

Abstract
Electro wetting-on-dielectric (EWOD) digital microfluidics (DMF) can be used to develop improved chemical screening platforms using 3-dimensional (3D) cell culture. Alginate hydrogels are one common method by which a 3D cell culture environment is created. This paper presents a study of alginate gelation on EWOD DMF and investigates designs to obtain uniform alginate hydrogels that can be repeatedly addressed by any desired liquids. A design which allows for gels to be retained in place during liquid delivery and removal without using any physical barriers or hydrophilic patterning of substrates is presented. A proof of concept screening platform is demonstrated by examining the effects of different concentrations of a test chemical on 3D cells in alginate hydrogels. In addition, the temporal effects of the various chemical concentrations on different hydrogel posts are demonstrated, thereby establishing the benefits of an EWOD DMF 3D cell culture and chemical screening platform using alginate hydrogels.
I. INTRODUCTION
The pharmaceutical drug development process involves exhaustive testing of potential drug candidates in laboratory cell culture studies and animal trials before moving on to human clinical trials.1 However, only one in ten drugs, that enter clinical trials, receives final FDA approval.2 This highlights the need for improved laboratory testing and chemical screening methods to identify failure candidates early, so that expensive and time consuming clinical trials can be avoided in later stages.
One method that could improve laboratory studies is by replacing conventional 2-dimensional (2D) cell culture based in-vitro testing with 3-dimensional (3D) cell culture. Cells cultured in 3D environments experience greater cell-cell and cell-matrix interactions that are more representative of the natural in-vivo situation than regular 2D environments.3,4 Thus, 3D cell cultures mimic physiological processes more accurately and are better predictors of how cells in the body would respond to potential drug candidates.5,6 Incorporating 3D cell culture into high throughput screening (HTS) procedures could therefore provide more relevant results.
The adoption of 3D cell culture for HTS has been slow despite the known benefits. This can be attributed to the increased difficulty in handling 3D scaffolds as well as the greater difficulty in imaging cells in 3D environments in a high throughput manner.7 Many microfluidic devices have been designed to attempt 3D cell culture by using hydrogels8,9 to provide a 3D environment to the cells and also to allow for subsequent exposure to candidate drugs to study the response.10–15 Microfluidics offers the advantages of increased automation and reagent savings, as well as smaller sample sizes which can improve the reaction and response time. However, developing screening systems with high order multiplexing capabilities using conventional microfluidics has its own limitations. As the number of targeted 3D cell culture sites increases, it becomes increasingly complex to individually address specific target sites without affecting neighboring locations.
Digital microfluidics (DMF) present advantages in such scenarios. Many studies have already demonstrated the efficiency of electro-wetting-on-dielectric (EWOD) based DMF in handling and micro-manipulating nanoscale volumes of liquids for various applications.16–20 Among these include cell culture21–23 and cell based assays.24,25 The application of EWOD DMF for 3D cell culture was first proposed by the authors in initial proof of concept work by using alginate hydrogels to provide a 3D environment.26,27 More recent studies have further developed the concept using thermally cross-linked hydrogels such as collagen, agarose, or Geltrex for 3D cell culture28 and for drug screening applications.29 However, depending on the kind of cells being cultured and studies being conducted, a wide variety of hydrogels have been used. Not all of these hydrogels are cross linked thermally. For DMF to become a suitable candidate as a versatile platform for carrying out 3D cell culture studies, its ability to handle various kinds of hydrogels with different methods of cross-linking should be demonstrated. Alginate hydrogels are one such type of hydrogels that are ionically cross linked and used for 3D cell culture due to its biocompatibility, low toxicity, and mild gelation methods. In addition, alginates can be tailored to yield custom stiffness and pore sizes.30 Alginates on its own tend to inhibit protein absorption and cell adhesion, though this can also be modified by the addition of specific peptide sequences.31 This allows alginate to be used as a blank slate, upon which researchers can specify the degree of cell-matrix interaction. Unmodified alginate can be used solely as a support matrix without getting involved in cell signaling processes, while appropriately selected peptide coupled alginates can be used to control the phenotype of cultured cells.32 Another reason why alginates are of interest for 3D cell culture studies is due to the reversible gelation mechanism. This allows for the recovery of cells that have been 3D cultured and subjected to desired chemical stimuli, for further analysis and characterization.33 Thus, alginates present themselves as a suitable candidate for incorporation into DMF platforms for 3D cell culture studies.
Calcium alginate hydrogels are commonly formed by the ionic cross-linking of sodium alginate with Ca2+ ions.34 Since gelation is very gentle as well as reversible, it has found numerous applications for cell trapping and immobilization.35–37 Calcium alginate hydrogels have also been widely documented for use in 3D cell culturing and tissue engineering.38 At the macroscale, alginate hydrogels have usually been formed by dropping sodium alginate into calcium chloride baths. This has been adapted at the microfluidic scale by using droplet microfluidics to generate microscale droplets of sodium alginate that are then delivered into calcium chloride solutions wherein they form calcium alginate hydrogel drops or beads.39–41 These beads are then collected and processed separately either using other microfluidic devices or by manual handling of the alginate gel beads. Others have used continuous microfluidic devices to create cell seeded alginate hydrogels by combining laminar flows of sodium alginate and calcium chloride to examine the cell response to chemical gradients.42,43 Still another approach that was demonstrated used spotters to create a micro-array of alginate gel spots but this approach could not address each gel spot individually and all gel spots were subject to same conditions.44 To date, the authors have not seen a complete integrated device capable of forming and sustaining discrete individually addressable cell seeded alginate hydrogels, multiplexing testing chemicals, and delivering these different combinations of test chemicals to individual 3D cell cultured structures. This is where the use of EWOD DMF is proposed since it is well suited for combining all these features in a single microfluidic device.
This paper investigates alginate hydrogel formation on EWOD DMF and proposes a design to allow for on demand formation of cell seeded alginate hydrogels. A proof of concept chemical screening application is then demonstrated by using a dimethyl sulfoxide (DMSO) as a sample test chemical. Different concentrations of DMSO are delivered to cell seeded calcium alginate gels and their effects on viability are measured. In addition, the ability to measure the temporal effects of chemical concentration is demonstrated by taking time lapse fluorescent images of cell seeded alginate hydrogels exposed to different concentrations of DMSO.
II. MATERIALS AND METHODS
A. Cell culture
Human breast cancer cells (MCF-7) obtained from American Type Culture Collection (ATCC) were maintained in culture medium (D-MEM/F12, Life Technologies, Carlsbad, CA), and supplemented with 4% fetal bovine serum, 2 mM l-glutamine, 100 μg/ml penicillin/streptomycin, and 0.01 mg/ml insulin. Cells were incubated in 25 cm2 T-flasks at 37 °C and 5% CO2 while being sub-cultured every 3–4 days at ∼80% confluency.
B. Alginate hydrogel precursor reagents and cell seeded sodium alginate preparation
In order to prepare 0.5% wt./vol. sodium alginate solutions, first stock solutions of sodium alginate were formed by dissolving low viscosity sodium alginate powder (Sigma Aldrich, St. Louis, MO—CAS 9005-38-3) in deionized water to obtain a concentrated 4% wt./vol. sodium alginate solution. This concentrated alginate solution was diluted in a 1:4 ratio with MCF-7 cells suspended in DMEM/F12 to obtain a 0.5% wt./vol. of cell seeded sodium alginate solution with a concentration of 0.5–1 × 106 cells/ml. Calcium chloride solutions in 100 mM were prepared by dissolving calcium chloride crystals (Sigma Aldrich) in DMEM/F12 culture media. In order to prevent biofouling,45 pluronic F-68 was added to all culture media containing reagents at 0.04% wt./vol. concentration.
C. Device fabrication and operation
EWOD DMF devices were fabricated at the University of Texas at Arlington's Nanofabrication facility in a manner similar to that used by Wijethunga et al.19 Briefly, a bottom EWOD DMF chip consists of patterned ITO electrodes on a glass substrate, 5 μm-thick dielectric layer of Su8-2005, and 150 nm-thick topmost hydrophobic layer of Teflon; whereas a top EWOD chip is formed with an ITO and a Teflon layer only. For devices where Teflon patterning was required, a simple Teflon lift off method as described by Eydelnant et al.46 was adopted. This consisted of patterning Shipley S1813 positive photoresist on the dielectric layer to cover the circular gel formation sites where Teflon was to be removed. After developing, the PR was weakened by UV flood exposure for 1 min, followed by spin coating 2% w/v Teflon AF solution at 2000 rpm. After soft baking for 2 min at 95 °C, the device was rinsed with acetone for 8 min. This caused the patterned photoresist to lift off from the dielectric layer and also removed the Teflon layer above it, resulting in circular patterned hydrophilic sites. This was followed by baking at 150 °C for 5 min to remove any remaining volatile solvents.
EWOD DMF bottom and top chips were assembled by means of 100 μm thick double sided kapton tape and mounted into a custom Plexiglas base holder. The Plexiglas base allowed for bottom EWOD DMF device contact pads to be aligned and in contact with z-directional zebra connectors (Fuji-Poly Industries, Carteret, NJ) that lined up with corresponding contact pads on a vertically aligned PCB board. The PCB in turn relayed these contact pads to a signal generator and amplifier that provided the driving AC voltages for the EWOD DMF device. The AC voltages driving each individual EWOD electrode were regulated by signals generated through a custom LabVIEW code and transmitted through a 96 channel data acquisition unit. By using pre-programmed sequences fed into the LabVIEW interface, automated operation of the DMF device was enabled.
D. Fluorescent dye preparation and staining protocol
Fluorescent dyes Hoechst 33342 (Life Technologies) was used to stain all the cells present in the hydrogels while Propidium Iodide (PI) (Life Technologies) was used to stain all the dead cells. H-33342 and PI were dissolved in sterile culture grade water to obtain final dye concentrations of 5 μg/ml of H-33342 and 8 μg/ml of PI.
E. DMSO preparation and dilution protocol
DMSO solutions of different concentrations were prepared on EWOD chips through a serial dilution protocol similar to that used by Park et al.47 A stock solution of DMSO was prepared by mixing sterile culture grade DMSO (Sigma Aldrich), deionized water, Hoechst-33342 and Propidium Iodide to obtain a 50% v/v DMSO solution with the same final dye concentration of H-33342 and PI as used to prepare the fluorescent dye solution described earlier. This stock solution was loaded into the EWOD reservoir. In order to obtain 25% DMSO solution, a 400 nl drop of 50% DMSO stock solution was dispensed from the reservoir and brought to a designated mixing zone where it was merged with another 400 nl drop of fluorescent dye in a 1:1 volume ratio. Mixing was carried out by completing 6 rotations of the merged drops (where one rotation comprised of moving the drop over 4 electrodes in a counter-clockwise pattern). This yielded 800 nl of a 25% solution of DMSO that was split into two 400 nl drops—one for delivery and the other for further dilution. Mixing the 400 nl 25% DMSO drop with another 400 nl drop of fluorescent dye in the same manner as discussed above (6 rotations) yielded a 12.5% DMSO solution from which one 400 nl drop was dispensed, while the other was sent to waste. In this manner, 400 nl drops of DMSO with concentrations of 50%, 25%, and 12.5% were obtained while the 0% concentration was a 400 nl control drop of fluorescent dye. By using the above protocol, the fluorescent dye concentration remained the same across all the drops and the only variable that changed was the percentage of DMSO.
F. Targeted chemical delivery experiment protocol
In order to carry out the targeted drug delivery experiments, MCF-7 cell seeded alginate hydrogels were created at designated tissue post sites. This was done by dispensing cell seeded sodium alginate drops and delivering them to designated gel formation sites. Calcium chloride drops (400 nl) was similarly dispensed from its parent reservoir and brought to the tissue post sites where they were merged with sodium alginate drops and gelation was allowed to occur. During this gelation process, simultaneous preparation of 400 nl drops with different concentrations of DMSO (0%, 12.5%, 25%, and 50%) was carried out in another area on the same chip using the DMSO dilution protocol as described earlier. After allowing sufficient time for gelation (∼7 min), excess liquid from the tissue posts was extracted and dispensed to waste. The 4 different DMSO solutions were then delivered to the target tissue posts and the whole chip was incubated for 30 min in a 37 °C, 5% CO2 humidified incubator following which fluorescence images were then taken using the protocol described below.
G. Fluorescence microscopy and cell counting
Fluorescence images were taken using fluorescence microscope (Olympus BX-51) to visualize the cells in the gel posts stained by the fluorescent dyes. H-33342 dye stained all the cells present blue when viewed under DAPI filters while dead cells were labelled red by PI dye when viewed under TRITC filters.
Fluorescent images thus obtained were combined using ImageJ (NIH), so that the dead cells labelled red were overlaid with all the cells present in the sample that were stained blue. The resulting image clearly allowed viable cells to be distinguished from dead cells.
For experiments where viability was measured, cell counting was carried out manually using combined fluorescent images and the cell counter plugin in ImageJ software. This yielded the total number of live and dead cells, which were then used to calculate the viability percentage. The targeted chemical delivery experiment was repeated 3 times and results were presented as viability percentage ±1 standard deviation.
III. RESULTS AND DISCUSSION
A. Alginate gelation on EWOD DMF
Since no detailed examination of alginate gel formation has been carried out on EWOD DMF to date, it is imperative to characterize the method of gel formation and establish a working protocol through which hydrogels can be reliably fabricated on chip. In order to do this, initial investigations were carried out on EWOD devices with individual electrode dimensions of 2 mm × 2 mm and a 100 μm spacer gap thickness which allowed for the creation and manipulation of droplets of 400 nl volume.
The basic protocol of gel formation on EWOD DMF consisted of merging two drops of cell seeded sodium alginate and calcium chloride in a 1:1 ratio. After allowing time for gelation, excess liquid was to be removed so that fresh liquid (culture media or any desired reagent/chemical) could be delivered to the cell seeded gel. This protocol is schematically depicted in Fig. 1.
FIG. 1.

Schematic of gelation protocol in EWOD DMF. (a)–(c) Dispensing of cell seeded sodium alginate solution droplet from the reservoir, (d)–(e) formation of calcium alginate gel post, and (f)–(g) extraction of excess liquid followed by addition of culture media/chemical reagent to the gel post. Blue arrows indicate direction of drop motion by EWOD electrode actuation. (h) A cell seeded alginate gel that was obtained on chip as a result of procedures (a)–(e) where the cells can be clearly seen contained within the gel boundary while surrounding the gel is excess liquid. Scale bar in (h) corresponds to 100 μm.
After merging the sodium alginate and calcium chloride drops and allowing 15 min for gelation, a clear 2 phase boundary could be seen consisting of an outer clear phase and an inner cell containing phase. The outer phase behaved as a liquid and responded well to electrode actuation. The cell-containing inner phase was unresponsive to electrode actuation though it could be dragged by the outer liquid phase when the outer liquid phase was moving. Even though the inner phase could be moved by the outer liquid phase, the cells within the inner phase remained fixed in place relative to other cells. This is in contrast to cells in the cell seeded liquid sodium alginate drops prior to merging, in which cells recirculate within the sodium alginate drop during drop motion. Thus, the lack of cell motion relative to other cells in the inner phase confirmed that this inner phase was behaving more like a solid than a liquid. This led to the conclusion that the inner phase was the calcium alginate hydrogel that had immobilized the cells and the outer phase was the excess liquid formed after gelation.
It was further observed that the method of merging the two drops affected the gel shapes. When sodium alginate drops were moved and merged with stationary calcium chloride drops as seen in Fig. 2(a), highly irregular gel shapes were formed. Such gels had a longer “tail” and covered more than one electrode. Whereas, when sodium alginate drops were kept stationary and calcium chloride drops were moved, irregularity in gel shapes were suppressed as seen in Fig. 2(b) which were well contained within a single electrode volume.
FIG. 2.

(a) and (b) Methods of merging sodium alginate and calcium chloride drops and the resulted gel shapes. (c) An example of irregular gel shapes when the merged sodium alginate—calcium chloride drops are actuated before complete gelation. Black dotted lines were added to figure to help identify the gel boundaries, while blue dotted lines indicate electrode boundaries. Scale bars represent 0.5 mm.
It was found that enough time should be provided for complete gelation. When the calcium chloride drop is merged with sodium alginate, gelation at the interface is almost instantaneous, resulting in the formation of a thin hydrogel layer. If the system is disturbed through EWOD actuation before sufficient time has been provided for complete gelation (15 min in this case), the liquid components of the system (calcium chloride and sodium alginate) respond to EWOD actuation while the thin layers of calcium alginate hydrogel already formed remains unresponsive. Due to this relative motion of the liquid components of the partly gelled system, new interfaces between sodium alginate and calcium chloride were created resulting in instantaneous gelation at the newly exposed interfaces. This had the effect of creating highly irregular gel shapes as shown in Fig. 2(c) where the gel system was disturbed after allowing only 5 min for gelation as opposed to the regular 15 min normally provided.
Removal of excess or waste liquid from a gel post is an essential step before new liquids are delivered. Otherwise, cell metabolism waste products would accumulate and lead to toxic conditions affecting viability. If liquid delivery to the gel post is continued without waste liquid removal, then liquid accumulation would occur. Large pools would eventually form around the gel rendering it ineffective from a chemical screening point of view since any chemicals delivered would be diluted to unknown concentrations.
Three different factors were identified as being responsible for difficulty in excess liquid removal in the EWOD DMF setup. The first is gel size. Gels formed of comparable size to the electrode dimensions preoccupy the fluidic paths, which prevents liquid from flowing around it to reach the adjacent electrode and being extracted from the gel. This led to a complete failure of excess liquid removal and resulted in a large pool of waste accumulation around a gel post.
The second factor affecting excess liquid removal is irregular gel shape. The irregular boundaries created pockets where excess liquid was trapped and left behind after partial removal of excess liquid (Fig. 2(c)). Although a major portion of excess liquid was removed, the remaining liquid trapped in pockets would serve to dilute any incoming chemicals and a variable chemical concentration gradient would be created locally, which hinders the accurate evaluation of chemical concentration and gradient effects on cell response.
The third factor identified for excess liquid removal was the need for gel anchoring. During excess liquid removal attempts, situations arose where the gel itself got carried away with the excess liquid, making it impossible to hold the gel in place. Under this scenario, it would be impossible to reliably address the gel posts and separate liquid from it in an automated fashion. Dragging the gels around would also possibly degrade the gel integrity and add additional variables such as shear stress to the cells, which would also affect the cell response.
Based on these factors identified, device design criteria were established as follows: In order to have better control over gel shapes, it is important to ensure that the sodium alginate gel precursor is kept stationary and only calcium chloride is moved during the gel formation stage. It is also essential to ensure that sufficient time has been provided for complete gelation since disturbing the system before complete gelation results in irregular shapes. To enable excess liquid removal, gels would have to be smaller in size than electrode dimensions. A method to create regular gel shapes would also have to be implemented to avoid the liquid pocket problem mentioned above. Also, means to anchor the gel or to hold it in place would have to be devised.
B. Designs for well controlled gel shapes
Based on the design criteria mentioned above, gelation sites smaller than a DMF electrode were considered. In addition, circular gels were identified as being an ideal shape to allow easy flow of liquid around the gels with minimal resistance. The circular shape would ensure that no pockets of liquid got trapped by the gel during excess liquid removal. The radial symmetry would also ensure that drug diffusion would occur uniformly through the gel.
Two different designs for more reliable gel formation and excess liquid separation were proposed. Device A (device with hydrophilic gel formation and anchoring sites) is shown in Fig. 3(a). Here, a smaller circular hydrophilic gel formation site (0.56 mm radius) was embedded in a larger 2 mm × 2 mm fluid delivery electrode pathway through which fluids would be delivered to and from the gel. Also, circular hydrophilic patterning of the gel formation site by Teflon lift off was carried out to ensure that circular gels were formed and anchored to prevent gel displacement during excess liquid extraction.
FIG. 3.

(a) Device A: Magnified section of EWOD layout used for hydrogel formation by using hydrophilic patterned sites (yellow circle) for gel formation. Blue lines show pathways for liquid delivery (calcium chloride, culture media, etc.) to the gel. (b) Device B: shows a magnified section of the EWOD mask design used for the encapsulation design. Blue dotted arrows show the pathways for fluid transport on 1.9 mm × 1.9 mm electrodes. Inset magnified image (c) shows the separator ring electrode (in green) that allows for on demand merging of the calcium chloride with sodium alginate. Blue arrows with numbers show how calcium chloride enters the encapsulating electrode (1) and then encapsulate the gel formation site (2). Actuation of the separator ring electrode causes calcium chloride to flow into and merge with the sodium alginate at the gel formation site as shown by red arrows (3). (b) and (c) Reprinted with permission from S. M. George and H. Moon, in 28th IEEE International Conference on Micro Electro Mechanical Systems (IEEE, 2015), pp. 443–446. Copyright 2015 IEEE.
In device B seen in Fig. 3(b), an encapsulation method was adopted.48 This method sought to form smaller circular gels and anchor them in place on the basis of intelligent electrode design rather than resorting to the use hydrophilic patterning. Here, a separator ring electrode (of thickness 70 μm with additional 15 μm space on either side from adjacent electrodes) was employed to separate the smaller circular gel formation site (radius 0.60 mm) from the encapsulating electrode (outer radius 1.25 mm) by a total distance of 100 μm. This would allow calcium chloride to fully surround the sodium alginate at the gel formation without merging. Once fully surrounded and encapsulated by calcium chloride, actuation of the separator ring electrode would cause the calcium chloride to merge on demand with sodium alginate. The encapsulation method of gel formation was expected to produce more symmetrical and uniform gel shapes since the instantaneous merging of the entire outer boundary of the sodium alginate drop with calcium chloride would immediately define the outer gel boundaries into a circular shape. This would thus eliminate any chance of irregular gels being formed due to premature electrode activation.
1. Hydrophilic gel site design (device A) testing
100 nl sodium alginate drops were dispensed from a reservoir and delivered to the hydrophilic gel formation site through a dedicated alginate transport pathway. A regular 88 Vrms at 1 kHz EWOD actuation voltage was used. Circular drops of sodium alginate (liquid) were formed due to circular hydrophilic sites. 400 nl calcium chloride was then brought to the gel formation sites and calcium alginate gelation was allowed to occur for ∼10 min (compared to 15 min provided for larger gels in previous experiment) before excess liquid separation was attempted. Repeated experiments revealed that the hydrophilic site for gel formation and anchoring was not a reliable approach. The hydrophilic patterning ensured that liquid always occupied the gel formation site as shown in Fig. 4(a), but it could not differentiate between sodium alginate and calcium chloride. When the intermediate electrode surrounding the gel formation site was turned on, a thin layer of calcium alginate hydrogel was instantly formed at the interface between sodium alginate and calcium chloride. The momentum of the incoming calcium chloride drop was transmitted through the thin calcium alginate hydrogel to the still liquid sodium alginate, causing the incomplete gelled system to be displaced out of the hydrophilic site and towards the adjacent electrode as seen in Fig. 4(b). Since gelation was occurring during this displacement stage, the final result was a very irregular triangular shaped gel. Although excess liquid separation was possible as evidenced in Figs. 4(c) and 4(d) (Multimedia view), the shape of the gel ensured that residual liquid (with volume ∼equal volume of the gel) remained trapped by the gel and the hydrophilic patterned site. This was already described as an undesirable phenomenon in the alginate gelation section.
FIG. 4.

In order to reduce the sodium alginate displacement by incoming calcium chloride, the entry velocity of calcium chloride into the intermediate electrode surrounding the gel formation site was reduced. This was accomplished by reducing the electrode actuation voltage since this is known to lower drop speed.49 By reducing the intermediate electrode voltage actuation from 88 to 53 Vrms, the velocity with which calcium chloride entered the intermediate electrode was reduced. This resulted in less displacement of sodium alginate by incoming calcium chloride as seen in Fig. 4(f) and a less irregular gel shape than that obtained in the previous case. However, the small amount of sodium alginate displacement still resulted in the gel being formed partly outside the hydrophilic gel formation site. This caused the entire gel to be carried away by the excess liquid during the excess liquid removal step as depicted in Figs. 4(g) and 4(h) (Multimedia view). The hydrophilic patterned site ensured that a drop of excess liquid remained in the place where the gel should have been secured.
The hydrophilic patterned gel formation site on their own did not result in an improvement in gel shape or gel anchoring and was deemed to be unsuitable for the purpose of a 3D tissue based chemical screening platform.
2. Encapsulation design (device B) testing
In order to the separator ring in the encapsulation design (device B, Fig. 3(b)) to work effectively, it is imperative that the sodium alginate dispensed at the gel formation site does not overflow its boundaries. If sodium alginate dispensed at the gel formation site is too large, then it could overflow beyond the separator ring and lie partly in the outer encapsulating electrode. This would render the separator ring ineffective since when the outer encapsulating electrode is actuated to bring in calcium chloride, the calcium chloride could merge with the sodium alginate before complete encapsulation resulting in non-uniform gel shapes.
In order to obtain precise volumes of sodium alginate contained within the gel formation site, protocol change was adopted. In previous designs, alginate was always dispensed from a separate reservoir and then delivered to the gel formation site. However, errors in dispensed volume using conventional reservoirs approaches 30%,50 so another approach had to be implemented as seen in Figs. 5(a)–5(c) (Multimedia view). Here, a large drop (∼530 nl) of sodium alginate was brought to the gel formation site to cover both the gel formation site and the encapsulating ring. By actuating both the inner gel formation site electrode as well as providing backing force through the main electrode pathways, an exact amount of sodium alginate (113 nl) could be dispensed at the gel formation. This precision dispensing at the gel formation site eliminated any possibility of sodium alginate flowing outside of its designated gel formation site during actuation of the encapsulating electrode or the separator ring.
FIG. 5.

Following the precision dispensing of sodium alginate, calcium chloride (400 nl volume) was brought to the encapsulating electrode where it fully encapsulated the sodium alginate at the gel formation site without merging in the process as seen in Figs. 5(d)–5(f). Once complete encapsulation was achieved, the separator ring was actuated which allowed for the controlled merging of the inner sodium alginate drop with the outer calcium chloride drop. Since the separator ring only separated the two drops by a total distance of 100 μm, there was little fluid flow and momentum transfer and the overall gel shape retained the circular shape of the gel formation site electrode. Also, no displacement of the sodium alginate was observed from the gel formation site and the resulting calcium alginate hydrogels always formed at the circular gel formation site. This ensured that the gel always remained in place during excess liquid removal and never got carried away with excess liquid. Since no barrier or hydrophilic patterning was required to retain the gel in place unlike other EWOD DMF devices reported for hydrogel based 3D cell culture and screening,28,29 this design proved to be easy to fabricate and implement and offers a simple solution not just for alginate hydrogels but any other hydrogel based system.
Using the improved encapsulation method, calcium alginate hydrogels could be formed of consistent and symmetrical circular shape between different gel formation sites on the same device as well as across multiple devices. The circular shape of the alginate hydrogel provided minimal resistance to the removal of excess liquid as seen in Figs. 5(g)–5(i). This new encapsulation method ensured that alginate hydrogels could be created in a convenient and fast manner requiring a gelation time of only 5–7 min and that excess liquids could be removed and new liquids added without loss of gel integrity or gel displacement.
C. Targeted chemical delivery
To demonstrate the targeted chemical delivery abilities of this proposed screening platform, four MCF-7 seeded calcium alginate hydrogels were formed on chip as per the improved encapsulation method. While gelation was taking place, different concentrations (0%, 12.5%, 25%, and 50%) of DMSO drops (400 nl volume) were formed on chip as per the method described in the Material and Methods section (Sec. II). Once the calcium alginate hydrogel gelation was complete, excess liquid was removed and discarded to waste and the 4 different DMSO concentration drops were delivered to 4 gels and incubated for 30 min. Fig. 6 shows one result from such an experiment where cells were seeded at a high density to allow better visualization of the variation in cell counts. Based on the fluorescence images, a clear difference in viability of cells is observed. Cell viability decreases as DMSO concentration increases.
FIG. 6.
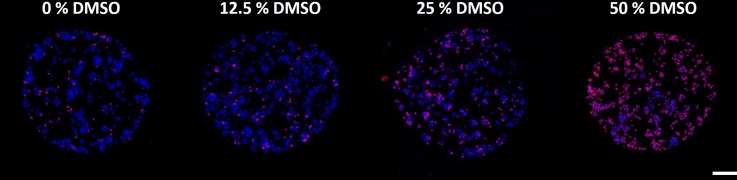
Effects of on chip DMSO delivery at 4 different concentrations (0%, 12.5%, 25%, and 50%) to MCF-7 cell seeded calcium alginate hydrogel post. Hydrogels shown here were seeded at high cell density to allow for easy visualization of the variation in viability. These gels were stained with Hoechst-33342 and Propidium Iodide and the resulting fluorescent images were superimposed such that pink dots represent dead cells and blue dots represent viable cells. A clear increase in cell death can be visually observed as DMSO concentration increases. Scale bar represents 100 μm. Reprinted with permission from S. M. George and H. Moon, in 28th IEEE International Conference on Micro Electro Mechanical Systems (IEEE, 2015), pp. 443–446. Copyright 2015 IEEE.
In order to determine the repeatability of this experimental procedure, 3 sets of the same experiment were carried out with cells seeded at lower density (in order to facilitate counting of cells). The variation in cell viability with exposure to 4 different concentrations of DMSO is shown in Fig. 7(a). The 0% DMSO concentration serves as the control gel to show the viability of cells formed on chip which are not exposed to any DMSO. The 12.5% DMSO gels showed a very slight decrease in viability compared to the control gels and this is to be expected since it is well known that exposure to low concentrations (∼10% DMSO) for short durations of time have a negligible effect on viability.
FIG. 7.

(a) Plot showing how cell viability varied based on exposure to DMSO at various concentrations for 30 min. Cell viability at 12.5% DMSO concentration is only nominally lower than the control post exposed to 0% DMSO. However, as DMSO concentration increases, a clear dip is seen in viability at 25% and at 50% DMSO, virtually all the cells have died. Error bars (in red) represent ±1 standard deviation, n = 3. (b) Plot showing time lapse variation in cell viability % during a single experiment (n = 1) as hydrogels are exposed to varying DMSO concentration (0%, 12.5%, and 25%) for 30, 60, 90, and 120 min. The cell seeded gel exposed to 0% DMSO shows almost no increase in cell death over time. The cell seeded gel exposed to 12.5% DMSO shows an initial increase in cell death over the first 60 min but then stabilizes with no major increase in cell death at 90 min and 120 min of exposure. However, the cell seeded gel exposed to 25% DMSO experience a sharp rate of cell death over the first 90 min before starting to taper off at 120 min. Error bars represent ±5% error to account for errors in cell counting. Reprinted, with permission, from S. M. George and H. Moon, in 28th IEEE International Conference on Micro Electro Mechanical Systems (IEEE, 2015), pp. 443–446. Copyright 2015 IEEE.
The gels exposed to 25% and 50% concentrations of DMSO show a marked decrease in viability with 25% DMSO concentrations showing a viability of 72.05% ± 0.85, while the 50% DMSO concentrations killing off all the cells in the 30 min exposure time.
One notable result is the high degree of repeatability with the significantly small standard deviations when compared with the relatively high level of variation in biological experiments. This is attributed to the high degree of automation and robustness of the platform ensuring that variations arising from manual handling between experiments were kept to a minimum.
In order to demonstrate the ability to note changes in response of cells to DMSO over time, cell seeded alginate gels were formed on chip. While gelation was allowed to happen, different concentrations of DMSO (0%, 12.5%, and 25%) were prepared on the same chip through serial dilution. Once gelation was complete and excess liquid was separated, the 3 different DMSO concentration drops were delivered to the tissue posts and the device was incubated. Fluorescent images were taken at 30 min intervals to measure the effect of DMSO over a span of 2 h on cell laden hydrogels. Fig. 7(b) shows the resultant viabilities obtained at each time step for each gel post exposed to DMSO concentration. From the graph, it can clearly be seen that there is minimal decrease in viability of the cells in the control gel exposed to 0% DMSO. However, a decrease in cell viability could be seen over the first 60 min for the 12.5% DMSO treated gel followed by no major increase in cell death at 90 min and 120 min. The 25% DMSO treated gel on the other hand showed a consistent increase in cell death at every 30 min interval for the first 90 min with the cell death rate starting to taper off at 120 min.
Such kinds of time lapse experiments provide valuable insight into the effect of chemicals over time on cells without having to carry out multiple experiments. If images were only taken at a single time interval, then the only conclusion drawn would be that an increase in cell death occurs based on DMSO concentration exposure. Repeating the experiment with different time spans would show that cell death increases over time as well. However, being able to monitor the variation in cell death during the same experiment over time reveals that cell death in the 12.5% DMSO post increases only over the first 60 min and after that remains more or less constant. This kind of observation relating the nature of cell death with both time and concentration would be hard to make using conventional experimental setups without having to carry out multiple experiments. The EWOD DMF platform provides a simple and convenient means to take rapid images of multiple gel posts over multiple time frames with ease allowing investigators the ability to observe the transient effect of chemicals on cells cultured in a 3D environment.
IV. CONCLUSIONS
Calcium alginate hydrogel gelation in EWOD DMF has been investigated and the factors affecting gel shape were identified. An encapsulation design that allowed consistent and stable alginate hydrogel gels during fluidic motion of reagent chemicals was developed and tested. The design changes implemented were successful and no extra steps in fabrication (such as creating physical barriers like weirs or chemical patterning) had to be taken to retain the gel in place during liquid delivery and removal. This encapsulation design offers potential not only for alginate based hydrogels but also can be extended to any hydrogel applications.
The encapsulation design was tested for use as a 3D cell culture platform for chemical screening and the differential effects of DMSO delivery on cell viability in alginate hydrogels were demonstrated. A time lapse experiment was also demonstrated showing how the effects of different chemical concentrations on cells could be tracked temporally. This showcases the potential for EWOD DMF to be used for hydrogel based cell culture and chemical screening.
ACKNOWLEDGMENTS
The authors acknowledge the support by National Science Foundation CAREER award (Grant No. ECCS-1254602).
References
- 1.Lombardino J. G. and Lowe J. A., Nat. Rev. Drug Discovery 3, 853 (2004). 10.1038/nrd1523 [DOI] [PubMed] [Google Scholar]
- 2.Hay M., Thomas D. W., Craighead J. L., Economides C., and Rosenthal J., Nat. Biotechnol. 32, 40 (2014). 10.1038/nbt.2786 [DOI] [PubMed] [Google Scholar]
- 3.Cukierman E., Pankov R., Stevens D. R., and Yamada K. M., Science 294, 1708 (2001). 10.1126/science.1064829 [DOI] [PubMed] [Google Scholar]
- 4.Hutmacher D. W., Nat. Mater. 9, 90 (2010). 10.1038/nmat2619 [DOI] [PubMed] [Google Scholar]
- 5.Pampaloni F., Reynaud E. G., and Stelzer E. H. K., Nat. Rev. Mol. Cell Biol. 8, 839 (2007). 10.1038/nrm2236 [DOI] [PubMed] [Google Scholar]
- 6.Gurski L. A., Petrelli N. J., Jia X., and Farach-carson M. C., Oncol. Issues 25, 20 (2010). [Google Scholar]
- 7.Zang R., Li D., Tang I.-C., Wang J., and Yang S.-T., Int. J. Biotechnol. Wellness Ind. 1, 31 (2012). 10.6000/1927-3037.2012.01.01.02 [DOI] [Google Scholar]
- 8.Khademhosseini A. and Langer R., Biomaterials 28, 5087 (2007). 10.1016/j.biomaterials.2007.07.021 [DOI] [PubMed] [Google Scholar]
- 9.Tibbitt M. W. and Anseth K. S., Biotechnol. Bioeng. 103, 655 (2009). 10.1002/bit.22361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toh Y.-C., Lim T. C., Tai D., Xiao G., van Noort D., and Yu H., Lab Chip 9, 2026 (2009). 10.1039/b900912d [DOI] [PubMed] [Google Scholar]
- 11.Lee J., Lilly G. D., Doty R. C., Podsiadlo P., and Kotov N. A., Small 5, 1213 (2009). 10.1002/smll.200801788 [DOI] [PubMed] [Google Scholar]
- 12.Goral V. N., Hsieh Y.-C., Petzold O. N., Clark J. S., Yuen P. K., and a Faris R., Lab Chip 10, 3380 (2010). 10.1039/c0lc00135j [DOI] [PubMed] [Google Scholar]
- 13.Lii J., Hsu W.-J., Parsa H., Das A., Rouse R., and Sia S. K., Anal. Chem. 80, 3640 (2008). 10.1021/ac8000034 [DOI] [PubMed] [Google Scholar]
- 14.Sung J. H. and Shuler M. L., Lab Chip 9, 1385 (2009). 10.1039/b901377f [DOI] [PubMed] [Google Scholar]
- 15.Xu F., Wu J., Wang S., Durmus N. G., Gurkan U. A., and Demirci U., Biofabrication 3, 034101 (2011). 10.1088/1758-5082/3/3/034101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon H., Cho S. K., Garrell R. L., and Kim C.-J., J. Appl. Phys. 92, 4080 (2002). 10.1063/1.1504171 [DOI] [Google Scholar]
- 17.Cho S. K., Moon H., and Kim C.-J., J. Microelectromech. Syst. 12, 70 (2003). 10.1109/JMEMS.2002.807467 [DOI] [Google Scholar]
- 18.Wheeler A. R., Moon H., Kim C.-J., Loo J. A., and Garrell R. L., Anal. Chem. 76, 4833 (2004). 10.1021/ac0498112 [DOI] [PubMed] [Google Scholar]
- 19.Wijethunga P. A. L., Nanayakkara Y. S., Kunchala P., Armstrong D. W., and Moon H., Anal. Chem. 83, 1658 (2011). 10.1021/ac102716s [DOI] [PubMed] [Google Scholar]
- 20.Nelson W. C. and Kim C.-J., J. Adhes. Sci. Technol. 26, 1747 (2012). 10.1163/156856111X599562 [DOI] [Google Scholar]
- 21.Barbulovic-Nad I., Au S. H., and Wheeler A. R., Lab Chip 10, 1536 (2010). 10.1039/c002147d [DOI] [PubMed] [Google Scholar]
- 22.Shih S. C. C., Barbulovic-Nad I., Yang X., Fobel R., and Wheeler A. R., Biosens. Bioelectron. 42, 314 (2013). 10.1016/j.bios.2012.10.035 [DOI] [PubMed] [Google Scholar]
- 23.Srigunapalan S., Eydelnant I. A., Simmons C. A., and Wheeler A. R., Lab Chip 12, 369 (2012). 10.1039/C1LC20844F [DOI] [PubMed] [Google Scholar]
- 24.Barbulovic-Nad I., Yang H., Park P. S., and Wheeler A. R., Lab Chip 8, 519 (2008). 10.1039/b717759c [DOI] [PubMed] [Google Scholar]
- 25.Bogojevic D., Chamberlain M. D., Barbulovic-Nad I., and Wheeler A. R., Lab Chip 12, 627 (2012). 10.1039/C2LC20893H [DOI] [PubMed] [Google Scholar]
- 26.George S. M. and Moon H., in ASME 2011 Summer Bioengineering Conference, Parts A and B ( ASME, 2011), p. 105. [Google Scholar]
- 27.George S. M. and Moon H., in 15th International Conference Miniaturized Systems for Chemistry and Life Science, Seattle, 2011. [Google Scholar]
- 28.Eydelnant I. A., Betty Li B., and Wheeler A. R., Nat. Commun. 5, 3355 (2014). 10.1038/ncomms4355 [DOI] [PubMed] [Google Scholar]
- 29.Au S. H., Chamberlain M. D., Mahesh S., Sefton M. V., and Wheeler A. R., Lab Chip 14, 3290 (2014). 10.1039/C4LC00531G [DOI] [PubMed] [Google Scholar]
- 30.West E. R., Xu M., Woodruff T. K., and Shea L. D., Biomaterials 28, 4439 (2007). 10.1016/j.biomaterials.2007.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowley J. A., Madlambayan G., and Mooney D. J., Biomaterials 20, 45 (1999). 10.1016/S0142-9612(98)00107-0 [DOI] [PubMed] [Google Scholar]
- 32.Lee K. Y. and Mooney D. J., Prog. Polym. Sci. 37, 106 (2012). 10.1016/j.progpolymsci.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunt N. C. and Grover L. M., Methods Mol. Biol. 1014, 201 (2013). 10.1007/978-1-62703-432-6_14 [DOI] [PubMed] [Google Scholar]
- 34.Kuo C. K. and Ma P. X., Biomaterials 22, 511 (2001). 10.1016/S0142-9612(00)00201-5 [DOI] [PubMed] [Google Scholar]
- 35.Orive G., Ponce S., Hernández R., Gascón A., Igartua M., and Pedraz J., Biomaterials 23, 3825 (2002). 10.1016/S0142-9612(02)00118-7 [DOI] [PubMed] [Google Scholar]
- 36.Braschler T., Johann R., Heule M., Metref L., and Renaud P., Lab Chip 5, 553 (2005). 10.1039/b417604a [DOI] [PubMed] [Google Scholar]
- 37.Manojlovic V., Djonlagic J., Obradovic B., Nedovic V., and Bugarski B., J. Chem. Technol. Biotechnol. 81, 505 (2006). 10.1002/jctb.1465 [DOI] [Google Scholar]
- 38.Augst A. D., Kong H. J., and Mooney D. J., Macromol. Biosci. 6, 623 (2006). 10.1002/mabi.200600069 [DOI] [PubMed] [Google Scholar]
- 39.Martinez C. J., Kim J. W., Ye C., Ortiz I., Rowat A. C., Marquez M., and Weitz D., Macromol. Biosci. 12, 946 (2012). 10.1002/mabi.201100351 [DOI] [PubMed] [Google Scholar]
- 40.Choi C.-H., Jung J.-H., Rhee Y. W., Kim D.-P., Shim S.-E., and Lee C.-S., Biomed. Microdevices 9, 855 (2007). 10.1007/s10544-007-9098-7 [DOI] [PubMed] [Google Scholar]
- 41.Yu L., Chen M. C. W., and Cheung K. C., Lab Chip 10, 2424 (2010). 10.1039/c004590j [DOI] [PubMed] [Google Scholar]
- 42.Wong A. P., Perez-Castillejos R., Christopher Love J., and Whitesides G. M., Biomaterials 29, 1853 (2008). 10.1016/j.biomaterials.2007.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johann R. M. and Renaud P., Biointerphases 2, 73 (2007). 10.1116/1.2746873 [DOI] [PubMed] [Google Scholar]
- 44.Lee M.-Y., Kumar R. A., Sukumaran S. M., Hogg M. G., Clark D. S., and Dordick J. S., Proc. Natl. Acad. Sci. U. S. A. 105, 59 (2008). 10.1073/pnas.0708756105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luk V. N., Mo G. C., and Wheeler A. R., Langmuir 24, 6382 (2008). 10.1021/la7039509 [DOI] [PubMed] [Google Scholar]
- 46.Eydelnant I. A., Uddayasankar U., Li B., Liao M. W., and Wheeler A. R., Lab Chip 12, 750 (2012). 10.1039/C2LC21004E [DOI] [PubMed] [Google Scholar]
- 47.Park S., Wijethunga P. A. L., Moon H., and Han B., Lab Chip 11, 2212 (2011). 10.1039/c1lc20111e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.George S. M. and Moon H., in 28th IEEE International Conference on Micro Electro Mechanical Systems ( IEEE, 2015), pp. 443–446. [Google Scholar]
- 49.Lu H.-W., Glasner K., Bertozzi A. L., and Kim C.-J., J. Fluid Mech. 590, 411 (2007). 10.1017/S0022112007008154 [DOI] [Google Scholar]
- 50.Nikapitiya J. Y., Ph.D thesis, University of Texas at Arlington, 2013. [Google Scholar]