

Abstract
A catalytic C–H alkylation using unactivated alkyl halides and a variety of arenes and heteroarenes is described. This ring-forming process is successful with a variety of unactivated primary and secondary alkyl halides, including those with β-hydrogens. In contrast to standard polar or radical cyclizations of aromatic systems, electronic activation of the substrate is not required. The mild, catalytic reaction conditions are highly functional group tolerant and facilitate access to a diverse range of synthetically and medicinally important carbocyclic and heterocyclic systems.
The cyclization of aromatic substrates with simple alkyl halides is an important approach to polycyclic carbocycles and heterocycles, most notably via Friedel–Crafts reactions.1 While these are valuable transformations in synthesis, the substrate scope is limited owing to the requirement of electron-rich aromatic substrates and the utilization of stoichiometric (or super-stoichiometric) amounts of strong Lewis acids. Radical-mediated homolytic aromatic substitutions offer an alternative approach, but often necessitate electron-poor aromatic substrates for efficient reactivity, with reductive dehalogenation commonly observed.2 Substitution of alkyl halides for alkyl xanthates (dithiocarbonates) in homolytic aromatic substitutions is an excellent option for addressing these limitations owing to the persistent radical effect, however this requires additional synthetic effort to access desired substrates and involves stoichiometric amounts of reactive peroxides (e.g., DLP, dilauroyl peroxide).3 Transition metal catalysts have proven useful in select aromatic C–H alkylations involving the cyclization of activated alkyl halides (α-halocarbonyls),4,5 or intermolecular reactions between aromatic substrates containing appended directing groups and unactivated alkyl halides.6,7 A general, catalytic ring-forming C–H alkylation of arenes and heteroarenes using unactivated alkyl halides has not been reported, yet would be highly enabling in the synthesis of polycyclic compounds.
We have recently reported a general approach to the palladium-catalyzed alkyl-Heck-type cross-coupling that our initial studies indicated proceeds via a hybrid organometallic-radical pathway (Scheme 1).8 We hypothesized that this approach to C–C bond formation using unactivated alkyl halides could be extended to aromatic substrates, constituting a mild, catalytic C–H alkylation with distinct advantages over Friedel–Crafts or standard radical-mediated homolytic aromatic substitutions. Herein we report the successful development of such a palladium-catalyzed ring-forming C–H alkylation, amenable to a diverse array of simple unactivated alkyl halide electrophiles, arenes, and heteroarenes.
Scheme 1. Palladium-Catalyzed Cyclizations of Unactivated Alkyl Halides.
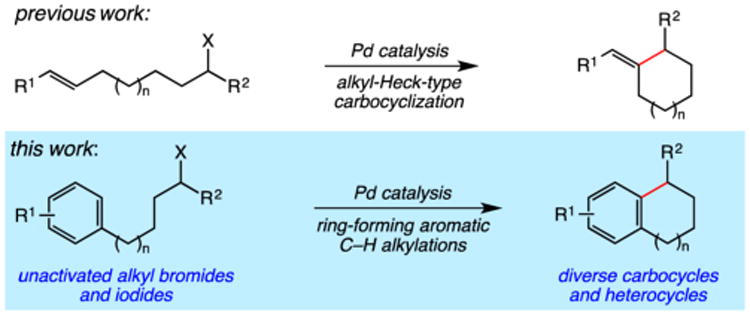
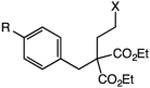
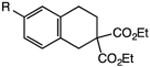
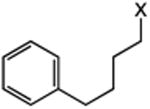
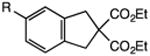
Our efforts commenced with the construction of a range of tetrahydronapthalenes and indanes via the catalytic alkylation of arenes using alkyl bromides and iodides (Table 1). The cyclizations of both unactivated alkyl bromides and alkyl iodides were successful using 10 mol % Pd(PPh3)4 as catalyst, although the reactions of alkyl bromides required elevated temperatures (130 °C) as compared to the alkyl iodides (100 °C). As demonstrated in entries 1–8, there are no limitations with respect to the electronic characteristics of the aromatic substrate, which is an important advantage over prior approaches to ring-forming C–H alkylations. Reactions involving alkyl bromides and iodides proceeded with similar efficiencies, and the tetrahydronapthalene products were isolated in good to excellent yield (59–92%, entries 1–8). While a number of substrates in Table 1 feature a malonate tether to assist in their preparation, this reaction does not require the Thorpe–Ingold effect to favor cyclization (entries 9 and 10). The reaction of ortho-substituted aromatic substrate 16 was successful, although a 50:50 mixture of product 17 and rearrangement product 18 was produced (entry 11).9 The indane framework was also easily accessed via 5-exo cyclization (entries 12–15). However, with neopentyl substrates (entries 12–14) PhtBu was substituted for dioxane as solvent to reduce the amount of undesired reductive dehalogenation byproducts formed.
Table 1. Palladium-Catalyzed C–H Alkylations Accessing Tetrahydronapthalenes and Indanes.
| entry | substrate | product | yield(%)a |
|---|---|---|---|
|
|
||
| 1 | 1: X = Br, R = H | 3: R = H | 91 |
| 2 | 2: X = I, R = H | 85 | |
| 3 | 4: X = Br, R = CF3 | 6: R = CF3 | 92 |
| 4 | 5: X = I, R = CF3 | 68 | |
| 5 | 7: X = Br, R = OMe | 9: R = OMe | 78 |
| 6 | 8: X = I, R = OMe | 66 | |
| 7 | 10: X = Br, R = CI | 12: R = Cl | 75 |
| 8 | 11: X = I, R = CI | 59 | |
|
|
||
| 9b | 13: X = Br | 15 | 89 |
| 10 | 14: X = I | 51 | |
| 11 |
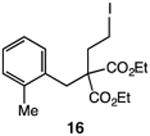
|
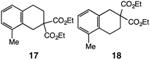
|
75 |
| 50:50 | |||
| 17:18 | |||
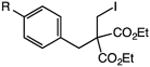
|
|
||
| 12c | 19: R = H | 20: R = H | 88d |
| 13c | 21: R = OMe | 22: R = OMe | 66d |
| 14c | 23: R = CI | 24: R = CI | 74d |
| 15 |
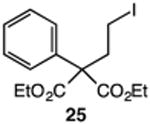
|
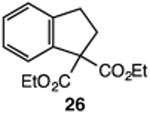
|
83 |
All reactions were performed with [substrate]0 = 0.5 M and 10 mol % Pd(PPh3)4 as catalyst. The reactions of alkyl bromides were performed in PhtBu at 130 °C with 2 equiv PMP (1,2,2,6,6-pentamethylpiperidine) as base. The reactions of alkyl iodides were performed in dioxane at 100 °C with 2 equiv K3PO4 as base.
Isolated yields.
K3PO4 used as base.
PhtBu used as solvent.
Calculated by 1H NMR spectroscopy of crude reaction mixtures using an internal standard.
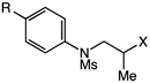
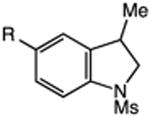
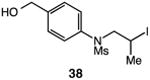
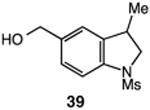
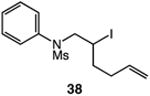
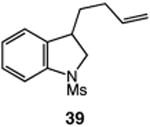
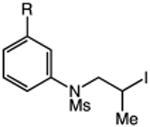
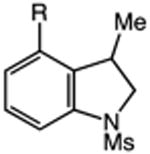
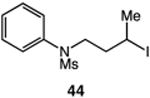
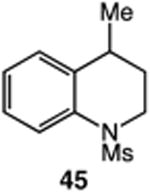
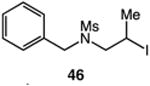
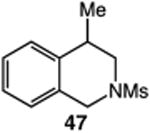
We next applied the catalytic C–H alkylation to the synthesis of a diverse set of synthetically and medicinally valuable heterocycles (Table 2). Cyclization of N-methanesulfonyl protected aniline substrates 27 (alkyl bromide) and 29 (alkyl iodide) delivered indoline product 28 in good yield (entries 1 and 2). As observed in the C–H alkylations of Table 1, these reactions did not require particular electronic modulation of the aromatic substrate as demonstrated in entries 1–4. The excellent functional group tolerance of this protocol is further exemplified by the successful reactions of entries 5–8, with substrates containing a boronic acid pinacol ester, ketone, alcohol, and alkene. We studied the ortho:para selectivity of the reaction using meta-substituted anilines 40 and 42 (entries 9 and 10). In both instances, the reactions produced a mixture of indoline products in high yield, with modest selectivity for the ortho functionalization products (2.4:1 and 2.0:1, ortho:para, respectively). Simple extension of the tether unit enabled access to the tetrahydroquinoline framework, as the reaction of substrate 44 provided N-methanesulfonyl 4-methyl-1,2,3,4-tetrahydroquinoline 45 in moderate yield (57%, entry 11). Changing the site of N-substitution in the tether permits facile access to the tetrahydroisoquinoline derivative (entry 12).10
Table 2. Synthesis of Diverse Polycyclic Heterocycles Using the Catalytic C–H Alkylation.
| Entry | Substrate | Product | yield(%)a |
|---|---|---|---|
|
|
||
| 1b | 27: X = Br, R = H | 28: R = H | 54 |
| 2 | 29: X = I, R = H | 82 | |
| 3 | 30: X = I, R = CF3 | 31: R = CF3 | 70 |
| 4 | 32: X = I, R = OMe | 33: R = OMe | 66 |
| 5 | 34: X = I, R = B(pin) | 35: R = B(pin) | 72 |
| 6 | 36: X = I, R = C(O)Me | 37: R = C(O)Me | 77 |
| 7 |
|
|
57 |
| 8 |
|
|
80 |
|
|
||
| 9 | 40: R = CF3 | 41: R = CF3 | 81: (2.4:1 o:p) |
| 10 | 42: R = Me | 43: R = Me | 91c(2.0:1 o:p) |
| 11 |
|
|
57 |
| 12 |
|
|
61 |
|
|
||
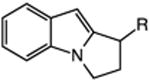
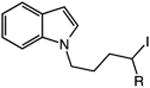
| 13 | 48: R = H | 49: R = H | 51c |
| 14 | 50: R = Me | 51: R = Me | 71c |
|
|
||
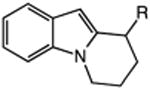
| 15 | 52: R = H | 53: R = H | 70c |
| 16 | 54: R = Me | 55: R = Me | 90c |
|
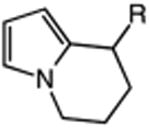
|
||
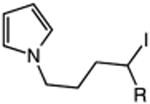
| 17 | 56: R = H | 57: R = H | 64c |
| 18 | 58: R = Me | 59: R=Me | 95c |
All reactions were performed with [substrate]0 = 0.5 M in dioxane at 100 °C with 10 mol % Pd(PPh3)4 and 2 equiv K3PO4 as base.
Isolated yields.
The reaction was performed at 130 °C in PhtBu.
Calculated by 1H NMR spectroscopy of crude reaction mixtures using an internal standard.
The catalytic C–H alkylation was also successful using indole and pyrrole heterocycles (entries 13–18). Reactions involving both primary and secondary alkyl iodides proceeded in good yields in these transformations. The reactions of indoles 48 and 50 afforded dihydro-1H-pyrrolo[1,2-a]indoles via 5-exo cyclization (entries 13 and 14). Extension of the methylene tether provided access to tetrahydropyrido[1,2-a]indoles via a 6-exo process (entries 15 and 16). The cyclizations of pyrrole substrates 56 and 58 successfully delivered tetrahydroindolizines 57 and 59 in 64% and 95% yield, respectively. Importantly, electronic activation of the indole or pyrrole nucleus—common to prior stoichiometric metal- or peroxide-mediated protocols for efficient cyclization—was not required using our mild, palladium-catalyzed approach. 2,3d
Following our synthetic studies, we performed a number of experiments to gain insight into the reaction mechanism (Scheme 2). The reaction of substrate 29 under standard conditions with 1 equiv of the persistent radical TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) yielded 60% of adduct 60 and no C–H alkylation product, consistent with the intermediacy of carbon-centered radicals. We additionally prepared enantioenriched (R)-29, which produced indoline 28 as a racemate, also consistent with a single-electron pathway rather than an SN2-type activation of the alkyl halide. This reaction was stopped at partial conversion to determine the enantiopurity of the remaining starting material. Interestingly, recovered 29 was also completely racemic. The observed stereoablation of 29 is consistent with a reversible single-electron activation of the alkyl halide substrate prior to cyclization. We have also performed an intermolecular competition experiment involving deuterated substrate 29-d5. No kinetic isotope effect was observed, demonstrating that C– H bond cleavage is not involved in the rate-determining step of the reaction.
Scheme 2. Studies Probing the Reaction Mechanism.

Our current mechanistic hypothesis for the C–H alkylation is shown in Scheme 3. The reaction is initiated by a reversible single-electron oxidative addition of the alkyl halide substrate, generating carbon-centered radical 61.11 The formation of the TEMPO adduct 60, and the generation of both racemic product and racemic starting material in a reaction involving an enantioenriched alkyl halide (Scheme 2) is consistent with a single-electron pathway. The carbon-centered radical then adds to the aromatic ring to generate a cyclohexadienyl radical intermediate 62. At this stage, rearomatization could occur via single-electron oxidation and loss of one proton with regeneration of the palladium(0) catalyst.12
Scheme 3. Plausible Mechanism for the Palladium-Catalyzed Ring-Forming C–H Alkylation.

In conclusion, we have developed a palladium-catalyzed approach to the direct ring-forming C–H alkylation of aromatic substrates using unactivated alkyl halides. The reaction is successful with both primary and secondary alkyl bromides and iodides, and efficiently delivers a diverse range of valuable carbocyclic and heterocyclic products. Electronic activation of the aromatic substrates is not required, significantly increasing the potential substrate scope of this process with respect to prior polar or radical-mediated ring-forming C–H alkylations. Furthermore, the mild, catalytic reaction conditions involved offer an attractive alternative to known stoichiometric Lewis acid or peroxide-mediated processes.
Supplementary Material
Acknowledgments
This work was supported by Award No. R01 GM107204 from the National Institute of General Medical Sciences and a UNC Chapel Hill SURF fellowship (P.T.B.).
Footnotes
Supporting Information: Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.(a) Hayashi R, Cook GR. In: Handbook of Cyclization Reactions. Ma S, editor. Vol. 2. Wiley-VCH; Weinheim: 2010. pp. 1025–1054. [Google Scholar]; (b) Rueping M, Nachtsheim BJ. Beilstein J Org Chem. 2010;6:6. doi: 10.3762/bjoc.6.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Bowman WR, Storey JMD. Chem Soc Rev. 2007;36:1803. doi: 10.1039/b605183a. [DOI] [PubMed] [Google Scholar]; (b) Allin SM, Barton WRS, Bowman WR, Bridge née Mann E, Elsegood MRJ, McInally T, McKee V. Tetrahedron. 2008;64:7745. [Google Scholar]; (c) Menes-Arzate M, Martínez R, Cruz-Almanza R, Muchowski JM, Osornio YM, Miranda LD. J Org Chem. 2004;69:4001. doi: 10.1021/jo0497048. [DOI] [PubMed] [Google Scholar]; (d) Artis DR, Cho IS, Jaime-Figueroa S, Muchowski JM. J Org Chem. 1994;59:2456. [Google Scholar]; (e) Vaillard SE, Schulte B, Studer A. In: Modern Arylation Methods. Ackermann L, editor. Wiley-VCH; Weinheim: 2009. pp. 475–511. [Google Scholar]
- 3.(a) Ly TM, Quiclet-Sire B, Sortais B, Zard SZ. Tetrahedron Lett. 1999;40:2533. [Google Scholar]; (b) Charrier N, Liu Z, Zard SZ. Org Lett. 2012;14:2018. doi: 10.1021/ol3005276. [DOI] [PubMed] [Google Scholar]; (c) Quiclet-Sire B, Zard SZ. Pure Appl Chem. 2011;83:519. [Google Scholar]; (d) Biechy A, Zard SZ. Org Lett. 2009;11:2800. doi: 10.1021/ol900996k. [DOI] [PubMed] [Google Scholar]
- 4.Liu C, Liu D, Zhang W, Zhou L, Lei A. Org Lett. 2013;15:6166. doi: 10.1021/ol403021p.Hennessy EJ, Buchwald SL. J Am Chem Soc. 2003;125:12084. doi: 10.1021/ja037546g. Intermolecular C–H functionalizations involving activated alkyl halides have also been achieved in certain instances: He RY, Zeng HT, Huang JM. Eur J Org Chem. 2014:4258.Loy RN, Sanford MS. Org Lett. 2011;13:2548. doi: 10.1021/ol200628n.Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CR. J Org Lett. 2010;12:3104. doi: 10.1021/ol101146f.Lapointe D, Fagnou K. Org Lett. 2009;11:4160. doi: 10.1021/ol901689q.
- 5.For an intramolecular C–H alkylation of arenes with unactivated neopentyl alkyl halides (no β-hydrogens): Beaulieu LPB, Roman DS, Vallée F, Charette AB. Chem Commun. 2012;48:8249. doi: 10.1039/c2cc33547f.
- 6.(a) Ackermann L, Novak P, Vicente R, Hofmann N. Angew Chem, Int Ed. 2009;48:6045. doi: 10.1002/anie.200902458. [DOI] [PubMed] [Google Scholar]; (b) Zhang YH, Shi BF, Yu JQ. Angew Chem, Int Ed. 2009;48:6097. doi: 10.1002/anie.200902262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shabashov D, Daugulis O. J Am Chem Soc. 2010;132:3965. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen Q, Ilies L, Nakamura E. J Am Chem Soc. 2011;133:428. doi: 10.1021/ja1099853. [DOI] [PubMed] [Google Scholar]; (e) Hofmann N, Ackermann L. J Am Chem Soc. 2013;135:5877. doi: 10.1021/ja401466y. [DOI] [PubMed] [Google Scholar]; (f) Song W, Lackner S, Ackermann L. Angew Chem, Int Ed. 2014;53:2477. doi: 10.1002/anie.201309584. [DOI] [PubMed] [Google Scholar]; (g) Ackermann L. Chem Commun. 2010;46:4866. doi: 10.1039/c0cc00778a. [DOI] [PubMed] [Google Scholar]
- 7.There are a limited number of intermolecular C–H functionalizations of select heteroarenes with unactivated alkyl halides: Xiao B, Liu ZJ, Liu L, Fu Y. J Am Chem Soc. 2013;135:616. doi: 10.1021/ja3113752.Vechorkin O, Proust V, Hu X. Angew Chem, Int Ed. 2010;49:3061. doi: 10.1002/anie.200907040.Wu X, See JWT, Xu K, Hirao H, Roger J, Hierso JC, Zhou J. Angew Chem, Int Ed. 2014;53:13573. doi: 10.1002/anie.201408355.
- 8.(a) McMahon CM, Alexanian EJ. Angew Chem, Int Ed. 2014;53:5974. doi: 10.1002/anie.201311323. [DOI] [PubMed] [Google Scholar]; (b) Bloome KS, McMahen RL, Alexanian EJ. J Am Chem Soc. 2011;133:20146. doi: 10.1021/ja2091883. [DOI] [PubMed] [Google Scholar]; (c) Bloome KS, Alexanian EJ. J Am Chem Soc. 2010;132:12823. doi: 10.1021/ja1053913. [DOI] [PubMed] [Google Scholar]
- 9.Product 18 is likely formed by an alkyl shift prior to rearomatization. For a proposed mechanism see the Supporting Information.
- 10.Common minor byproducts in the catalytic alkylations include dehydrohalogenation and alkyl halide reduction products. As an example in this particular reaction tetrahydroisoquinoline 47 is produced in 72% yield along with 10% of a dehydrohalogenation and 5% of an alkyl halide reduction byproduct (as calculated by 1H NMR spectroscopy of the crude reaction mixture using an internal standard).
- 11.(a) Hartwig J. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito, CA: 2010. pp. 301–320. [Google Scholar]; (b) Matyjaszewski K, Xia J. Chem Rev. 2001;101:2921. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 12.There are two alternative mechanistic possibilities: (a) direct electron transfer to the substrate from the cyclohexadienyl radical or (b) electron transfer to the substrate from a radical anion formed from deprotonation of the cyclohexadienyl radical (base-promoted homolytic aromatic substitution HAS). We consider the former unlikely owing to the highly endergonic electron transfer involved. Oxidation potential of the cyclohexadienyl radical −0.1 V versus SCE see: Bhatia K, Schuler RH. J Phys Chem. 1974;78:2335. Reduction potential of s-butyl iodide −2.1 V versus SCE, see: Rondinini S, Mussini PR, Muttini P, Sello G. Electrochim Acta. 2001;46:3245. With respect to a base-mediated HAS these reactions typically require either a strong base (e.g., KOtBu) or appreciably electron-poor aromatic substrates neither of which are characteristic of the reactions herein. For further discussion see: Studer A, Curran DP. Angew Chem, Int Ed. 2011;50:5018. doi: 10.1002/anie.201101597. We thank a reviewer for suggesting these mechanistic variants.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.