

Abstract
The Notch pathway plays multiple key roles in tumorigenesis, and its signaling components have therefore aroused great interest as targets for emerging therapies. Here we show that inhibition of Notch, using a soluble receptor Notch1 decoy, unexpectedly caused a remarkable increase in liver metastases from neuroblastoma and breast cancer cells. Increased liver metastases were also seen after treatment with the γ-secretase inhibitor PF-03084014. Transgenic mice with heterozygous loss of Notch1 demonstrated a marked increase in hepatic metastases, indicating that Notch1 signaling acts as metastatic suppressor in the liver microenvironment. Inhibition of DLL1/4 with ligand-specific Notch1 decoys increased sprouting of sinusoidal endothelial cells into micrometastases, thereby supporting early metastatic angiogenic growth. Inhibition of tumor-derived JAG1 signaling activated hepatic stellate cells, increasing their recruitment to vasculature of micrometastases, thereby supporting progression to macrometastases. These results demonstrate that inhibition of Notch causes pathological activation of liver stromal cells, promoting angiogenesis and growth of hepatic metastases. Our findings have potentially serious implications for Notch inhibition therapy.
INTRODUCTION
The four transmembrane Notch receptors and five membrane-bound ligands, DLL1, DLL3, DLL4, JAG1 and JAG2, classically function in development and differentiation, but also play a critical role in cancer (1–4). Aberrant Notch activation was first discovered in T-cell acute lymphoblastic leukemia (1) and later found in a variety of solid tumors (2–5). Notch functions in tumor angiogenesis are also well documented, with DLL4 highly expressed in tumor vasculature (6,7).
Consequently, targeting Notch pathway components is currently a focus of preclinical and clinical research (8–14). Yet the widespread functions and highly pleiotropic nature of Notch raises the possibility of unanticipated effects on host tissues. For example, γ-secretase inhibitors (GSI), which prevent cleavage and activation of Notch receptors, cause serious gastrointestinal toxicity due to induction of goblet cell hyperplasia, a direct result of Notch inhibition (15). DLL4 inhibition in animal studies can cause aberrant activation of endothelial cells (ECs), leading to formation of vascular tumors (16).
Here we show that inhibition of Notch signaling causes a remarkable increase in spontaneous liver metastasis from neuroblastoma and breast cancer cells. Similarly, heterozygous loss of Notch1 in host animals leads to a marked increase in liver metastasis. Our data indicates that this effect is due to decreased Notch activation in liver sinusoidal endothelial cells (SECs) and hepatic stellate cells (HSCs). Our findings demonstrate that perturbing Notch signaling can induce pathological activation of hepatic stromal cells, leading to the growth of metastatic deposits.
MATERIALS and METHODS
Cell culture
The NGP cell line was obtained from Garrett Brodeur, Children’s Hospital of Philadelphia, and authenticated by short tandem repeat profiling. SH-SY5Y and MDA-MB-231 cell lines were obtained from ATCC, BALB/c SECs from CellBiologics, and human HSCs from ScienCell Research.
Lentiviral production and transfection
NGP was stably transfected with pLKO.1 Notch1 shRNA lentiviral plasmid (Sigma-Aldrich) as described (14). For other transfections, lentiviral plasmid pCCL encoding Fc, N11–36-decoy, N11–24-decoy, N11–13-decoy or N110–24-decoy, were co-transfected with other plasmids (pCCL-GFP, pVSVG, pPRE, pRSV-rev) in HEK293T cells by Fugene (Promega).
Animals
Rag2/II2rg double knockout (Rag2−/−, Il2rg−/−) in a C57BL/6JxC57BL/10SgSnAi background (Taconic, model 4111), were crossed to a pan-eGFP expressing mouse (C57BL/6-Tg(CAG-EGFP)1Osb/J, model 003291, Jackson). Resulting F1s were back-crossed to Rag2/II2rg double knockout to obtain Rag2−/−, Il2rg−/−, eGFP+ mice. These were then back-crossed to the Rag2/II2rg double knockout for 9 generations. Notch1+/− mice (17), were crossed with Rag2−/−, Il2rg−/−, eGFP+ mice. Notch1+/−, Rag2−/−, Il2rg−/−, eGFP+/− mice were then backcrossed with Rag2−/−, Il2rg−/−, eGFP+/+ mice to generate Notch1+/−, Rag2−/−, Il2rg−/−, eGFP+/+ mice and Notch1+/+ Rag2−/−, Il2rg−/−, eGFP+/+ control mice.
Tumor xenografts
Procedures were approved by Columbia University IACUC. For intrarenal tumors, 4–6 week old female nude mice (Taconic) were anesthetized with ketamine and xylazine, an incision made at the left flank, and 106 cells injected into the renal parenchyma. For intracardiac liver metastases studies, 105–106 cells were injected into the left cardiac ventricle, and bioluminescence imaging obtained after 20min to monitor distribution of tumor cells.
GSI
18 days after implantation of NGP cells, mice were randomized for GSI treatment. PF-003084014 (provided by Pfizer), formulated in 0.5%methylcellulose (vehicle) was administered orally by gavage at 125mg/kg b.i.d. for 10 days.
Bioluminescence analysis
Mice were injected intraperitoneally with 75mg/kg D-Luciferin (PerkinElmer), anesthetized with isoflurane, and imaged with a Xenogen IVIS200. For survival studies, mice were sacrificed when bioluminescence flux reached a threshold value 6x109 (photons/sec). For ex vivo liver imaging, mice were injected with D-Luciferin, sacrificed and the liver dissected, imaged and bioluminescence measured.
Assessment of liver metastasis
Livers were fixed in 4% paraformaldehyde, paraffin-embedded, sectioned (5μm) at 50μm intervals, and H&E stained. Diameters of metastatic nodules from 3 non-consecutive sections were measured. For quantification by bioluminescence, a liver piece was homogenized in lysis buffer, centrifuged, supernatant mixed with LARII reagent (Promega), and bioluminescence measured with a luminometer and normalized to the liver piece weight.
Circulating tumor cells (CTC)
Blood was collected by cardiac puncture, lysed by centrifugation, supernatant mixed with LARII reagent and bioluminescence measured with a luminometer.
Quantification of vasculature
Vascular parameters were determined as previously described (14,18). For antibodies see Supplementary Table S1.
Migration Assay
HSCs expressing GFP were seeded (1.5x104 cells/well) in the upper chamber of CytoSelect™ 24-Well Cell Migration plate (8μm pore-size, CellBiolabs). NGP cells expressing ligand decoys or Jag1-siRNA transfected were seeded to the upper chamber (1.5x104 cells/well). RPMI1640+10%FBS was added to the lower chamber. After 48hr, migrating HSCs were counted by fluorescence microscopy from 8 random fields from 3 inserts. To verify inhibition of Notch signaling, HSC-GFP cells were FACS sorted and cell lysates immunoblotted for Hey1 and Hes1.
Statistical Analysis
All statistical analysis was performed using Prism5 software (GraphPad). Survival was determined by Log-rank (Mantel-Cox). Data were analyzed for normality by the D’Agostino-Pearson omnibus K2 test. Normal data were analyzed by unpaired t-test or ANOVA with post-hoc analysis by Tukey’s Multiple Comparison test. Non-normal data were analyzed nonparameterically with Mann-Whitney or Kruskal-Wallis test with post-hoc analysis by Dunn’s Multiple comparison test. Bioluminescence data was transformed Y=log(Y), then analyzed by ANOVA, with post-hoc analysis by Tukey’s Multiple Comparison test.
RESULTS
Combined blockade of Notch and VEGF prolongs survival but increases liver metastasis
We previously demonstrated that combining Notch and VEGF blockade disrupted angiogenesis (14), and therefore hypothesized that this would translate into prolonged survival in comparison to VEGF blockade alone. The Notch1 decoy (N1D) is composed of EGF repeats 1–36, and blocks Notch activation by both DLL and JAGGED ligands (13). The neuroblastoma cell line NGP expressing N1D (NGP-N1D) or LacZ (NGP-LacZ) was implanted into the left kidney of nude mice, and treated with the anti-VEGF antibody bevacizumab (BV). We monitored tumor size in vivo using bioluminescence and sacrificed animals at a predetermined photon flux. Combined Notch and VEGF blockade (NGP-N1D+BV) prolonged median survival as compared to BV only (NGP-LacZ+BV)(Fig. 1A, 43 vs. 36 days, P=0.048). There was no difference in the tumor weight between NGP-N1D+BV compared to NGP-LacZ+BV (1.88±0.95 vs. 1.46±0.91 gm, mean ± stddev).
Figure 1. Combined Blockade of Notch and VEGF prolongs survival but increases liver metastasis.

(A) NGP-LacZ (n=30), or NGP-N1D (n=27) were treated with BV twice weekly, and sacrificed at a flux of 6x109 photons/sec. P=0.048, Log-Rank (Mantel-Cox). (B) Bioluminescent images at days 35 and 50. Contralateral signal (red arrows) indicates metastatic disease. (C) Livers from NGP-N1D+BV mice display multiple nodules. (D) Liver metastatic burden was quantified by measuring bioluminescence of liver homogenates, NGP-N1D+BV (n=23), NGP-LacZ+BV (n=19). ***P<0.001. (E) Histologic examination of liver metastases (arrows). Bar, 400μm. (F) Metastatic foci/mm2, NGP-N1D+BV (n=18) NGP-LacZ+BV (n=23). **P<0.01. (G) Diameter of liver metastasis, with each point on the scatter plot an individual lesion. NGP-LacZ+BV (n=79, from 23 tumors); NGP-N1D+BV (n=254, from 18 tumors). ****P<0.0001. (H) Quantification of CTC. NGP-LacZ+BV (n=14), NGP-N1D+BV (n=22), P=n.s.
Unexpectedly, at day 35 we noted bioluminescent signal contralateral to the primary tumors in NGP-N1D+BV mice, with increasing signal at day 50 (Fig. 1B). Necropsy of NGP-N1D+BV mice demonstrated multiple liver nodules (Fig. 1C). In comparison, few NGP-LacZ+BV mice had gross liver nodules (17/28 vs. 4/25, respectively, P=0.0017, Fisher’s exact test). Metastatic burden as measured by bioluminescence of liver homogenates was 10-fold higher in NGP-N1D+BV (Fig. 1D, P<0.001). The number and size of hepatic metastases was increased (Fig. 1E), with metastatic foci increased in NGP-N1D+BV (Fig. 1F; mean 11.2 vs. 2.8 metastatic foci/mm2, P<0.01), with parallel increases in metastases’ mean diameter (Fig. 1G; 744 vs. 195μm, P<0.0001). Thus, combined Notch and VEGF inhibition while prolonging survival, paradoxically led to increased liver metastasis.
Combined Notch and VEGF blockade does not increase circulating tumor cells
We have previously demonstrated that combined blockade of Notch and VEGF decreased the vasculature of neuroblastoma xenografts and disrupted the interaction of ECs with pericytes (14). Similar results were obtained when the vasculature of the primary tumor of the survival experiment was examined. There was a 23% decrease in the EC marker endomucin in NGP-N1D+BV tumors (Supplementary Fig. S1A, P<0.05). While almost all endomucin(+) vessels had adjacent αSMA(+) pericytes (96% for NGP-LacZ+BV, 94% NGP-N1D+BV), the pericytes in NGP-N1D+BV often appeared dissociated from the ECs (Supplementary Fig. S1B). This apparent disruption of tumor vasculature suggested that Notch blockade might facilitate the entry of tumor cells into the circulation, thus promoting liver metastasis. We therefore quantified CTCs by bioluminescence from blood obtained at time of sacrifice (Fig. 1H). There was no difference in CTCs, indicating that concurrent Notch and VEGF inhibition does not increase tumor cell intravasation.
Notch blockade is sufficient to promote liver metastasis
To determine if Notch blockade alone could promote liver metastasis, we implanted NGP-LacZ or NGP-N1D intrarenally. NGP-N1D tumors had increased liver homogenate bioluminescence (40-fold, P<0.05), metastatic foci (9.2-fold, P<0.05), and diameters (0.75 vs. 0.17mm, P<0.01)(Supplementary Fig. S2A–C),
To determine whether increased metastasis was due to an effect on the primary tumor or the liver, we injected NGP-LacZ or NGP-N1D cells into the left cardiac ventricle (19). We also treated with BV or placebo to determine if VEGF blockade contributed to liver metastases. Intracardiac injection of NGP-N1D resulted in a markedly higher liver metastatic burden by ex vivo liver bioluminescence (Fig. 2A), and by total liver flux compared to NGP-LacZ (Fig. 2B, 113-fold, P<0.05). Treatment with BV did not alter the metastatic pattern, with NGP-N1D+BV having increased liver metastases compared to NGP-LacZ+BV (Fig. 2B 93-fold, P<0.05). Quantitation of liver homogenate bioluminescence yielded similar results with increased metastases for N1D and N1D+BV (Supplementary Fig. S3A). Notch blockade resulted in significantly larger metastases (Fig. 2C,D; 3.00 and 2.91mm, NGP-N1D and NGP-N1D+BV, vs. 1.55 and 1.51mm, NGP-LacZ and NGP-LacZ+BV). These results demonstrate that Notch inhibition is sufficient to promote liver metastases, and does not require concurrent VEGF blockade.
Figure 2. Blockade of Notch is sufficient to promote liver metastases.

NGP-LacZ or N1D cells were injected in the left ventricle, and treated with either placebo or BV; NGP-LacZ (n=9), NGP-N1D (n=13), NGP-LacZ+BV (n=13), NGP-N1D+BV (n=14). All mice were killed at 7 weeks. (A) Ex vivo imaging of livers. (B) Quantification of total flux (photon/sec) for NGP-LacZ (n=9), NGP-N1D (n=13), NGP-LacZ+BV (n=13), NGP-N1D+BV (n=14). *P<0.05. (C) Histologic examination of liver metastases. Bar, 400μm. (D) Quantification of liver metastases diameters: NGP-LacZ (n=12, from 9 tumors), NGP-N1D (n=35, from 13 tumors), NGP-LacZ+BV (n=17, from 13 tumors), NGP-N1D+BV (n=55, from 14 tumors). *P<0.05; **P<0.01. (E) Ex vivo imaging of livers, 6 weeks after intracardiac injection of SH-SY5Y-GFP (n=13) or SH-SY5Y-N1D (n=10). (F) Quantification of total flux (photon/sec) by ex vivo liver bioluminescence. ***P<0.001. (G) Quantification of liver metastases diameter for SH-SY5Y-GFP (n=15, from 1 tumor, 11 had none), SH-SY5Y-N1D (n=117, from 8 tumors, 3 had none). ****P<0.0001. (H) Ex vivo imaging of livers, 5 weeks after intracardiac injection of MDA-MB-231-Fc (n=8) or MDA-MB-231-N1D (n=9) (I) Quantification of total flux (photon/sec) by ex vivo liver bioluminescence. *P<0.05.
To determine whether the effect of Notch blockade on liver metastasis was cell line-specific, we expressed the N1D in the MYCN-non-amplified neuroblastoma cell line SH-SY5Y (SH-SY5Y-N1D). Intracardiac injection of SH-SY5Y-N1D resulted in markedly increased liver metastatic burden (Fig. 2E,F; 1,065-fold, P<0.001), and larger metastases (Fig. 2G, 2.03 vs 0.92mm, P<0.0001), compared with SH-SY5Y-GFP.
To determine whether this effect was tumor-type specific, we engineered the breast cancer cell line MDA-MB-231 to express the N1D (MDA-MB-231-N1D). Intracardiac injection of MDA-MB-231-N1D resulted in increased liver metastasis as compared to controls (Fig. 2H), with a 2.1-fold increase in liver flux (Fig. 2I, *P<0.05), and a 11.7-fold increase in liver homogenate bioluminescence (Supplementary Fig. S3B, P<0.05).
To determine if Notch blockade promotes metastasis to other organs, we examined the spleen, kidney, and bone marrow after intracardiac injection of NGP-N1D or SH-SY5Y-N1D and the lung after intracardiac injection of MDA-MB-231-N1D (Supplementary Fig. S4A–C). There was no difference in bioluminescence with Notch blockade in the organs evaluated, indicating that the promotion of metastasis by Notch blockade is specific to the liver.
GSI PF-03084014 promotes liver metastasis
Our results demonstrate that cell lines engineered to express the N1D have increased liver metastasis. We also utilized a pharmacological approach to inhibit Notch signaling, with the GSI PF-03084014 (20), which has completed Phase1 testing in adults with advanced solid tumors (21). Treatment of NGP intrarenal tumors began 18 days after implantation, with PF-003084014 administered for 10 days, and sacrificed 7 days later. PF-003084014 did not inhibit the growth of the primary tumors (Fig. 3A). Liver metastases, however, were significantly increased in mice treated with PF-003084014, with a 38-fold higher total liver flux compared to vehicle treated mice (Fig. 3B,C, P<0.01). These results demonstrate that GSI inhibition of Notch signaling can promote liver metastases.
Figure 3. Liver metastasis is promoted by GSI but not by Notch1 knockdown.
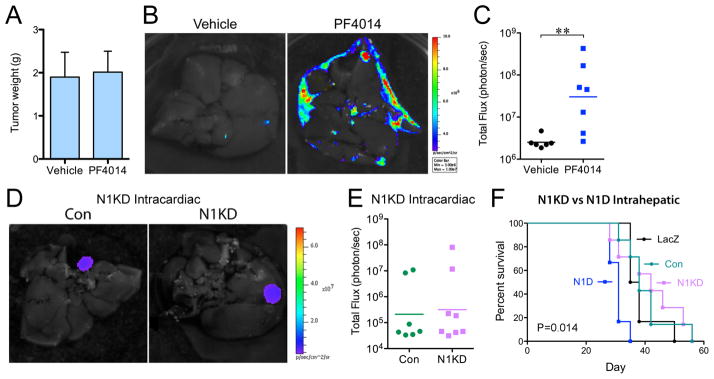
(A) Intrarenal NGP tumors were treated 18 days after implantation, with PF-003084014 (n=7) or Vehicle (n=6) for 10 days, and sacrificed 7 days later. P=n.s. (B) Ex vivo imaging of livers. (C) Quantification of total flux (photon/sec) by ex vivo liver bioluminescence. ***P<0.01. (D) Ex vivo imaging of livers after intracardiac injection of NGP-Con (n=7) and NGP-N1KD (n=8). (E) Quantification of total flux (photon/sec) by ex vivo liver bioluminescence. P=n.s. (F) NGP-Con (n=7), NGP-N1KD (n=7), NGP-LacZ (n=6), NGP-N1D (n=6); were implanted into the left lobe of the liver, and mice sacrificed when flux reached 6x109 photons/sec. Log-Rank (Mantel-Cox), P=0.0144.
Notch inhibition does not affect prometastatic characteristics of tumor cells
The increased liver metastases could be due to a direct effect on tumor cells or indirectly on the liver. We asked whether N1D inhibition of autocrine or paracrine Notch signaling in tumor cells promoted prometastatic characteristics. NGP expresses Notch1, while SH-SY5Y expresses Notch1, 2 and 3, and both cell lines express DLL4, JAG1, and JAG2 (Supplementary Fig. S5A). Yet despite these expression patterns, N1D did not affect in vitro proliferation, invasion, or migration of either NGP or SH-SY5Y (Supplementary Fig. S5B,C).
Knockdown of Notch1 in tumor cells does not promote liver metastases
To determine if loss of Notch signaling in tumor cells was responsible for liver metastasis, we used shRNA to knockdown Notch1 (N1KD) in NGP (Supplementary Fig. S6A). N1KD, similar to N1D, decreased expression of Notch-responsive genes Hes1 and Hey1, but did not affect proliferation in vitro (Supplementary Fig. S6B). In vivo, intracardiac injection of NGP-N1KD did not increase liver metastasis (Fig. 3D,E), indicating that inhibition of Notch signaling in tumor cells is not responsible for promoting liver metastasis.
NGP-N1D and NGP-N1KD cells were also directly implanted in the left lobe of the liver, and growth monitored by bioluminescence. NGP-N1D intrahepatic tumors progressed significantly faster (Fig. 3F; median survival 31 days, P=0.0144) than NGP-N1KD (42 days), or control NGP-LacZ and NGP-Con tumors (37 and 38 days). This suggests that inhibition of Notch in the liver, but not in tumor cells, promotes the growth of metastatic lesions.
Notch inhibition increases vascularity of liver metastases
One potential target of Notch blockade is the host vasculature. We segregated liver metastases into small (<300μm) versus large (>300μm) lesions, reasoning that progression could be enhanced by alterations in vasculature at either early or late stages. In small metastases (<300μm) of NGP-LacZ+BV, there was a paucity of intrametastatic vessels as demonstrated by a lack of endomucin (Fig. 4A) and ICAM-1 (not shown). In contrast, invading vascular sprouts were common seen in NGP-N1D+BV metastases (Fig. 4A). Quantifying the incidence of vascularized metastases, 74% of NGP-N1D+BV metastases were endomucin(+) as compared to 21% in NGP-LacZ+BV (P<0.0001). These small metastases lacked α-smooth muscle actin (αSMA) staining (not shown), suggesting that in this early angiogenic phase vessels were not yet invested by pericytes. In examining tumor cell proliferation within metastases, we noted a 3.8-fold increase (P<0.0001) in NGP-N1D+BV compared to NGP-LacZ+BV (Fig. 4A). Examination of large metastases (>300μm) demonstrated increased vasculature in NGP-N1D+BV as seen with endomucin (Fig. 4B, 3-fold, P<0.05), collagen IV (Supplementary Fig. S7A), and the pericyte markers NG2 (Fig. 4B, P<0.01), and αSMA (not shown).
Figure 4. Notch inhibition increases vascularity of liver metastases.

(A) Top panel: small liver metastases (<300μm) immunostained for endomucin. Arrows indicate vascular sprouts. Liver metastases with endomucin(+) sprouts were counted, P<0.0001, Fisher’s exact test. Bar, 50μm. Bottom panel: Proliferation determined by phosphorylated histone H3 (pH3). Mean±stddev. ****P<0.0001. (B) Large metastases (>300 μm) immunostained for endomucin (top panel) and NG2 (bottom panel). Green autofluorescence distinguishes RBC within metastases. Mean±stddev. *P<0.05; **P<0.01. Bar, 100μm. (C) Liver metastases from the NGP intracardiac experiment were immunostained for endomucin (top panel), and αSMA (bottom panel). Mean±stddev. *P<0.05; **P<0.01; ***P<0.001. Bar, 200μm. Nuclei shown by DAPI. Dashed line outlines metastases.
Liver metastases formed after NGP intracardiac injections were predominantly large (>300μm, Fig. 2D). NGP-N1D liver metastases (NGP-N1D or NGP-N1D+BV) displayed enhanced angiogenic vasculature by collagen IV (Supplementary Fig. S7B), endomucin (Fig. 4C) and ICAM-1 (not shown), and αSMA (Fig. 4C), compared to NGP-LacZ or NGP-LacZ+BV. Similar results were obtained after intracardiac injection of SH-SY5Y-N1D, with Notch blockade markedly increasing vascularity by ICAM-1 (Supplementary Fig. S7C, 13-fold, P<0.0001) and α SMA (Supplementary Fig. S7C, 12-fold, P<0.0001). Thus, Notch blockade increases liver metastasis angiogenesis both at the initial step of SEC sprouting and the later step of pericyte recruitment.
Notch inhibition activates HSCs
HSCs are often considered liver pericytes, and can contribute to tumor angiogenesis (22). Increased recruitment of NG2/αSMA(+) cells to the metastatic vasculature suggests the possibility that Notch blockade affects HSCs. Desmin has been widely used as a marker for quiescent HSCs (23,24), with αSMA and NG2 commonly regarded as typifying activated HSCs (10). We therefore performed double-label IHC for desmin and αSMA. In normal mouse liver, desmin(+) HSCs are seen in peri-sinusoidal locations and are αSMA(−) indicating quiescence (Supplementary Fig. S8A). After intracardiac injection of NGP cells, quiescent desmin(+)/αSMA(−) HSCs are seen within adjacent liver in NGP-LacZ and NGP-N1D (Supplementary Fig. S8B). Desmin and αSMA signals were scant within NGP-LacZ metastases. In contrast, markedly increased desmin(+)/αSMA(+) signal was detected within NGP-N1D metastases. These results suggest that Notch inhibition activates HSCs, leading to their recruitment into metastases.
Decrease in host Notch1 promotes liver metastasis
To evaluate the role of host Notch1 signaling in liver metastasis, mice with heterozygous deletion of Notch1 (17) were crossed to immunodeficient Rag2−/−, Il2rg−/−, eGFP+/+ mice (Fig. 5A). SECs of Notch1+/− livers had a 3-fold higher proliferation rate as seen with BrdU (Fig. 5B, P<0.0001), and a 14-fold increase in ICAM-1 expression as compared to littermate Notch1+/+ (Fig. 5B, P<0.0001).
Figure 5. Decrease in host Notch1 signaling promotes liver metastases.

(A) Schema to produce Notch1+/−, Rag2−/−, Il2rg−/−, eGFP+/+ mice. (B) Livers from Notch1+/+ (n=2) and Notch1+/− (n=2) mice were examined for: BrdU (top panel). Mean±stddev. ****P<0.0001. Bar, 50μm. ICAM-1 (bottom panel). Mean±stddev. ****P<0.0001. Bar, 200μm (C) Ex vivo imaging of livers 8 weeks after intracardiac injection of 105 NGP cells into Notch1+/+ (n=4), or Notch1+/− (n=5) mice. (D) Quantification of total flux (photon/sec) by ex vivo liver bioluminescence. **P<0.01. (F) Ex vivo imaging of livers 3 weeks after intracardiac injection of 106 NGP cells into Notch1+/+ (n=2) or Notch1+/− (n=3) mice. Mean±stddev. ****P<0.0001. Bar, 100μm.
To determine if deficient Notch1 signaling would promote liver metastasis, Notch1+/− and Notch1+/+ mice had intracardiac injection of NGP (105 cells) and were sacrificed at 8 weeks. Notch1+/− mice had markedly increased liver metastatic burden compared to littermate Notch1+/+ mice (Fig. 5C), with a 711-fold increase in liver flux (Fig. 5D, P<0.01). Histologic examination demonstrated large metastases in Notch1+/− mice (2.79±1.82mm, mean ± stddev), but a paucity of metastases in Notch1+/+ mice (solitary metastasis 0.17mm), which precluded further analysis of the vasculature.
To increase the number of metastatic liver lesions in Notch1+/+ controls, intracardiac injection of 10-fold more NGP cells (106) was performed and mice sacrificed after 3 weeks. ICAM-1(+) vessels in liver metastases from Notch1+/− mice were increased 16-fold compared to Notch1+/+ controls (Fig. 5E, P<0.0001). Thus, deficient host Notch1 signaling is permissive for liver metastasis and increases early angiogenesis.
Notch regulates the ICAM-1 expression in SECs
Our results demonstrate increased ICAM-1(+) vasculature in the livers of Notch1+/− mice and in the hepatic metastases from Notch1+/− mice and N1D expressing tumors. To determine if expression of ICAM-1 in SECs is regulated by Notch1, SECs were isolated from mouse liver. mSECs express Notch1, 2, 4, and DLL1, DLL4, JAG1, and JAG2 (Supplementary Fig. S9A). Notch1-knockdown increased the expression of ICAM-1 in mSEC, but not in human umbilical vein endothelial cells (HUVEC) (Supplementary Fig. S9B). Conversely, expression of Notch1-intracellular active domain (N1IC)(25), decreased the expression of ICAM-1 (Supplementary Fig. S9C). These results demonstrate that ICAM-1 is regulated in SEC by Notch1.
Notch inhibition increases ICAM-1 but not the retention of tumor cells
ICAM-1 has previously been shown to promote the arrest of tumor cells in liver sinusoidal endothelium (26). We therefore speculated that attenuation of Notch signaling, by up-regulating ICAM-1, might increase tumor cell retention within the liver. To address this possibility, we utilized adenovirus expressing either N1D (AdN1D) or control Fc (AdFc). Injection of AdN1D into nude mice, leads to hepatocyte infection, increased liver ICAM-1 expression (Supplementary Fig. S10A,B), and secretion of N1D into the circulation (Supplementary Fig. S10E). 3 days after AdFc or AdN1D infection, 105 NGP cells were injected intracardially, and 24hr later mice were sacrificed and liver metastasis quantified. (Supplementary Fig. S10C). There was no difference in bioluminescence between AdFc and AdN1D, indicating that Notch inhibition, despite increasing ICAM-1, does not increase the retention of tumor cells within the liver. If, however, mice are sacrificed 6 weeks later, increased liver metastasis is observed (34-fold, P<0.05, Supplementary Fig. S10D), suggesting that Notch inhibition affects a later step in metastasis.
Blockade of both DLL and JAGGED is required for progression of liver metastases
We have recently developed Notch1-decoy variants (Fig. 6A): N11–24-decoy containing EGF repeats 1–24, blocks both DLL and JAGGED ligands; N11–13-decoy containing EGF repeats 1–13, specific for DLL ligands; and N110–24-decoy containing EGF repeats 10–24, specific for JAGGED ligands (27). NGP cells expressing these variants were generated (Supplementary Fig. S11A). N11–24-decoy, N11–13-decoy, or N110–24-decoy, similar to full-length N1D, did not affect NGP proliferation in vitro (Supplementary Fig. S11B).
Figure 6. Blockade of DLL and JAGGED ligands promotes liver metastases.

(A) N11–24-decoy containing EGF repeats 1–24, blocks DLL and JAGGED ligands; N11–13-decoy containing EGF repeats 1–13, blocks DLL ligands; and N110–24-decoy containing EGF repeats 10–24, blocks JAGGED ligands. (B) Quantification of total flux (photon/sec) by ex vivo liver bioluminescence 3 weeks after intracardiac injection of 106 cells for NGP-Fc (n=10), NGP-N11–24-decoy (n=10), N11–13-decoy (n=10), N110–24-decoy (n=10). *P<0.05; ***P<0.001. (C) Diameter of liver metastasis, with each point on the scatter plot an individual metastasis. N11–24-decoy (n=87, from 7 tumors) and N11–13-decoy (n=72, from 6 tumors). *P<0.05. (D) Liver metastases were immunostained for ICAM-1 (upper panel). Mean±stddev. P=n.s. Bar, 100μm. αSMA (middle panel). Mean±stddev. *P<0.05. Bar, 100μm. Double immunostaining (bottom panel) for desmin (green) and αSMA (red), demonstrated co-localization (arrows), inset (2x magnification). Bar, 50μm.
To determine the roles of DLL and JAGGED in liver metastasis, intracardiac injection of Notch1-decoy variant expressing NGP cells was performed and mice sacrificed at 3 weeks. Significantly increased liver metastatic burden (Fig. 6B) was detected with NGP-N11–24, as compared to either NGP-Fc (25-fold, P<0.05) or NGP-N110–24 (74-fold, P<0.001). Histologic examination showed scant metastasis for NGP-Fc or NGP-N110–24 (1 metastasis seen in multiple liver sections), whereas multiple metastases were seen with NGP-N11–24-decoy and NGP-N11–13-decoy (Fig. 6C). Metastatic diameter, however, was significantly higher for NGP-N11–24-decoy compared to NGP-N11–13-decoy (296 vs 246μm, P<0.05), with an increase in very large metastases (>600μm, P=0.03, Fisher’s exact test).
IHC demonstrated no difference in ICAM-1(+) vasculature between NGP-N11–24 and NGP-N11–13 (Fig. 6D). This suggests that DLL1/4 blockade promotes initial vascularization with ICAM-1(+) SECs. NGP-N11–24 metastases, however, displayed a 3.1-fold increase in αSMA(+) vasculature (Fig. 6D). Double-label IHC for desmin and αSMA demonstrated co-localization in NGP-N11–24 metastases, but not in NGP-N11–13 metastases. Thus, DLL inhibition promoted development of early liver metastases by SECs, while the ability of NGP-N11–24 to block JAGGED increased activation and recruitment of HSC to the metastatic vasculature.
Blockade of DLL ligands increases sprouting of mSECs
Our in vivo results indicate that blockade of DLL, but not JAGGED, induced initial sprouting of vessels into hepatic metastases. To examine DLL specificity for sprouting, we performed in vitro assays with mSECs. Dextran beads were coated with SECs expressing N1-decoy variants, and assessed for sprout formation. On day 3, SECs expressing N11–24 or N11–13-decoy markedly increased sprouting per bead and sprout length (Fig. 7A–C), compared with either Fc or N110–24-decoy (similar results day 6, not shown). This DLL-specific effect on sprouting is not due to a selective effect on SEC proliferation, as both N11–13 and N110–24 increase proliferation (Supplementary Fig. S11C). Thus, SEC sprouting is mediated by inhibition of DLL-mediated signaling.
Figure 7. DLL Blockade promotes SEC sprouting while JAGGED blockade promotes HSC migration.

(A) Fibrin bead assay with mSEC transfected with Fc, N11–24-decoy, N11–13-decoy, and N110–24-decoy. Arrows indicate sprouts. Bar, 200 μm. (B) Sprouts per bead, (C) Sprout length, Mean±stddev. *P<0.05; **P<0.01; ***P<0.001. (D) In the top-chamber of a Boyden chamber, HSC-GFP cells were co-cultured with NGP cells expressing Notch1 decoys. (E) migration of HSC-GFP measured at 48h. Mean±stddev. *P<0.05; **P<0.01. (F) Immunoblot of Hes1, Hey1, αSMA, and β-actin of FACS sorted HSCs. (G) Migration of HSC-GFP, at 48h, co-cultured with NGP or NGP-siJag1. Mean±stddev. ***P<0.001. (H) Model of Notch blockade effects on hepatic microenvironment. Quiescent: SECs are maintained in quiescence by Notch1 signaling. HSCs in the space of Disse are maintained in quiescence by Notch2/3 signaling from JAG1 expressing hepatocytes (28). SEC Proliferation and Sprouting: Soluble N1D inhibits DLL4 activation of Notch1 in SECs, leading to expression of ICAM-1 and proliferation of SECs, and subsequent sprouting into tumor micrometastases (<300μm). HSC Recruitment: N1D blocks tumor JAG1, promoting HSC activation, (indicated by αSMA(+) expression), migration, and recruitment to the vasculature of macrometastases (>300 μm).
JAG1 blockade increases HSC migration
Our in vivo results suggest that blockade of JAGGED ligands promotes HSC recruitment, raising the possibility that JAG1 expressed on NGP cells may interact with Notch receptors on HSCs (28). GFP labeled-HSCs (which express Notch2/3, data not shown), were co-cultured with NGP cells expressing N1-decoy variants and migration assessed in a Boyden chamber assay (Fig. 7D). Only decoys that blocked JAGGED, N11–24-decoy and N110–24-decoy, but not DLL, increased HSC migration (Fig. 7E). FACS isolation of HSC-GFP cells, after co-culture with NGP cells, demonstrated that blockade of JAGGED, inhibited Notch signaling, as shown by decreased Hes1 and Hey1 (Fig. 7F). Blockade of JAGGED also resulted in αSMA expression in HSCs, suggesting an activated state. The role of tumor-derived JAG1 in suppressing HSC activation is supported by increased migration of HSCs when JAG1 is knocked-down in co-cultured NGP cells (Fig. 7G, Supplementary Fig. S12A,B). Thus, loss of tumor-derived JAG1 promotes activation and recruitment of HSCs into the vasculature of liver metastases.
DISCUSSION
Given its important role in tumorigenesis, the Notch pathway has aroused great interest as a therapeutic target. Employing intrarenal and intracardiac metastasis models with different Notch inhibitors, used in preclinical and clinical studies, we have shown that inhibition of Notch signaling in liver stromal microenvironment promotes metastatic growth. Notch blockade produced significantly larger and more highly vascularized hepatic metastatic lesions in neuroblastoma cell lines NGP and SH-SY5Y, and the breast cancer cell line MDA-MD-231. In these studies, we uncover a novel function of Notch, and show that inhibition of Notch signaling increases metastatic angiogenesis to support growth and enlargement.
Our results demonstrate that Notch inhibition is sufficient to promote liver metastases and increased vascularization, and is not limited by concurrent VEGF blockade. As bevacizumab binds selectively to human but not murine VEGF, angiogenesis in liver metastases is not dependent on tumor-derived VEGF. VEGF blockade, however, by slowing growth of the primary tumor and prolonging survival, may lead to an apparent increase in liver metastatic burden by extending the time period in which metastases can grow.
We have previously shown that the N1D impairs tumor angiogenesis (13,14). In tumor cells, however, N1D did not alter proliferation, migration and invasion. Furthermore, N1KD did not increase liver metastases when injected via intracardiac or intrahepatic routes. Therefore, inhibition of intrinsic Notch signaling within neuroblastoma cells appears to have no effect on inherent tumorigenic or metastatic properties. Notch1 deficiency in Notch1+/− mice promoted similarly enlarged liver metastatic lesions formed by NGP cells injected intracardially. Therefore, we conclude that Notch signaling in host liver cells acts as a metastasis suppressor.
There is emerging evidence that Notch1 signaling acts to maintain quiescence and differentiation of SECs. Conditional Notch1-knockout mice show the development of nodular regenerative hyperplasia, a disease associated with persistent increase in SEC proliferation (29). Similarly, DLL4 blockade has been shown to induce abnormal SEC activation and vascular tumorigenesis (16). In the present study we have utilized Notch1 decoys; N1D and N11–24-decoy that act as antagonists of both DLL and JAGGED ligands, or N11–13 and N110–24-decoys that selectively block DLL and JAG, respectively (27). DLL blockade, by N11–24 and N11–13-decoys, promoted development of small metastases with extensive ICAM-1 staining. SEC-expressing N11–24 and N11–13-decoys demonstrated increased sprouting in in vitro fibrin bead assays. Consistent with these findings, Notch1 deficiency in Notch1+/− mice leads to SEC proliferation. Thus, blockade of DLL-Notch1 signaling enhances SEC proliferation and sprouting, supporting early angiogenesis and growth of micrometastases within the liver (Fig. 7G).
The activation of SEC due to Notch1 inhibition has previously been reported in mice with Notch1 loss of heterozygosity (30). Vascular tumors were found predominantly in livers, but not in other organs including kidney, spleen, heart, and lung. Increased angiogenic activity was found only in liver SEC in those mice. Similarly, Notch1 deletion in MxCre Notch1lox/lox mice resulted in persistent and cell-autonomous proliferation of liver SECs only, that ultimately led to hepatic angiosarcomas (29). Consistent with these studies, we also observed predominant liver metastasis in our in vivo mice models. Our data also shows that knockdown of Notch1, in vitro, induces ICAM-1 in SECs but not in HUVECs. The reason for the selectivity of Notch1 inhibition is not known. However, the unique characteristics of liver SEC as well as specific anatomical and environmental factors may render them more proliferative upon Notch1 inhibition.
Liver metastasis progression is a multistep process that begins with the arrest of tumor cells in sinusoids, with ICAM-1 promoting the adhesion of tumor cells to liver sinusoidal endothelium (26). Our in vivo tumor cell liver adhesion assays showed that pretreatment with N1D adenovirus elevated liver ICAM-1, but did not increase the retention of tumor cells in the liver. Therefore, attenuation of Notch signaling appears to affect another step of early survival or growth of tumor cells, rather than adhesion.
A role for Notch signaling in maintaining HSCs in a quiescent state has also been proposed (28). Cancer cells by releasing paracrine factors can create a prometastatic microenvironment within the liver, promoting the activation of HSCs (26,31). Our results indicate that tumor-derived JAG1 has the opposite effect, suppressing the activation of HSCs. Blockade of both JAGGED and DLL (N11–24-decoy), produced larger metastases with increased HSC recruitment and activation, compared to DLL blockade only (N11–13-decoy). Consistent with these results, our in vitro studies demonstrated that HSCs in which tumor-derived JAGGED was blocked, displayed increased migration and evidence of an activated state. Similarly, increased HSC migration was observed when HSCs were co-cultured with NGP cells in which JAG1 was silenced. Our studies thus suggest that inhibition of tumor-derived JAG1 signaling disrupts Notch-mediated quiescence, promoting HSC activation and migration, and subsequent vascular maturation and enlargement of micrometastases (Fig. 7G).
In some settings, DLL4 and JAG1 can exert opposing effects on angiogenesis (32). DLL4-mediated Notch signaling inhibits the sprouting of endothelial tip cells in growing blood vessels. In contrast, JAG1 overexpression inhibits DLL4 signaling in ECs and increases sprouting angiogenesis (32). In our studies, however, we have found that the Notch ligands in the liver have distinct rather than opposing effects, with both acting to inhibit metastatic progression.
Our findings have potentially serious implications for Notch inhibition therapy. Due to the critical role of this pathway in regulating of tumor angiogenesis, Notch components have emerged as an attractive potential target, and Notch inhibitors are entering clinical cancer trials. Our study shows that treatment with N1-decoys or the GSI PF-03084014 increase hepatic metastasis. These findings raise the concern that Notch signaling blockade could disrupt normal liver homeostasis and quiescence by pathological activation of SECs and HSCs, creating a host microenvironment favorable for the metastatic growth of cancer cells.
Supplementary Material
Acknowledgments
We thank Arul Thirumoorthi, Minji Kim, Shuobo Zhang, and Jason Mitchell for technical assistance, and Robert Schwabe for assistance with HSC experiments.
Grant Support
These studies were supported by NIH 5R01CA124644 (Yamashiro), 3R01CA124644-03S1 (Kadenhe-Chiweshe), R01CA136673 (Shawber, Kitajewski), Pediatric Cancer Foundation (Kandel, Yamashiro), Sorkin Fund (Kandel), tay-bandz Foundation (Yamashiro), Children’s Neuroblastoma Cancer Foundation (Hernandez), and Eisai Incorporated (Kitajewski).
Footnotes
Disclosure of Potential Conflicts of Interest
Authors have licensed patents related to the work described herein: “Human NOTCH1 decoys” (WO2013052607; T. Kangsamaksin, C.J. Shawber, J.K. Kitajewski), “Composition of humanized NOTCH fusion proteins and methods of treatment” (US Patent 20110008342 A1; C.J. Shawber, J.K. Kitajewski), and “Notch-based Fusion Proteins and Uses Thereof” (US Patent 7662919 B2; C.J. Shawber, J.K. Kitajewski). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Other authors disclosed no potential conflicts of interest.
Authors’ Contributions
Conception and design: D. Banerjee, S.L. Hernandez, A. Kadenhe-Chiweshe, C.J. Shawber, J.K. Kitajewski, J.J. Kandel, D.J. Yamashiro
Development of methodology: D. Banerjee, S.L. Hernandez, A. Garcia, T. Kangsamaksin, C.J. Shawber, J.K. Kitajewski, J.J. Kandel, D.J. Yamashiro
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): D. Banerjee, S.L. Hernandez, A. Garcia, T. Kangsamaksin, E. Sbiroli, J. Andrews, L. Forrester, N. Wei, A. Kadenhe-Chiweshe, D.J. Yamashiro
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): D. Banerjee, S.L. Hernandez, J.J. Kandel, D.J. Yamashiro
Writing, review, and/or revision of the manuscript: D. Banerjee, S.L. Hernandez, C.J. Shawber, J.K. Kitajewski, J.J. Kandel, D.J. Yamashiro
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C.J. Shawber, J.K. Kitajewski, J.J. Kandel, D.J. Yamashiro
Study supervision: J.J. Kandel, D.J. Yamashiro
References
- 1.Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–61. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 2.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–76. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 3.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–7. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 4.Hopfer O, Zwahlen D, Fey MF, Aebi S. The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer. 2005;93:709–18. doi: 10.1038/sj.bjc.6602719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y, De Marco MA, Graziani I, Gazdar AF, Strack PR, Miele L, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67:7954–9. doi: 10.1158/0008-5472.CAN-07-1229. [DOI] [PubMed] [Google Scholar]
- 6.Mailhos C, Modlich U, Lewis J, Harris A, Bicknell R, Ish-Horowicz D. Delta4, an endothelial specific notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation. 2001;69:135–44. doi: 10.1046/j.1432-0436.2001.690207.x. [DOI] [PubMed] [Google Scholar]
- 7.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–7. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 8.Takebe N, Nguyen D, Yang SX. Targeting notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther. 2014;141:140–9. doi: 10.1016/j.pharmthera.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deangelo DJ, Stone RM, Silverman LB, Stock W, Attar EC, Fearen I, et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol. 2006;24:abstr 6585. [Google Scholar]
- 10.Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012;30:2307–13. doi: 10.1200/JCO.2011.39.1540. [DOI] [PubMed] [Google Scholar]
- 11.Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol. 2012;30:2348–53. doi: 10.1200/JCO.2011.36.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol. 2011;29:3529–34. doi: 10.1200/JCO.2011.35.7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008;68:4727–35. doi: 10.1158/0008-5472.CAN-07-6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez SL, Banerjee D, Garcia A, Kangsamaksin T, Cheng WY, Anastassiou D, et al. Notch and VEGF pathways play distinct but complementary roles in tumor angiogenesis. Vascular Cell. 2013;5:17. doi: 10.1186/2045-824X-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–63. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 16.Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature. 2010;463:E6–7. doi: 10.1038/nature08751. [DOI] [PubMed] [Google Scholar]
- 17.Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–19. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 18.Huang J, Frischer JS, New T, Kim ES, Serur A, Lee A, et al. TNP-470 promotes initial vascular sprouting in xenograft tumors. Mol Cancer Ther. 2004;3:335–43. [PubMed] [Google Scholar]
- 19.Olaso E, Salado C, Egilegor E, Gutierrez V, Santisteban A, Sancho-Bru P, et al. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology. 2003;37:674–85. doi: 10.1053/jhep.2003.50068. [DOI] [PubMed] [Google Scholar]
- 20.Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010;9:1618–28. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- 21.Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, et al. A Phase I, Dose-finding Study in Patients With Advanced Solid Malignancies of the Oral Gamma-Secretase Inhibitor PF-03084014. Clin Cancer Res. 2015;21:60–7. doi: 10.1158/1078-0432.CCR-14-0607. [DOI] [PubMed] [Google Scholar]
- 22.Hellerbrand C. Hepatic stellate cells--the pericytes in the liver. Pflugers Archiv. 2013;465:775–8. doi: 10.1007/s00424-012-1209-5. [DOI] [PubMed] [Google Scholar]
- 23.Yokoi Y, Namihisa T, Kuroda H, Komatsu I, Miyazaki A, Watanabe S, et al. Immunocytochemical detection of desmin in fat-storing cells (Ito cells) Hepatology. 1984;4:709–14. doi: 10.1002/hep.1840040425. [DOI] [PubMed] [Google Scholar]
- 24.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beatus P, Lundkvist J, Oberg C, Lendahl U. The notch 3 intracellular domain represses notch 1-mediated activation through Hairy/Enhancer of split (HES) promoters. Development. 1999;126:3925–35. doi: 10.1242/dev.126.17.3925. [DOI] [PubMed] [Google Scholar]
- 26.Van den Eynden GG, Majeed AW, Illemann M, Vermeulen PB, Bird NC, Hoyer-Hansen G, et al. The multifaceted role of the microenvironment in liver metastasis: biology and clinical implications. Cancer Res. 2013;73:2031–43. doi: 10.1158/0008-5472.CAN-12-3931. [DOI] [PubMed] [Google Scholar]
- 27.Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, et al. Notch decoys that selectively block Dll/Notch or Jagged/Notch disrupt angiogenesis by unique mechanisms to Inhibit tumor growth. Cancer Discov. 2014;5:1–16. doi: 10.1158/2159-8290.CD-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawitza I, Kordes C, Reister S, Haussinger D. The niche of stellate cells within rat liver. Hepatology. 2009;50:1617–24. doi: 10.1002/hep.23184. [DOI] [PubMed] [Google Scholar]
- 29.Dill MT, Rothweiler S, Djonov V, Hlushchuk R, Tornillo L, Terracciano L, et al. Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology. 2012;142:967–77. e2. doi: 10.1053/j.gastro.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Turkoz A, Jackson EN, Corbo JC, Engelbach JA, Garbow JR, et al. Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. J Clin Invest. 2011;121:800–8. doi: 10.1172/JCI43114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vidal-Vanaclocha F. The prometastatic microenvironment of the liver. Cancer Microenviron. 2008;1:113–29. doi: 10.1007/s12307-008-0011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–35. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.