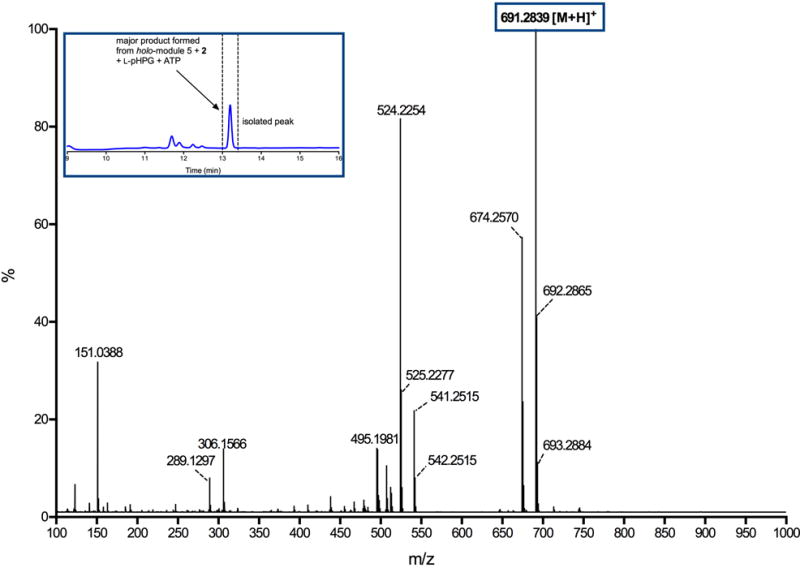
The major product observed from the reaction containing L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2), holo-module 5, ATP and L-pHPG was isolated over multiple injections by HPLC (peak isolated indicated in inset HPLC trace) and then characterized by HRMS (ESI+). The exact mass ion corresponding to the M+H ion of pro-nocardicin G was observed. Exact mass calculated for pro-nocardicin G: C33H39N8O9: 691.2835; Found: 691.2839 [M+H]+.
Extended Data Figure 3. Negative experiments resulting from incubation of holo-module 5 with alternate substrates.
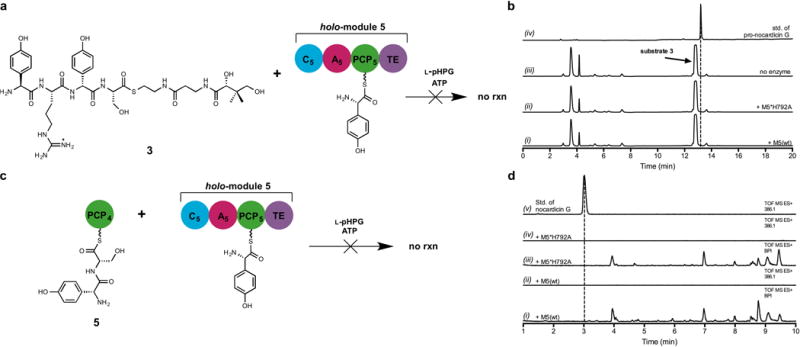
a. Schematic of incubation of the tetrapeptidyl substrate L-pHPG-L-Arg-D-pHPG-L-Ser-S-pantetheine (3) with holo-module 5 supplemented with ATP and L-pHPG. Pro-nocardicin G was not produced. b. HPLC traces of products obtained after incubation of L-pHPG-L-Arg-D-pHPG-L-Ser-S-pantetheine (3) and holo-module 5 [+M5(wt)] supplemented with ATP and L-pHPG. Pro-nocardicin G was not observed as verified by comparison to authentic standard. Additionally, it was noted that substrate 3 was not consumed over the duration of the experiment. (i) HPLC trace of the unbound products resulting from incubation of wild-type holo-module 5 reaction with L-pHPG-L-Arg-D-pHPG-L-Ser-S-pantetheine (3), L-pHPG and ATP. (ii) HPLC trace of the unbound products resulting from incubation of holo-M5*H792A reaction with L-pHPG-L-Arg-D-pHPG-L-Ser-S-pantetheine (3), L-pHPG and ATP. (iii) HPLC trace of incubation of L-pHPG-L-Arg-D-pHPG-L-Ser-S-pantetheine (3), L-pHPG and ATP without enzyme. The peak corresponding to L-pHPG-L-Arg-D-pHPG-L-Ser-S-pantetheine (3) substrate is indicated. (iv) HPLC trace of authentic standard of pro-nocardicin G. c. Schematic of incubation of the dipeptidyl substrate D-pHPG-L-Ser-S-PCP4 (5) with holo-module 5, L-pHPG and ATP. Nocardicin G, the corresponding expected product, was not observed. d. LC-MS traces of products obtained after incubation of D-pHPG-L-Ser-S-PCP4 (5) and holo-module 5 [+M5(wt)] supplemented with ATP and L-pHPG. The corresponding β-lactam product was not observed as verified by comparison to authentic standard. (i) Total ion chromatogram of the unbound products resulting from wild-type holo-module 5 reaction with D-pHPG-L-Ser-S-PCP4 (5), L-pHPG and ATP (ii) Extracted ion chromatogram of the wild-type reaction in trace i, the 386.1 m/z ion, corresponding to [M+H] of nocardicin G was not observed. (iii) Total ion chromatogram of unbound products from holo-M5*H792A control reaction with D-pHPG-L-Ser-S-PCP4 (5), L-pHPG and ATP. (iv) Extracted ion chromatogram of the wild-type reaction in trace iii. (v) Extracted ion chromatogram of authentic standard of nocardicin G.
Extended Data Figure 4. Mass spectrometric comparison of apo to holo conversion of PCP4 with Sfp and D-pHPG-L-Ser-S-coenzyme A (4) forming D-pHPG-L-Ser-S-PCP4 (5).
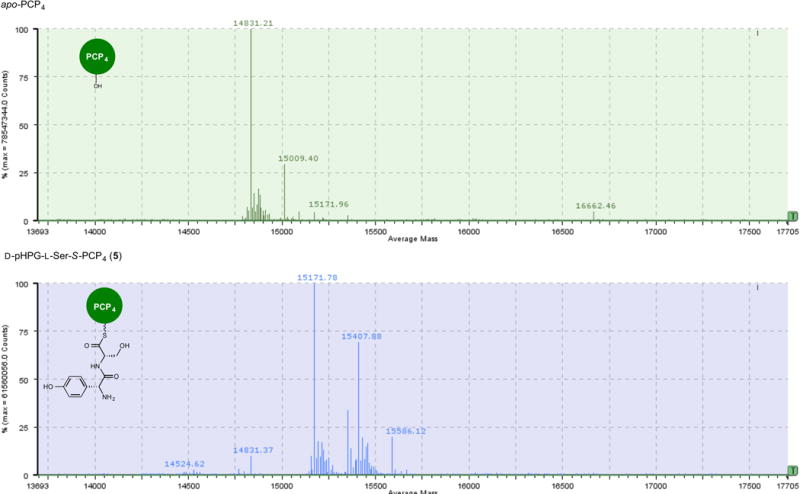
Top: mass spectrum (ESI+) of apo-PCP4. Bottom: mass spectrum (ESI+) of D-pHPG-L-Ser-S-PCP4 (5) derived from treatment of apo-PCP4 with Sfp and corresponding synthetic dipeptidyl-CoA substrate 4. Owing to diketipiperazine22,31 formation and slow hydrolysis to the unloaded holo-PCP4, the half-life of 4 is approximately 20 min.
Extended Data Figure 5. Multiple sequence alignment of homologous NRPS C domains and the NocB C5 domain.
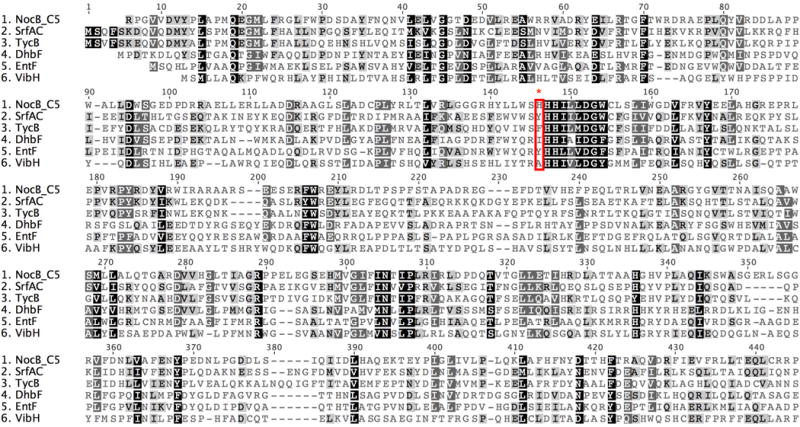
The alignment was performed using Clustal2 with gap opening and extending penalties of 10 and 0.1 respectively. The alignment included the N-terminal C fragment of EntF, the bacillibactin synthetase DhbF, the tyrocidine synthetase TycB, the surfactin synthetase SrfAC and the naturally free-standing condensation enzyme VibH. Highlighted in a red box is H790 uniquely present in the NocB C5 domain along with the corresponding residues present in the depicted C domains. The NocB C5 domain is N-terminal to the consensus catalytic motif, HHxxxDG, present in all canonical condensation domains.
Extended Data Figure 6. HPLC comparative analysis of the reactions catalysed by holo-module 5 and holo-M5*H790A with L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2).
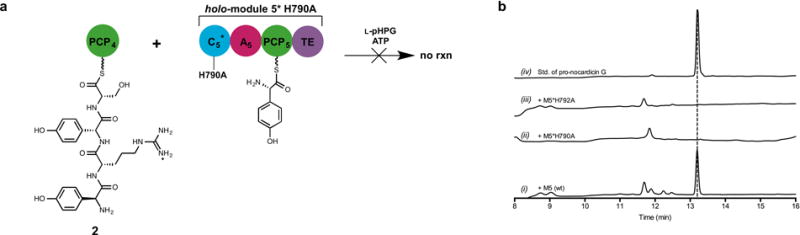
a. Schematic of incubation of the tetrapeptidyl substrate L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2) with holo-M5*H790A supplemented with ATP and L-pHPG. Pro-nocardicin G was not produced. b. HPLC comparison of unbound products from reaction mixtures containing L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2) and either holo-module 5 or M5*H790A variant supplemented with ATP, L-pHPG, indicating the formation of β-lactam product only from the wild-type reaction. (i) HPLC trace of the unbound products resulting from incubation of wild-type holo-module 5 reaction with L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2), L-pHPG and ATP. (ii) HPLC trace of the unbound products resulting from incubation of holo-M5*H790A reaction with L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2), L-pHPG and ATP. Pro-nocardicin G was not observed. (iii) HPLC trace of the unbound products resulting from incubation of holo-M5*H792A reaction with L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2), L-pHPG and ATP. (iv) HPLC trace of authentic standard of pro-nocardicin G.
Extended Data Figure 7. Mass spectral comparison of apo to holo conversion of PCP4 with Sfp and L-pHPG-L-Arg-D-pHPG-dehydroalanyl-S-3′-dephospho coenzyme A (6) forming L-pHPG-L-Arg-D-pHPG-dehydroalanyl-S-PCP4 (7).
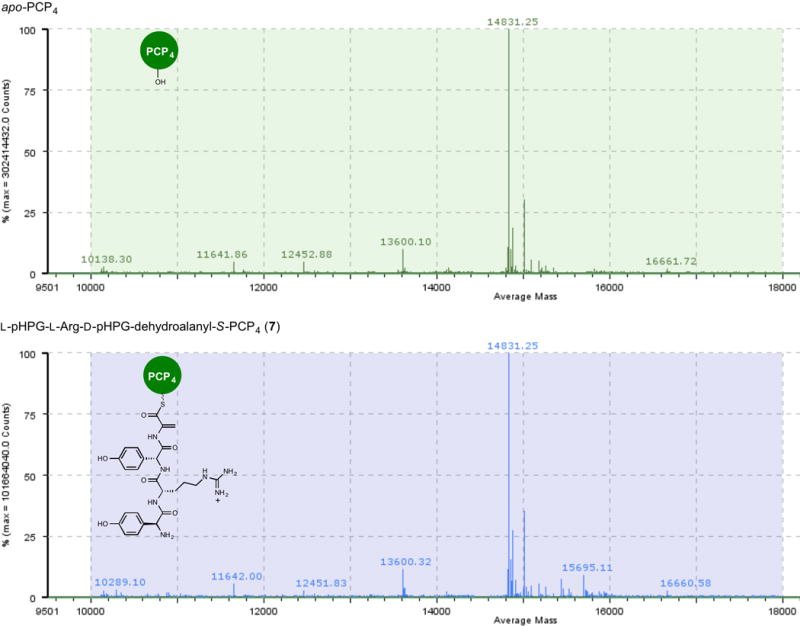
Top: mass spectrum (ESI+) of apo-PCP4 Bottom: mass spectrum (ESI+) of L-pHPG-L-Arg-D-pHPG-dehydroalanyl-S-PCP4 (7) derived from treatment of apo-PCP4 with Sfp and corresponding synthetic eliminated tetrapeptidyl-CoA substrate 6.
Extended Data Figure 8. Mass spectrum of major product generated from reaction catalysed by holo-module 5 and L-pHPG-L-Arg-D-pHPG-dehydroalanyl-S-PCP4 (7) supplemented with ATP and L-pHPG.
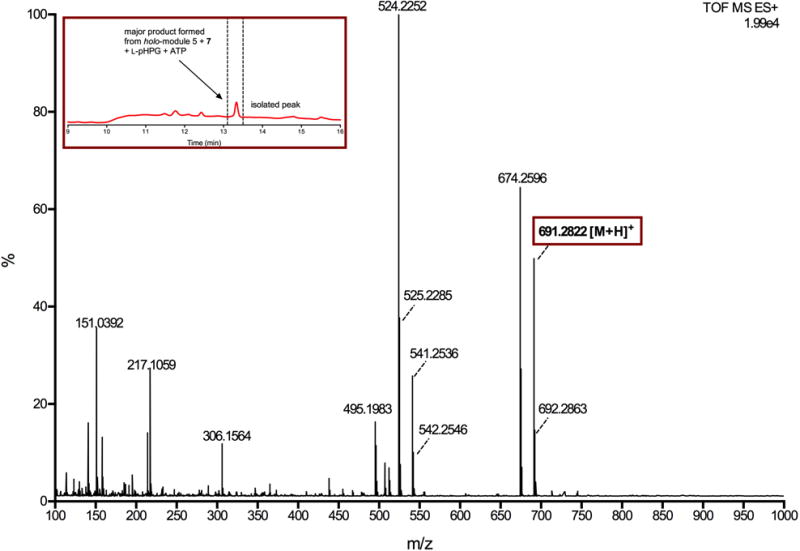
The major product from the reaction containing L-pHPG-L-Arg-D-pHPG-dehydroalanyl-S-PCP4 (7) was isolated over multiple injections by HPLC (peak indicated in inset HPLC trace) and then determined by HRMS (ESI+). The exact mass ion corresponding to the M+H ion of pro-nocardicin G was observed. Exact mass calculated for C33H39N8O9: 691.2835; Found: 691.2822 [M+H]+.
Extended Data Figure 9. MS-MS spectrometric comparison of holo-module 5 synthesised pro-nocardicin G from L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2) and L-pHPG-L-Arg-D-pHPG-dehydroalanyl-S-PCP4 (7).
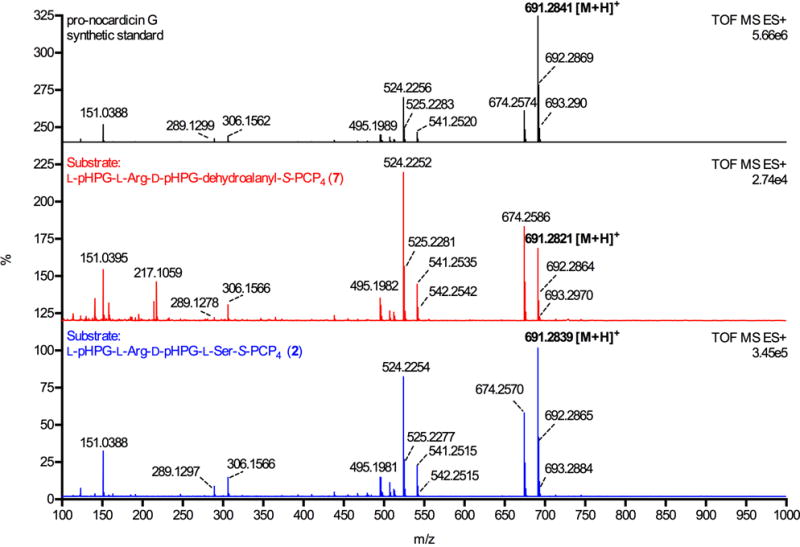
Fragmentation pattern of holo-module 5 biosynthetic pro-nocardicin G from serine-containing tetrapeptide substrate L-pHPG-L-Arg-D-pHPG-L-Ser-S-PCP4 (2, bottom) is directly comparable to the MS-MS fragmentation pattern of holo-module 5 synthesised pro-nocardicin G from the dehydroalanine-containing substrate L-pHPG-L-Arg-D-pHPG-dehydroalanyl-S-PCP4 (7, middle). For comparison, the mass spectrum of synthetic pro-nocardicin G is shown at top.
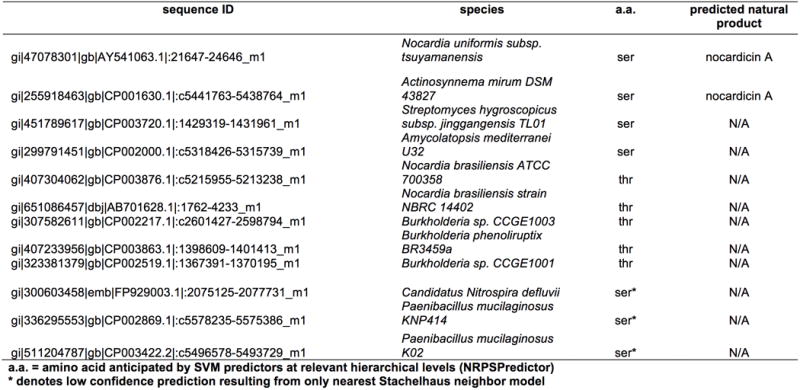
Extended Data Table 1. Condensation Domains with a HHHxxxDG motif and an upstream predicted serine- or threonine-activating A Domain.
Condensation domains with a HHHxxxDG motif and an upstream putative serine- or threonine- activating A domain were identified through a tblastn search on the NCBI nucleotide database. An initial search was done using the A4PCP4C5 tri-domain from the nocardicin gene cluster as a query resulting in 25532 hits from 7164 sequences under an E-value threshold of 1E-20. These hits were then filtered to exclude sequences with query coverages of < 75%, yielding 3834. All sequences were translated and parsed for HHHxxxDG motifs. The subsequent 37 sequences were submitted to NRPSPredictor232,33 to identify upstream predicted serine or threonine A domains. After removing duplicate sequences, a final set of 12 remained. From the final set of sequences, only those with high confidence predictions of upstream serine- or threonine-specific A domains were aligned with the nocardicin module 5 condensation domain. Analysis revealed that all sequences in the alignment displayed characteristic motifs predominantly observed in the DCL subtype34,35 of condensation domains despite the fact that nocardicin C5 exhibits LCL binding.
|
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grant AI014937. We are indebted to Drs. D.W. Udwary (bioinformatics), K.A. Moshos, J.W. Labonte (chemistry), and R-f. Li (molecular biology) for advice and discussion. We thank Prof. C.T. Walsh (Harvard University) for the pET29-Sfp expression plasmid, and Drs. I.P. Mortimer for HRMS data and C. Moore for help with difficult NMR acquisitions.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author contributions: C.A.T. and N.M.G developed the hypothesis and designed the study. N.M.G. and D.H.L. performed syntheses and biochemical experiments reported. All authors analyzed and discussed the results. N.M.G and C.A.T prepared the manuscript.
The authors declare no competing financial interests.
Author Information: Reprints and permissions information is available at www.nature.com/reprints. Readers are welcome to comment on the online version of the paper.
References
- 1.Walsh CT, O’Brien RV, Khosla C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew Chem Int Ed. 2013;52:7098–7124. doi: 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banko G, Demain AL, Wolfe S. δ-(L-α-Aminoadipyl)-L-cysteinyl-D-valine synthetase (ACV synthetase): A Multifunctional Enzyme with Broad Substrate Specificity for the Synthesis of Penicillin and Cephalosporin Precursors. J Am Chem Soc. 1987;109:2858–2860. [Google Scholar]
- 3.Buller AR, Townsend CA. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc Natl Acad Sci USA. 2013;110:E653–61. doi: 10.1073/pnas.1221050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galletti P, Giacomini D. Monocyclic β-lactams: new structures for new biological activities. Curr Med Chem. 2011;18:4265–4283. doi: 10.2174/092986711797200480. [DOI] [PubMed] [Google Scholar]
- 5.Hamad B. The antibiotics market. Nat Rev Drug Discov. 2010;9:675–676. doi: 10.1038/nrd3267. [DOI] [PubMed] [Google Scholar]
- 6.Demain AL. Antibiotics: Natural products essential to human health. Med Res Rev. 2009;29:821–842. doi: 10.1002/med.20154. [DOI] [PubMed] [Google Scholar]
- 7.McKenna M. Antibiotic resistance: the last resort. Nature. 2013;499:394–396. doi: 10.1038/499394a. [DOI] [PubMed] [Google Scholar]
- 8.Livermore DM. Has the era of untreatable infections arrived? J Antimicrob Chemother. 2009;64:i29–36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- 9.Burzlaff NI, et al. The reaction cycle of isopenicillin N synthase observed by X-ray diffraction. Nature. 1999;401:721–724. doi: 10.1038/44400. [DOI] [PubMed] [Google Scholar]
- 10.Bachmann BO, Li R, Townsend CA. β-Lactam synthetase: a new biosynthetic enzyme. Proc Natl Acad Sci USA. 1998;95:9082–9086. doi: 10.1073/pnas.95.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freeman MF, Moshos KA, Bodner MJ, Li R, Townsend CA. Four enzymes define the incorporation of coenzyme A in thienamycin biosynthesis. Proc Natl Acad Sci USA. 2008;105:11128–11133. doi: 10.1073/pnas.0804500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidsen JM, Townsend CA. In vivo characterization of nonribosomal peptide synthetases NocA and NocB in the biosynthesis of nocardicin A. Chem Biol. 2012;19:297–306. doi: 10.1016/j.chembiol.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davidsen JM, Bartley DM, Townsend CA. Non-ribosomal propeptide precursor in nocardicin A biosynthesis predicted from adenylation domain specificity dependent on the MbtH family protein NocI. J Am Chem Soc. 2013;135:1749–1759. doi: 10.1021/ja307710d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reeve AM, Breazeale SD, Townsend CA. Purification, characterization, and cloning of an S-adenosylmethionine-dependent 3-amino-3-carboxypropyltransferase in nocardicin biosynthesis. J Biol Chem. 1998;273:30695–30703. doi: 10.1074/jbc.273.46.30695. [DOI] [PubMed] [Google Scholar]
- 15.Townsend CA, Brown AM. Nocardicin A: biosynthetic experiments with amino acid precursors. J Am Chem Soc. 1983;105:913–918. [Google Scholar]
- 16.Hubbard BK, Thomas MG, Walsh CT. Biosynthesis of L-p-hydroxyphenylglycine, a non-proteinogenic amino acid constituent of peptide antibiotics. Chem Biol. 2000;7:931–942. doi: 10.1016/s1074-5521(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 17.Townsend CA, Wilson BA. The role of nocardicin G in nocardicin A biosynthesis. J Am Chem Soc. 1988;110:3320–3321. [Google Scholar]
- 18.Gaudelli NM, Townsend CA. Epimerization and substrate gating by a TE domain in β-lactam antibiotic biosynthesis. Nat Chem Biol. 2014;10:251–258. doi: 10.1038/nchembio.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh CT, et al. Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr Opin Struct Biol. 2001;5:525–534. doi: 10.1016/s1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 20.Salituro GM, Townsend CA. Total Syntheses of (−)-Nocardicins A–G: a Biogenetic Approach. J Am Chem Soc. 1990;112:760–770. [Google Scholar]
- 21.Gunsior M, et al. The biosynthetic gene cluster for a monocyclic β-Lactam antibiotic, nocardicin A. Chem Biol. 2004;11:927–938. doi: 10.1016/j.chembiol.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Belshaw PJ. Aminoacyl-CoAs as probes of condensation domain selectivity in nonribosomal peptide synthesis. Science. 1999;284:486–489. doi: 10.1126/science.284.5413.486. [DOI] [PubMed] [Google Scholar]
- 23.Quadri LEN. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 24.Roche ED, Walsh CT. Dissection of the EntF condensation domain boundary and active site residues in nonribosomal peptide synthesis. Biochemistry. 2003;42:1334–1344. doi: 10.1021/bi026867m. [DOI] [PubMed] [Google Scholar]
- 25.Townsend CA, Brown AM, Nguyen LT. Nocardicin A: stereochemical and biomimetic studies of monocyclic β-lactam formation. J Am Chem Soc. 1983;105:919–927. [Google Scholar]
- 26.Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure. 2007;15:781–792. doi: 10.1016/j.str.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 27.Tahlan K, Jensen SE. Origins of the β-lactam rings in natural products. J Antibiot. 2013;66:401–410. doi: 10.1038/ja.2013.24. [DOI] [PubMed] [Google Scholar]
- 28.Sattely ES, Walsh CT. A latent oxazoline electrophile for N-O-C bond formation in pseudomonine biosynthesis. J Am Chem Soc. 2008;130:12282–12284. doi: 10.1021/ja804499r. [DOI] [PubMed] [Google Scholar]
- 29.Silverman RB. The Organic Chemistry of Enzyme-Catalyzed Reactions. Academic Press; San Diego: 2002. [Google Scholar]
- 30.Arnison PG, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for Methods and Extended Data figure legends
- 31.Ehmann DE, Trauger JW, Stachelhaus T, Walsh CT. Aminoacyl-SNACs as small-molecule substrates for the condensation domains of nonribosomal peptide synthetases. Chem Biol. 2000;7:765–772. doi: 10.1016/s1074-5521(00)00022-3. [DOI] [PubMed] [Google Scholar]
- 32.Röttig M, et al. NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Research. 2011;39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs) Nucleic Acids Research. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clugston SL, Sieber SA, Marahiel MA, Walsh CT. Chirality of peptide bond-forming condensation domains in nonribosomal peptide synthetases: the C5 domain of tyrocidine synthetase is a (D)C(L) catalyst. Biochemistry. 2003;42:12095–12104. doi: 10.1021/bi035090+. [DOI] [PubMed] [Google Scholar]
- 35.Rausch C, Hoof I, Weber T, Wohlleben W, Huson DH. Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol Biol. 2007;7:78. doi: 10.1186/1471-2148-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.