

Abstract
During last decades, changes have been observed in the frequency of different histological subtypes of lung cancer - one of the most common causes of morbidity and mortality - with a declining proportion of squamous cell carcinomas and an increasing proportion of adenocarcinomas, particularly in developed countries. This suggests the emergence of new etiological factors and mechanisms including those defining the lung microenvironment promoting tumor growth. Assuming that the lung is the main portal of entry for broadly used nanomaterials and their established pro-inflammatory propensities, we hypothesized that nanomaterials may contribute to changes facilitating tumor growth. Here we report that an acute exposure to single-walled carbon nanotubes (SWCNT) induces recruitment and accumulation of lung-associated myeloid-derived suppressor cells (MDSC) and MDSC-derived production of TGF-β resulting in up-regulated tumor burden in the lung. The production of TGF-β by MDSC requires their interaction with both SWCNT and tumor cells. We conclude that pulmonary exposure to SWCNT favors the formation of a niche that supports ingrowth of lung carcinoma in vivo via activation of TGF-β production by SWCNT-attracted and pre-sensitized MDSC.
Keywords: lung carcinoma, Transforming growth factor beta, myeloid-derived suppressor cells, single-walled carbon nanotubes, premalignant niche, nanomaterials
Introduction
Currently, lung cancer is one of the most common causes of morbidity and mortality, although in 1912, it was described as “one of the rarest forms of cancer” (1). It accounts for 14% of all cancer diagnoses and up to 30% of all cancer deaths. It is the leading cause of cancer death in the developed world whereby carcinoma of the lung and bronchus is the most impactful compared to the other forms of cancer (2). Etiologically lung cancer is strongly associated with the environmental exposure, with tobacco smoking and industrial air pollution being the major risk factors. Two main types of pulmonary carcinoma related to the exposure to cigarette and industrial smoke are squamous cell carcinoma (SCC) and small cell neuroendocrine carcinoma. These tumors usually develop from bronchial mucosa of conductive airways, where smoke particles deposit and lead to carcinogenesis. With the current efforts towards smoking cessation and a better control of industrial smoke exhausts, one would expect a decline of lung cancer prevalence. Instead, the lung cancer incidence keeps increasing (3). Moreover, although incidence of SCC appears to have decreased, the incidence of another type of lung cancer – adenocarcinoma, has been escalating in industrially developed countries in contrast to still preserved prevalence of SCC in the developing countries and regions (4, 5). This tumor accounts for up to 50% of newly diagnosed lung cancer cases and the reasons for its recently increased incidence remain poorly defined. Although the most frequently observed histological type of lung cancer among smokers is SCC, among never smokers, adenocarcinomas are more frequent than SCC and comprise the majority of tumors (6). Over 75% of these malignancies develop from the deep portions of the lung (the acinar structures).
These epidemiological data suggest that entirely new, possibly environmental, factors got engaged in pulmonary carcinogenesis. One of possible candidates are nanoparticles (NP) with an estimated ~1,300 kinds of nanomaterials currently either in use or in testing for potential commercial applications ranging from electronic (e.g., advanced memory chips) to industrial (e.g., coatings or composites) to biomedical fields (e.g., drug delivery systems, diagnostics) (7). While the risk of adverse effects of nanomaterials on the human body has still to be investigated (8), their ability to induce robust inflammatory response and fibrosis have been firmly established (9-11). The highest risk arises from occupational exposure via chronic inhalation of NP. For instance, carbon nanotubes (CNT) could be released as dust into the atmosphere during disposal or lower temperature incineration of lithium-ion secondary batteries and synthetic textiles (12). It was estimated that there could be between 20,000 to 100,000 natural and man-made NP per cubic centimeter in the everyday environment (13). Analysis of indoor and outdoor environmental NP revealed that their most significant fraction were carbonaceous NP, particularly CNT (14). Lung is the major entry portal and target organ for airborne CNT which can migrate into the alveolar interstitial compartment of the lung (9, 15). In contrast to tobacco smoke particulates, CNT - due to their small size - can deposit deep in the lung and even reach the pleura (16). In spite of several enzymatic pathways for their biodegradation by inflammatory cells (17, 18), CNT are relatively biopersistent (19). Tragically, among 60,000-70,000 exposedresponders during the World Trade Center (WTC) disaster, generated as a result of the combustion of fuel in the presence of carbon and metals, CNT were found in the lungs of WTC patients suffering from pulmonary diseases (20).
Recently, we have demonstrated that a single pulmonary exposure to single-walled CNT (SWCNT) accelerated growth of lung cancer in mice (21). We further uncovered that depletion of CNT-induced accumulation of myeloid-derived suppressor cells (MDSC) prevented accelerated growth of lung carcinoma. However, the key question about molecular mechanisms of this phenomenon has not been established. Based on bioinformatics analysis of previously obtained proteomics (22) and transcriptomics data as well as existing known factors of MDSC recruitment/function and tumor initiation/growth/progression/metastasis, we hypothesized that TGF-β is the most likely candidate for: (a) nanotubes-induced changes in the lung, (b) MDSC-mediated support of tumor growth, and (c) MDSC functioning in the tumor microenvironment. Therefore, we examined the role of aberrant TGF-β signaling on increased homing of MDSC and accelerated tumor progression upon SWCNT exposure.We report that CNT up-regulate TGF-β production by tumor-activated MDSC and support immunosuppressive protumorigenic microenvironment in the lungs of CNT-exposed animals. Consequently, TGF-β-deficiency completely abrogated the CNT-induced acceleration of cancer growth in the lung. We provide evidence that CNT are required for upregulated production of TGF-β by MDSC induced by tumor cells. In turn, TGF-β produced by activated MDSC was responsible for suppression of T cell activation and proliferation. Overall, our results identify the airborne SWCNT as a potential risk factor of accelerated progression of lung carcinoma in vivo and uncovered immunological mechanisms underlying this effect.
Materials and Methods
Animals
Pathogen-free C57BL6/J mice (7–8 wk old) and Tgfb1tm1Doe/J mice from Jackson Labs (Bar Harbor, ME, USA) were individually housed and acclimated for 2 weeks. Animals were supplied with water and food ad libitum and housed under controlled light, temperature and humidity conditions. All animal studies were conducted under a protocol approved by the Institutional Animal Care and Use Committee.
Preparation of SWCNT
SWCNT (CNI Inc., Houston, TX) were produced by the high pressure CO disproportionation process (HiPco) technique employing CO in a continuous-flow gas phase as the carbon feedstock and Fe(CO)5 as the iron-containing catalyst precursor, and purified by acid treatment to remove metal contaminates. Chemical analysis trace metal (iron) in SWCNT was performed at the Chemical Exposure and Monitoring Branch (DART/NIOSH, Cincinnati, OH) using nitric acid dissolution and inductively coupled plasma-atomic emission spectrometry (ICP-AES) as described earlier (21). The mean diameter and surface area of SWCNT were 1-4 nm and 1040 m2/g. Surface area was determined by Brunauer, Emmett and Teller (BET) analysis, and diameter and length was measured by transmission electron microscopy (TEM). The chemical cutting of SWCNT was performed as reported previously (21). TEM determined the length distribution: 228 ± 77 nm. Atomic force microscopy and diffuse reflectance infrared Fourier Transform spectroscopy were utilized to determine structure and purity of prepared SWCNT as described (21). Stock suspensions (1mg/ml) were prepared before each experiment in PBS and pH was adjusted to 7.0; suspensions were sonicated for 5 min and sterilized by autoclaving. Stock suspensions were diluted to achieve required concentrations and sonicated (three one-min cycles) before use.
Tumor cells, cell lines and experimental procedures
Lewis lung carcinoma cells (LLC) and BEAS-2B cells (human bronchial epithelial cell line), were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). LLC cells were maintained in RPMI 1640 medium supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, 10% heat-inactivated FBS, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate (Invitrogen Life Technologies, Inc, Grand Island, NY, USA). BEAS-2B cells were cultured in DMEM (5% FBS) and exposed to 0.06, 0.12, 0.24 mg/ml of SWCNT for 18h.
Wild type (WT) and TGF-β-deficient mice were exposed to CNT by pharyngeal aspiration (21), shown to induce alterations in the lungs similar to those caused by inhalation in special chambers (9). Briefly, after anesthesia with ketamine/xylazine (62.5 and 2.5 mg/kg s.c.), each mouse was placed on a board in a near-vertical position and a suspension of CNT (80 μg/mouse in saline) was placed posterior in the throat to be aspirated into the lungs. Seven-eight animals per group were utilized for the in vivo studies and all experiments were independently repeated at least three times. In control groups, both WT and TGF-β-deficient mice were exposed to a single aspiration of PBS.
Forty eight hours after CNT or PBS exposure, mice were inoculated with 3 × 105 LLC cells (300 μl PBS) via the tail vein. Twenty one days later, the animals were sacrificed and the number of visible pulmonary metastases was determined using a dissection microscope. Lungs were weighted, and right lobes were fixed in 10% formaldehyde for histopathology evaluation of H&E stained specimens (21).
Evaluation of MDSC
For pulmonary MDSC isolation and analysis (48 h after CNT exposure), mouse lungs were dispersed using 2% collagenase A and 0.75% DNAse I (Roche Diagnostics GmbH, Mannheim, Germany) in RPMI 1640 medium supplemented with 10% FBS at 20°C for 1 h. Spleens were harvested, grounded, and filtered through a 70 μm cell strainer. Red blood cells were lysed with lysing buffer (155 mM NH4Cl in 10 mM Tris-HCl buffer pH 7.5, 25°C) for 3 min. After RBC lysis, cells were washed, labeled with anti-CD11b, anti-Ly6G, anti-Ly6C, anti-Gr-1 and anti-CD45 antibodies (Biolegend Inc., San Diego, CA, USA) directly conjugated to FITC, PE, PE/Cy7, APC/Cy7 or Alexa700, and analyzed by flow cytometry (BD LSR II instrument, BD Biosciences, San Jose, CA, USA). Data on MDSC counts in the lung and spleen are presented as the percentage of total cells and CD45+ cells in the tested tissue.
CD11b+ Gr-1+ MDSC were isolated from the lungs of WT and TGF-β-deficient mice exposed to SWCNT or saline by magnetic cell sorting using a mouse MDSC Isolation Kit (MACS, Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s instructions. Syngeneic T cells were isolated from the spleens of WT mice using T cell enrichment columns and activated with ConA (5 h, 2.5 μg/ml, Sigma, St. Louis, MO, USA) for 24 h. For functional studies, isolated MDSC were co-cultured with pre-activated syngeneic splenic T cells (1:5 ratio) for 48 h and the levels of IL-2 production by T cells were assessed by ELISA (R&D Systems Inc., Minneapolis, MN). T cell proliferation was measured by uptake of 3H-thymidine (1 μCi/well, 5 Ci/mmol; DuPont-NEN, Boston, MA, USA) pulsed for 16-18 h after two days in culture. Cells were harvested on GF/C glass fiber filters (Whatman Intl. Ltd, Maidstone, UK) using MACH III microwell harvester (Tomtec, Hamden, CT, USA). 3H-thymidine incorporation was determined on MicroBeta TRILUX liquid scintillation counter (WALLAC, Finland) and expressed as count per minute (cpm). Alterations of T cell proliferation by MDSC are shown as Index of Stimulation (IS) calculated as a ratio of treated to non-treated (control) T cells.
Production of TGF-β by MDSC was determined by assessing the levels of TGF-β1 in cell-free supernatants by ELISA (R&D Systems, Minneapolis, MN, USA).
Bioinformatics based identification of CNT-induced and MDSC-mediated factors facilitating accelerated growth of lung cancer. The list of mediators that play a key role in MDSC recruitment/function was created based on the published data (23, 24). The list of genes encoding cytokines, chemokines and growth factors associated with tumor initiation/progression/growth/ metastasis in lung cancer was generated through the use of IPA software tool (Ingenuity® Systems, www.ingenuity.com). Finally, the list of inflammatory mediators differentially up-regulated (>1.5 folds in at least two different concentrations) upon SWCNT exposure was composed based on the data obtained from microarray analysis studies of BEAS-2B cells.
High Density Oligonucleotide Array Expression Analysis
Total RNA from BEAS-2B cells cultured with medium or SWCNT were isolated using RNeasy (Qiagen, Valencia, CA). Further sample preparation for microarray analysis was carried out as described by the manufacturer (Affymetrix, Santa Clara, CA). Briefly, total RNA (12 μg) was used for the preparation of double stranded cDNA using an oligonucleotide (dT)24 primer with a T7 RNA polymerase promoter sequence at its 5' end.Following second strand synthesis, a labeled cRNA transcript was generated from the cDNA in an in vitro transcription (IVT) reaction using Enzo BioArray high yield RNA transcript labeling (Enzo Diagnostics Inc., Farmingdale, NY).The labeled antisense cRNA was purified using RNeasy (Qiagen) and each cRNA sample was fragmented (94°C for 35 min) in the presence of Tris-acetate (40 mM, pH 8.1), potassium acetate (100 mM), and magnesium acetate (30mM).
The fragmented cRNA was mixed with eukaryotic hybridization control oligonucleotides (20X; BioB, BioC, BioD, cre at 1.5, 5, 25 and 100 pM, respectively), control oligonucleotide B2, herring sperm DNA (10 mg/ml), acetylated BSA (50 mg/ml) and hybridization buffer (2X) to form the hybridization cocktail. This cocktail, was heated (99°C for 5 min; and 45°C for 5 min) prior to introduction to microarray cartridge (U133A). Hybridization was allowed to proceed (45°C in rotisserie oven set at 60 rpm) for 16 h. Following hybridization, the arrays were washed and stained using the GeneChip™ fluidics station protocol EukGE-WS2. Following washing and staining, probe arrays were scanned using Gene Array® 2500 scanner (Affymetrix). ImageGene 3.0 software (BioDiscovery Inc.) was used to quantify, correct for background noise and normalize signals from hybridization chip. Thus obtained data were filtered based on signal intensity and detection calls. To identify genes differentially expressed between control and SWCNT exposed groups, we used univariate F-test (after log2 conversion of the mRNA expression abundance, FPKM) between control and exposed groups with a p value cut off of 0.05 and false discovery rate below 0.05 using statistical package R.
Statistical analysis
Results were analyzed using one-way ANOVA and Student unpaired t-test with Welch's correction for unequal variances. All experiments were repeated at least twice and the results are presented as the means ± SEM. P values of < 0.05 were considered to be statistically significant.
Results
Bioinformatics analysis of inflammatory mediators common to CNT exposure and MDSC-driven immuno-suppression in lung cancer
Multifactorial processes of tumor progression and metastasis are strongly associated with chronic inflammation whereby several cytokines, chemokines and growth factors produced by cancer cells as well as modified immune cells, including MDSC, play essential roles. Based on the available literature we used bioinformatics to generate lists of key cytokines, chemokines and growth factors associated with (a) MDSC recruitment and function, (b) initiation, progression, metastasis and growth of lung tumors, and (c) SWCNT exposures as summarized on Figure 1. A total of three factors – macrophage colony stimulating factor (M-CSF), granulocyte/macrophage colony stimulating factor (GM-CSF), andTGF-β - were common to all three selected categories. This analysis suggests that the up-regulation of GM-CSF, M-CSF and TGF-β upon SWCNT exposure may lead to increased accumulation of MDSC in the lung and accelerated growth of lung cancer. To explore mechanisms of CNT in pulmonary metastatic tumor growth, we chose to experimentally assess the effects of TGF-β.
Figure 1. Venn diagram of cytokines, chemokines and growth factors that (I) affect MDSC generation, recruitment, expansion and function, (II) facilitate lung cancer growth, progression and metastasis; (III) are upregulated (>1.5 folds) upon SWCNT exposure.

The list of mediators that play a key role in MDSC recruitment/function was created based on the published data (23, 24). The list of genes encoding cytokines, chemokines and growth factors associated with tumor initiation/progression/growth/ metastasis in lung cancer was generated through the use of IPA software tool (Ingenuity® Systems, www.ingenuity.com). Finally, the list of inflammatory mediators differentially up-regulated (>1.5 folds in at least two different concentrations) upon SWCNT exposure was composed based on the data obtained from microarray analysis studies of BEAS-2B cells.
Accelerated tumor growth induced by SWCNT is blocked in TGF-β-deficient mice
Injection of LLC cells to control WT mice caused multiple bilateral pulmonary tumor nodules by day 21. Histologically, these tumors showed characteristic features of poorly-differentiated non-small cells carcinoma. Exposure of WT mice to SWCNT prior to tumor cell injection resulted in significant acceleration of tumor growth revealed by up to 5-fold increase in the weight of the lungs (the overall tumor burden), up to 2.5-fold elevation of the number of visible pulmonary macro-metastasis, and up to 3-fold increase in the total area of tumor nodules upon histopathological evaluation of the lung tissues (Fig.2, p<0.05). SWCNT did not change the typical morphological features of tumor, but accelerated tumor growth and the appearance of intratumoral necrosis zones, associated with rapid tumor growth.
Figure 2. TGF-β deficiency aborts SWCNT-induced accelerated growth of lung carcinoma.

Wild type (WT) and TGF-β-deficient (TGF-β def) mice received SWCNT by pharyngeal aspiration 48 h prior to i.v. tumor cell injection and sacrificed 3 weeks later. The lungs were collected for determining the weight (a), visualization of macroscopic tumor nodules (b), and histochemical analysis of micrometastases after H&E staining (c). The average weight of the lungs, the number of tumor nodules, and the average collective areas of lung carcinoma on tissue slides were calculated from 3 independent experiments (5-6 mice/group in each) and are shown for LLC and CNT/LLC groups in both WT and TGF-β-deficient mice. LLC, mice received a single aspiration of PBS and 48 h later were injected i.v. with LLC tumor cells; CNT/LLC, mice were pre-treated with SWCNT 48 h prior to administration of LLC cells. *, p<0.05 (Student t-test, N=3).
In control TGF-β-deficient mice, injection of LLC cells led to the appearance of pulmonary metastases, similar in size and morphology to those in WT mice (Fig.2a, b, c). However, the effect of CNT on tumor growth was completely abrogated in TGF-β-deficient animals: neither the average lung weight (Fig.2a), nor the numbers of detectable tumor nodules (Fig.2b) were altered in TGF-β-deficient mice after exposure to CNT. Histopathological examination of H&E stained tissue specimens confirmed the absence of difference in the total area of tumor nodules between CNT-exposed and saline-exposed TGF-β-deficient mice receiving tumor cells after exposure (Fig.2c). These data indicate that exposure of SWCNT promoted establishment and growth of lung carcinoma in mice in vivo in a TGF-β-dependent manner.
TGF-β deficiency does not abrogate acute accumulation of MDSC in the lung after a single pulmonary exposure to SWCNT
Because depletion of MDSC induced by CNT exposure prior to administration of tumor cells completely abrogated increase in lung cancer growth (21), it is likely that MDSC may form the premalignant niches supporting initial survival and growth of tumor cells in the lungs. Assuming that MDSC contribute to TGF-β-mediated metastasis (25), it is possible that CNT may induce early MDSC accumulation in TGF-β-deficient mice. To test this, we determined the accumulation of MDSC in the lungs 48 h after SWCNT aspiration because the elimination of this acute MDSC response in TGF-β-deficient animals might explain the abrogation of the tumor-promoting activity of CNT in Tgfb1tm1Doe mice shown in Figure 2. However, the results showed that TGF-β is not involved in the initial recruitment of MDSC in the lungs and lymphoid tissues by acute SWCNT exposure (Fig.3). In fact, similar to WT mice (21), SWCNT significantly (p<0.05) up-regulated homing of granulocytic MDSC in lymphoid and non-lymphoid tissues in TFG-β-deficient animals: elevated up to 3-fold numbers of CD11b+Ly6G+Ly6Clow MDSC were detected in the lungs and spleens of SWCNT-treated tumor-free animals 48 h after CNT administration (Fig.3a,b). These data indicate that TGF-β was not involved in SWCNT-induced recruitment of MDSC to the lungs to form the premalignant niches. Thus, TGF-β plays a different role – possibly participating in MDSC functions facilitating CNT-induced enhanced growth of lung carcinoma.
Figure 3. TGF-β deficiency does not abrogate acute accumulation of MDSC in the lung and spleen after a single pulmonary exposure to SWCNT.

Wild type (WT) and Tgfb1tm1Doe mice received SWCNT by pharyngeal inhalation and the levels of monocytic CD11b+Ly6GnegLy6Chigh (area 1) and granulocytic CD11b+Ly6G+Ly6Clow/neg (area 2) MDSC were assessed by flow cytometry in the lymphoid tissues and lungs. The results of flow cytometry analysis of MDSC accumulation in tumor-free Tgfb1tm1Doe mice 48h after SWCNT aspiration are shown from a representative experiment (a) (CD11b+ gated cells are shown). Statistical analysis of data from 3 independent experiments (5-6 mice/group in each) is shown as the mean ± SEM and demonstrates accumulation of polymorphonuclear MDSC in different tissues in tumor-free mice 48 h after SWCNT aspiration. *, p<0.05 (Student t-test, N=3) (b). Control, mice received a single aspiration of PBS. CNT, mice received SWCNT once by pharyngeal inhalation.
SWCNT pre-sensitize MDSC to tumor-induced activation of the TGF-β-mediated immune suppressive activity
One of the potential – although not fully characterized – immunosuppressive mechanisms of MDSC is the production of TGF-β (26, 27). To assess the role of MDSC-derived TGF-β in CNT-induced MDSC-mediated immunosuppressive premalignant niche in the lung, pulmonary MDSC were isolated from control and SWCNT-exposed WT and Tgfb1tm1Doe mice, treated with control and LLC-conditioned media, co-cultured with pre-activated syngeneic T cells, and then T cell activity was assessed by IL-2 production (Fig. 4a). Analysis of MDSC-mediated inhibition of T cell activation (Fig. 4b) reveled that only MDSC harvested from WT SWCNT-exposed mice, but not from saline-exposed mice, and pre-incubated with tumor-conditioned medium were able to suppress T cells ex vivo up to 2-fold (p<0.05). Importantly, the immunosuppressive effect of MDSC was abrogated if they were isolated from TGF-β-deficient mice. These results suggest that MDSC exposed to the SWCNT-modified lung microenvironment for even less than 48 h in vivo are sensitive to lung carcinoma-induced emergence of the immunosuppressive potential mediated by TGF-β.
Figure 4. TGF-β mediates inhibition of T cells by lung MDSC isolated from SWCNT-exposed mice and treated with lung cancer-derived factors in vitro.

Experimental protocol (a): WT and Tgfb1tm1Doe mice received SWCNT or saline by pharyngeal inhalation and 48 h later MDSC were isolated from the lungs as described in Materials and Methods. MDSC were then treated with LLC-conditioned medium and control medium for 24 h, washed and co-cultured with ConA pre-activated syngeneic T cells. Inhibition of T cell activation was determined in 48 h by assessing the levels of IL-2 in cell-free supernatants by ELISA (b). The results are shown as the mean ± SEM (triplicate measurements for each mouse, 5-6 mouse/group, two independent experiments). *, p<0.05 versus all other groups (one-way ANOVA). Control (cntr), MDSC from saline-treated mice; CNT, MDSC from SWCNT exposed mice; Tum, MDSC treated with LLC-conditioned medium; T cells, control T cells cultured w/o MDSC.
Although these results provide strong evidence that tumor-derived factors play a key role in activating the immunosuppressive phenotype of CNT-pre-sensitized MDSC, they were based on the in vitro treatment of MDSC with the LLC-conditioned medium. To explore the in vivo role of tumor cells in activating pulmonary MDSC, we further isolated MDSC from saline- and SWCNT- exposed mice bearing lung cancer. Tumor-free mice served as a control. The design and results of these studies are shown in Figure 5 and demonstrate that in vivo generated lung MDSC required in vivo interaction with tumor cells to acquire an immunosuppressive phenotype assessed by their ability to suppress proliferation of syngeneic T cells. Furthermore, the ability of MDSC to inhibit T cells was aborted if pulmonary MDSC were harvested from TGF-β-deficient animals (Fig.5, p<0.05). Together, the in vitro and in vivo data suggest that MDSC attracted to the lungs by SWCNT exposure are more sensitive to the activation in the lung cancer microenvironment than control pulmonary MDSC, and that the ability of lung MDSC to inhibit T cells is mediated by TGF-β.
Figure 5. SWCNT predispose pulmonary tumor-associated MDSC to inhibit T cell proliferation via TGF-β1 pathways in vivo.

Experimental protocol (a): WT and Tgfb1tm1Doe mice received SWCNT or saline by pharyngeal inhalation and LLC cells i.v. 48 h later. MDSC were isolated from the lungs 2 weeks later and co-cultured with ConA pre-activated syngeneic T cells. Inhibition of T cell proliferation was determined in 48 h by 3H-thymidine incorporation and expressed as Index of Stimulation (b). The results are shown as the mean ± SEM (triplicate measurements for each mouse, 5 mouse/group, N=2). *, p<0.05 versus all other groups (one-way ANOVA). Control (cntr), saline inhalation; CNT, SWCNT inhalation; Tum, injection of LLC; T cells, control T cells cultured w/o MDSC.
This encouraged us to further examine direct effects of CNT on MDSC and TGF-β production by MDSC. To this end, MDSC were directly treated with SWCNT in vitro and TGF-β1 expression was evaluated. We found that, although CNT and LLC did not markedly increased TFG-β levels in MDSC cultures (189.2±19.6 ng/ml vs 201.1±18.2 ng/ml and 229.4±24.4 ng/ml in control, CNT-treated and LLC-treated cells, respectively), concomitant treatment of MDSC with SWCNT and LLC-conditioned medium significantly increased TGF-β in MDSC cultures (313.7±21.2 ng/ml, p<0.05) (Figure 6). Thus, SWCNT augment TGF-β expression by MDSC induced by lung cancer cells.
Figure 6. Exposure of MDSC to SWCNT and tumor-derived factors up-regulates TGF-β expression.
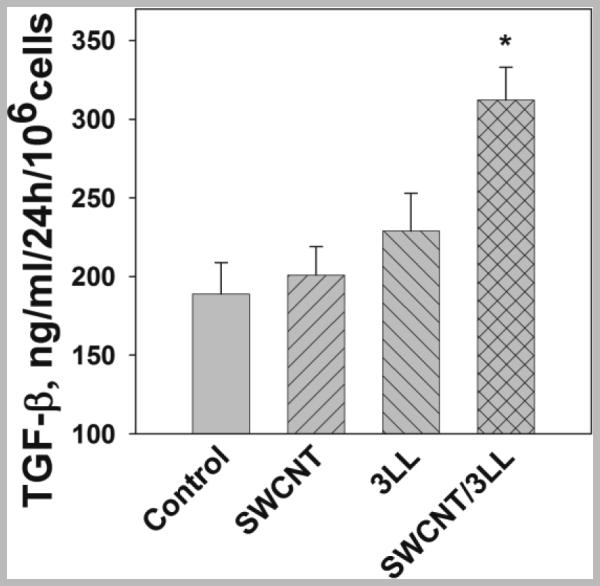
MDSC were isolated from the bone marrow and cultured with saline (cntr), SWCNT (6.25 μg/ml), LLC conditioned medium (25% v/v) overnight. MDSC were then collected, washed and cultured for 24 h. The levels of TGF-β1 in cell-free medium were assessed by ELISA.*, p<0.05 vs cntr (ANOVA, triplicate wells, N=3).
Overall, we established a potential mechanism of CNT-induced enhanced growth of lung carcinoma, which is mediated by an immediate recruitment and accumulation of MDSC in the lungs and formation of specific microenvironmental niches that support tumor cell growth. Although TGF-β was not involved in the initial recruitment of MDSC to the lung by SWCNT, its deficiency completely blocked accelerated tumor progression in the lungs after SWCNT exposure. CNT-triggered changes in the lung microenvironment encouraged accumulation of MDSC that were sensitive to tumor-induced activation leading to TGF-β-dependent suppression of T cells.
Discussion
A huge diversity of carbonaceous nanomaterials is on the way to multiple industrial and biomedical applications; therefore, understanding their toxicity is critical for their commercialization (28). Among those, CNT have been reported to have several adverse effects on cellular and organismal levels, which may be predictive of detrimental human health effects upon exposures (29-31). For instance, in vitro studies indicate that single- and multi-walled CNT may be genotoxic, demonstrating a potential carcinogenic potential. Chronic exposure to CNT was shown to induce DNA damage and increase mutation frequency in mouse embryonic cells and human epithelial cells (32, 33). CNT induce mitotic abnormality with one rather than two mitotic spindle poles, a potential mechanism the impaired cell division (34). Many studies have reported that CNT can induce apoptosis, DNA damage, and activation of major regulatory pathways - MAPKs, AP-1, NF-κB, and Akt - all of which recapitulate key molecular events involved in asbestos-induced lung cancer (31, 35). Moreover, Pacurari et al. reported that MWCNT exposure affects a subset of lung cancer prognostic biomarkers in mouse lungs (36) and Wang et al. showed that chronic exposure to SWCNT in vitro caused malignant transformation of human lung epithelial cells (37).
In the context of tumorogenesis and tumor/host relationships, important are the effects of CNT on pulmonary microenvironment. Several animal studies demonstrated that exposure to CNT induced acute and chronic inflammatory response, chronic granulomatous reaction and substantial interstitial fibrosis in the lungs (9, 11, 16, 29, 30). Fibrosis may be viewed as a precursor to lung cancer. In fact, pulmonary scarring and lung cancer were found to occur to the same lung regions and extend over time, indicating that lung cancer could originate from lung scarring (38). Overall, there are two potential pro-tumorigenic mechanisms associated with CNT exposure: i) direct carcinogenic effects on pulmonary cells and ii) alteration of inflammatory pulmonary milieu making it conducive to cancer development. Surprisingly, the second mechanism - acute CNT effects on host-tumor interactions and growth of tumors has not been yet considered.
Previously we have reported that CNT promote tumor establishment and lung tumor growth by altering the tissue microenvironment, specifically by facilitating early MDSC accumulation in the lungs, as depletion of MDSC abrogated pro-tumor effect of acute exposure to CNT (21). It is likely that inflammatory MDSC induced by CNT support the initial tumor cell homing in the lung and then become polarized into tumor-associated MDSC. The latter are known to block the development of antitumor immunity hence facilitate tumor growth by producing specific growth factors and cytokines that also stimulate intratumoral neoangiogenesis (39). However, the specific mechanisms through which SWCNT affect MDSC-tumor cell interactions and induce immunosuppressive activity have not been deciphered. Bioinformatics analysis of previously published data revealed that 3 growth factors, GM-CSF, G-CSF, and TGF-β are common to CNT, formation, progression of lung cancer, and accumulation and immunosuppressive function of MDSC (Fig. 1). These growth factors have been shown to be expressed in tumor lesions (40). While M-CSF stimulates the differentiation of granulocytes or macrophages (41), GM-CSF and TGF-β have been reported to have a strong impact on MDSC expansion (40, 42, 43). Thus it is possible that all three of the factors - GM-CSF, M-CSF and TGF-β - are involved in the accelerated tumor growth after SWCNT exposure. Notably, TGF-β has been identified as a metastasis promoter (44). Given a recently reported up-regulation of TGF-β in the lungs of mice after chronic inhalation of multiwalled CNT (45), we speculated that TGF-β could be responsible for the increased homing of MDSC and accelerated tumor progression responses upon SWCNT exposure.
Here we uncovered the key function of TGF-β in SWCNT-induced and MDSC-mediated formation of premalignant niches and acceleration of lung carcinoma establishment in vivo (Fig. 2). TGF-β is involved in many biological processes, including cell proliferation, differentiation, apoptosis, embryogenesis as well as cancer (46, 47). Alterations in TGF-β signaling have significant effects on tumor initiation and progression (46) and the lack of TGF-β signaling may promote collective cancer cell invasion and tumor progression (48). One of the identified mechanisms of TGF-β-dependent tumor promotion is the recruitment of MDSC to the tumor microenvironment (25). TGF-β is also an important suppressive cytokine produced by MDSC for inhibiting the antitumor immunity (26). TGF-β may also suppress VEGF-mediated angiogenesis in cancer metastasis (49). Several reports showed that blocking of TGF-β by an antibody or soluble receptor inhibited tumor growth (50-52). Moreover, TGF-β can suppress antitumor immune responses and create a local environment of immune tolerance (46). Elevated levels of serum TGF-β1 and its association with metastases were independently detected in patients with different types of cancer (53). Further, in lung cancer, increased expression of TGF-β was correlated with advanced stages of disease malignancy, metastases with decreased survival rates (54). There are several mechanisms through which TGF-β can accelerate growth of lung carcinoma. Up-regulated levels of TGF-β upon SWCNT exposure could (a) participate directly in recruiting/expanding MDSC, (b) contribute to the suppression of T-cell responses by activating/differentiating MDSC to secrete IL-10 and TGF-β, (c) contribute to indirect suppression by inducing Tregs through membrane binding on MDSC (39, 55). However, the mechanisms of induction of TGF-β expression and the source of this cytokine in tumor models have not been understood, except for the fact that tumor cells are known to produce this cytokine.
Here, we show that TGF-β is not involved in the initial recruitment and accumulation of MDSC to the lungs induced by SWCNT (Fig. 3), although TGF-β deficiency abrogates the pro-tumor effect of CNT mediated by MDSC. This suggests that TGF-β released by MDSC in the lung may be involved in CNT-induced up-regulation of tumor growth. Experimentally, this has been examined using the ex vivo model systems where MDSC isolated from control and CNT-exposed mice were treated with LLC-conditioned medium and then tested for their ability to suppress proliferation of T lymphocytes. We demonstrated that the in vivo exposure to SWCNT rendered lung MDSC sensitive to tumor-induced expression of TGF-β, which was responsible for the inhibition of T cell activity by MDSC. This was further supported by the results obtained in another ex vivo model where MDSC were harvested from tumor-bearing CNT-exposed mice and shown to inhibit T cell proliferation in TGF-β-dependent manner. In our tumor model, although LLC cells can produce TGF-β, this source of the cytokine cannot play a critical role in the negative regulation of tumor immunosurveillance because LLC-bearing TGF-β deficient mice were resistant to SWCNT-induced acceleration of tumor establishment. Considering that TGF-β produced by CD11b+Gr-1+ cells is necessary for down-regulation of cytotoxic T cell-mediated tumor immunosurveillance (27) and our data demonstrating direct effect of CNT on tumor-induced TGF-β production by MDSC, our combined findings uncovered a new pathway through which acute pulmonary exposure to CNT changes the interaction between myeloid regulatory cells and malignant cells to benefit tumor growth and metastasis in vivo.
Acknowledgements
We would like to thank Mariana T. Farcas for technical support. This work was supported in part by NIOSH OH008282, NTRC 927ZJHF and 939011K, NIEHS R01ES019304, NIH RO1 CA154369, NANOSOLUTIONS FP7 309329.
Footnotes
Conflict of Interest: The Authors declare that there is no conflict of interest.
Disclaimer: The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
REFERENCES
- 1.Adler I. Primary malignant growths of the lung and bronchi. Longmans, Green and Company; New York: 1912. [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Dresler C. The changing epidemic of lung cancer and occupational and environmental risk factors. Thoracic surgery clinics. 2013;23:113–22. doi: 10.1016/j.thorsurg.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Alberg AJ, Ford JG, Samet JM. Epidemiology of lung cancer: ACCP evidence-based clinical practice guidelines. Chest. (2nd) 2007;132:29S–55S. doi: 10.1378/chest.07-1347. [DOI] [PubMed] [Google Scholar]
- 5.Dey A, Biswas D, Saha SK, Kundu S, Kundu S, Sengupta A. Comparison study of clinicoradiological profile of primary lung cancer cases: an Eastern India experience. Indian journal of cancer. 2012;49:89–95. doi: 10.4103/0019-509X.98930. [DOI] [PubMed] [Google Scholar]
- 6.Samet JM, Avila-Tang E, Boffetta P, Hannan LM, Olivo-Marston S, Thun MJ, et al. Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin Cancer Res. 2009;15:5626–45. doi: 10.1158/1078-0432.CCR-09-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jud C, Clift MJ, Petri-Fink A, Rothen-Rutishauser B. Nanomaterials and the human lung: what is known and what must be deciphered to realise their potential advantages? Swiss medical weekly. 2013;143:w13758. doi: 10.4414/smw.2013.13758. [DOI] [PubMed] [Google Scholar]
- 8.Roco MC. Environmentally responsible development of nanotechnology. Environmental science & technology. 2005;39:106A–12A. doi: 10.1021/es053199u. [DOI] [PubMed] [Google Scholar]
- 9.Shvedova AA, Kisin E, Murray AR, Johnson VJ, Gorelik O, Arepalli S, et al. Inhalation vs. aspiration of single-walled carbon nanotubes in C57BL/6 mice: inflammation, fibrosis, oxidative stress, and mutagenesis. American journal of physiology. 2008;295:L552–65. doi: 10.1152/ajplung.90287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shvedova AA, Kisin ER, Murray AR, Mouithys-Mickalad A, Stadler K, Mason RP, et al. ESR evidence for in vivo formation of free radicals in tissue of mice exposed to single-walled carbon nanotubes. Free radical biology & medicine. 2014;73:154–65. doi: 10.1016/j.freeradbiomed.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Shvedova AA, Yanamala N, Kisin ER, Tkach AV, Murray AR, Hubbs A, et al. Long-term effects of carbon containing engineered nanomaterials and asbestos in the lung: one year postexposure comparisons. American journal of physiology. 2014;306:L170–82. doi: 10.1152/ajplung.00167.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Köhler A, Som C, Helland A, Gottschalk F. Studying the potential release of carbon nanotubes throughout the application lifecycle. Journal of Cleaner Production. 2008;16:927–37. [Google Scholar]
- 13.NGOs” NCB. Nanoparticles In The Atmosphere. 2010 Available from: http://www.nanocap.eu/Flex/Site/Download047b.pdf.
- 14.Murr LE, Garza KM. Natural and anthropogenic environmental nanoparticulates : Their microstructural characterization and respiratory health implications. Atmos Environ. 2009;43:2683–92. [Google Scholar]
- 15.Mercer RR, Scabilloni J, Wang L, Kisin E, Murray AR, Schwegler-Berry D, et al. Alteration of deposition pattern and pulmonary response as a result of improved dispersion of aspirated single-walled carbon nanotubes in a mouse model. American journal of physiology. 2008;294:L87–97. doi: 10.1152/ajplung.00186.2007. [DOI] [PubMed] [Google Scholar]
- 16.Porter DW, Hubbs AF, Mercer RR, Wu N, Wolfarth MG, Sriram K, et al. Mouse pulmonary dose- and time course-responses induced by exposure to multi-walled carbon nanotubes. Toxicology. 2010;269:136–47. doi: 10.1016/j.tox.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 17.Andon FT, Kapralov AA, Yanamala N, Feng W, Baygan A, Chambers BJ, et al. Biodegradation of single-walled carbon nanotubes by eosinophil peroxidase. Small. 2013;9:2721–9. doi: 10.1002/smll.201202508. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kagan VE, Kapralov AA, St Croix CM, Watkins SC, Kisin ER, Kotchey GP, et al. Lung macrophages "digest" carbon nanotubes using a superoxide/peroxynitrite oxidative pathway. ACS nano. 2014;8:5610–21. doi: 10.1021/nn406484b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donaldson K, Murphy FA, Duffin R, Poland CA. Asbestos, carbon nanotubes and the pleural mesothelium: a review of the hypothesis regarding the role of long fibre retention in the parietal pleura, inflammation and mesothelioma. Particle and fibre toxicology. 2010;7:5. doi: 10.1186/1743-8977-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu M, Gordon RE, Herbert R, Padilla M, Moline J, Mendelson D, et al. Case report: Lung disease in World Trade Center responders exposed to dust and smoke: carbon nanotubes found in the lungs of World Trade Center patients and dust samples. Environmental health perspectives. 2010;118:499–504. doi: 10.1289/ehp.0901159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shvedova AA, Tkach AV, Kisin ER, Khaliullin T, Stanley S, Gutkin DW, et al. Carbon Nanotubes Enhance Metastatic Growth of Lung Carcinoma via Up-Regulation of Myeloid-Derived Suppressor Cells. Small. 2013;9:1691–1695. doi: 10.1002/smll.201201470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teeguarden JG, Webb-Robertson BJ, Waters KM, Murray AR, Kisin ER, Varnum SM, et al. Comparative proteomics and pulmonary toxicity of instilled single-walled carbon nanotubes, crocidolite asbestos, and ultrafine carbon black in mice. Toxicol Sci. 2011;120:123–35. doi: 10.1093/toxsci/kfq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ribechini E, Greifenberg V, Sandwick S, Lutz M. Subsets, expansion and activation of myeloid-derived suppressor cells. Med Microbiol Immun. 2010;199:273–81. doi: 10.1007/s00430-010-0151-4. [DOI] [PubMed] [Google Scholar]
- 24.Sevko A, Umansky V. Myeloid-Derived Suppressor Cells Interact with Tumors in Terms of Myelopoiesis, Tumorigenesis and Immunosuppression: Thick as Thieves. J Cancer. 2013;4:3–11. doi: 10.7150/jca.5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182:240–9. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 27.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. The Journal of experimental medicine. 2003;198:1741–52. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker H, Herzberg F, Schulte A, Kolossa-Gehring M. The carcinogenic potential of nanomaterials, their release from products and options for regulating them. International journal of hygiene and environmental health. 2011;214:231–8. doi: 10.1016/j.ijheh.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Lam CW, James JT, McCluskey R, Hunter RL. Pulmonary toxicity of single-wall carbon nanotubes in mice 7 and 90 days after intratracheal instillation. Toxicol Sci. 2004;77:126–34. doi: 10.1093/toxsci/kfg243. [DOI] [PubMed] [Google Scholar]
- 30.Shvedova AA, Kisin ER, Mercer R, Murray AR, Johnson VJ, Potapovich AI, et al. Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice. American journal of physiology. 2005;289:L698–708. doi: 10.1152/ajplung.00084.2005. [DOI] [PubMed] [Google Scholar]
- 31.Pacurari M, Yin XJ, Zhao J, Ding M, Leonard SS, Schwegler-Berry D, et al. Raw single-wall carbon nanotubes induce oxidative stress and activate MAPKs, AP-1, NF-kappaB, and Akt in normal and malignant human mesothelial cells. Environmental health perspectives. 2008;116:1211–7. doi: 10.1289/ehp.10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabet L, Bussy C, Amara N, Setyan A, Grodet A, Rossi MJ, et al. Adverse effects of industrial multiwalled carbon nanotubes on human pulmonary cells. Journal of toxicology and environmental health. 2009;72:60–73. doi: 10.1080/15287390802476991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu L, Chang DW, Dai L, Hong Y. DNA damage induced by multiwalled carbon nanotubes in mouse embryonic stem cells. Nano letters. 2007;7:3592–7. doi: 10.1021/nl071303v. [DOI] [PubMed] [Google Scholar]
- 34.Sargent LM, Hubbs AF, Young SH, Kashon ML, Dinu CZ, Salisbury JL, et al. Single-walled carbon nanotube-induced mitotic disruption. Mutation research. 2011;745:28–37. doi: 10.1016/j.mrgentox.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirano S, Fujitani Y, Furuyama A, Kanno S. Uptake and cytotoxic effects of multi-walled carbon nanotubes in human bronchial epithelial cells. Toxicology and applied pharmacology. 2010;249:8–15. doi: 10.1016/j.taap.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 36.Pacurari M, Qian Y, Porter DW, Wolfarth M, Wan Y, Luo D, et al. Multi-walled carbon nanotube-induced gene expression in the mouse lung: association with lung pathology. Toxicology and applied pharmacology. 2011;255:18–31. doi: 10.1016/j.taap.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Luanpitpong S, Castranova V, Tse W, Lu Y, Pongrakhananon V, et al. Carbon nanotubes induce malignant transformation and tumorigenesis of human lung epithelial cells. Nano letters. 2011;11:2796–803. doi: 10.1021/nl2011214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu YY, Pinsky PF, Caporaso NE, Chatterjee N, Baumgarten M, Langenberg P, et al. Lung cancer risk following detection of pulmonary scarring by chest radiography in the prostate, lung, colorectal, and ovarian cancer screening trial. Archives of internal medicine. 2008;168:2326–32. doi: 10.1001/archinte.168.21.2326. discussion 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature reviews. 2012;12:253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilcox RA. Cancer-Associated Myeloproliferation: Old Association, New Therapeutic Target. Mayo Clin Proc. 2010;85:656–63. doi: 10.4065/mcp.2010.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Developmental and comparative immunology. 2004;28:509–54. doi: 10.1016/j.dci.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Baniyash M. Chronic inflammation, immunosuppression and cancer: new insights and outlook. Seminars in cancer biology. 2006;16:80–8. doi: 10.1016/j.semcancer.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 43.Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1-bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Tr. 2010;123:39–49. doi: 10.1007/s10549-009-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drabsch Y, ten Dijke P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer metastasis reviews. 2012;31:553–68. doi: 10.1007/s10555-012-9375-7. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell LA, Lauer FT, Burchiel SW, McDonald JD. Mechanisms for how inhaled multiwalled carbon nanotubes suppress systemic immune function in mice. Nature nanotechnology. 2009;4:451–6. doi: 10.1038/nnano.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nature reviews Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 47.Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Current opinion in cell biology. 2009;21:166–76. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 48.Matise LA, Palmer TD, Ashby WJ, Nashabi A, Chytil A, Aakre M, et al. Lack of transforming growth factor-beta signaling promotes collective cancer cell invasion through tumor-stromal crosstalk. Breast Cancer Res. 2012;14:R98. doi: 10.1186/bcr3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geng L, Chaudhuri A, Talmon G, Wisecarver JL, Wang J. TGF-Beta suppresses VEGFA-mediated angiogenesis in colon cancer metastasis. PloS one. 2013;8:e59918. doi: 10.1371/journal.pone.0059918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Won J, Kim H, Park EJ, Hong Y, Kim SJ, Yun Y. Tumorigenicity of mouse thymoma is suppressed by soluble type II transforming growth factor beta receptor therapy. Cancer research. 1999;59:1273–7. [PubMed] [Google Scholar]
- 51.Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. The Journal of clinical investigation. 2002;109:1607–15. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. The Journal of clinical investigation. 2002;109:1551–9. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. The lancet oncology. 2013;14:e218–28. doi: 10.1016/S1470-2045(12)70582-X. [DOI] [PubMed] [Google Scholar]
- 54.Hasegawa Y, Takanashi S, Kanehira Y, Tsushima T, Imai T, Okumura K. Transforming growth factor-beta 1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer. 2001;91:964–71. [PubMed] [Google Scholar]
- 55.Poschke I, Kiessling R. On the armament and appearances of human myeloid-derived suppressor cells. Clin Immunol. 2012;144:250–68. doi: 10.1016/j.clim.2012.06.003. [DOI] [PubMed] [Google Scholar]