

Abstract
Research over the past decade has demonstrated substantial interactions between the circadian system and the processes through which alcohol affects behavior and physiology. Here we summarize the results of our collaborative efforts focused on this intersection. Using a combination of in vivo and in vitro approaches, we have shown that ethanol affects many aspects of the mammalian circadian system, both acutely as well as after chronic administration. Conversely, we have shown circadian influences on ethanol consumption. Importantly, we are beginning to delve into the cellular mechanisms associated with these effects. We are also starting to form a picture of the neuroanatomical bases for many of these actions. Finally, we put our current findings into perspective by suggesting new avenues of inquiry for our future efforts.
1. Introduction: The synergy of complementary approaches
Before reviewing our research on alcohol-circadian system interactions, it is worth noting the various techniques we have used, and the benefits derived from, combining in vivo and in vitro approaches. Using both approaches in parallel generates extremely useful data on the effects of ethanol and on the mammalian circadian system, taking into account their respective advantages and disadvantages.
In general, in vivo studies offer the advantage of being able to assess how the intact organism reacts to specific stimuli through the measurement of a wide array of physiological and behavioral parameters. For example, studying circadian rhythms in vivo readily allows simultaneous measurements of multiple behavioral parameters, including entrainment to environmental stimuli, free-running period, phase-angle of entrainment, phase-shifting responses, and the duration of activity (or inactivity). Likewise, in vivo studies of ethanol allow behavioral testing of ethanol effects, daily patterns of ethanol consumption, and monitoring differential effects of ethanol on multiple physiological outputs. Studies of intact animals can also combine behavioral analyses with other techniques such as microdialysis, multi-unit neuronal activity recordings, telemetric recording, conditional gene manipulation and optigenetics.
Conversely, in vitro approaches, and specifically the acute brain slice preparation that we use, have different strengths. This methodology allows for fine control over both the location and timing of stimulus application, which can be especially important for investigating rapidly-changing cellular events. It can simplify pharmacological experiments, since issues such as animal toxicity, metabolism, and brain penetration are avoided, and you can easily combine pharmacological and electrical stimulations within the same brain area. A variety of techniques to assess cell activity can be used in addition to the extracellular recordings we use, including ionophoresis, patch-clamp and whole-cell electrophysiology, and Ca2+-imaging. With respect to studying the circadian clock located in the suprachiasmatic nucleus (SCN), while behavioral and neuronal activity are both outputs of the clock, the neuronal activity rhythm is a more direct and fundamental output, and therefore has less potential to be modulated by other factors. And in general, using deafferented brain slices eliminates modulatory input by other brain regions, whether investigating circadian or ethanol responses.
There are also techniques that can be used in both in vivo and in vitro preparations. For example, conditional regulation of gene expression and optigenetics can be implemented under both approaches, with minor modifications. And analogous stimuli (e.g., light pulses vs. glutamate application) can be applied under the two conditions to investigate photic-input processes in the SCN.
Thus, there are significant advantages in bringing both in vivo and in vitro approaches to bear on the neural processes underlying the effects of ethanol on the functioning of the mammalian circadian system. Importantly, we have found that the experiments conducted in vivo and in vitro yield complementary data, essentially providing a cross-check of the results without unnecessary duplication. Importantly, novel conclusions are often possible only through comparing the results of both sets of experiments. Thus, integrating these different experimental approaches in a single study offers a robust and reliable means to advance this developing area of ethanol-circadian research.
2. Acute ethanol inhibits photic phase resetting of the SCN circadian clock through actions within the SCN
When we started investigating the effects of ethanol on the circadian system, the existing literature was quite sparse. There were scattered studies assessing physiological rhythms in humans during or after ethanol (acute or chronic) consumption (Rojdmark et al., 1993;Kuhlwein et al., 2003;Adinoff et al., 1991;Danel and Touitou, 2003;Danel et al., 2006). Studies on chronic ethanol modulation of circadian rhythms in rodents reported inconsistent effects (Rosenwasser, 2004;Rosenwasser et al., 2005a,c;Seggio et al., 2007;Holloway et al., 1993;Mistlberger and Nadeau, 1992;Chen et al., 2004;Rajakrishnan et al., 1999). On the other hand, there was a general consensus that ethanol impairs sleep, and that sleep problems often contribute to relapse drinking (Landolt and Gill, 2001;Schwartz et al., 1997;Roehrs et al., 1996;Roehrs and Roth, 2001;Clark et al., 1998;Brower et al., 2001). Because of the strong circadian modulation of sleep processes, it seemed worthwhile to more systematically assess ethanol interactions with the SCN circadian system.
A. Ethanol inhibits glutamate-induced phase delays and phase advances in vitro
In light of ethanol's well known inhibitory actions on glutamate signaling (Grosshans and Browning, 2001;Hoffman, 2003;Allgaier, 2002), we started by focusing on ethanol modulation of glutamate-induced phase resetting of the SCN clock in vitro. In much the way long-term potentiation in hippocampal brain slices mimics memory formation in vivo, applying glutamate to SCN brain slices mimics photic phase resetting of in vivo behavioral rhythms. As a quick note, in all of our in vitro experiments we have used adult male C57BL/6 mice housed under 12:12 light-dark (LD) conditions with food and water available ad libitum unless specifically mentioned. Brain slices are prepared during early subjective day and maintained in vitro as described in detail (Prosser, 2001;Prosser, 2003;Prosser et al., 2008). These conditions allow us to maintain the tissue for up to 3 days in vitro in serum-free medium. We and others have demonstrated that the circadian clock continues to generate 24 h rhythms in neuronal activity, metabolism, neurotransmitter release, etc. in vitro that parallel those expressed in vivo (Newman et al., 1988;Gillette and Reppert, 1987;Ding and Gillette, 1993;Newman et al., 1992). We assess clock phase by recording SCN spontaneous electrical activity, which exhibits a highly stable, robust circadian rhythm (Prosser, 2003;Ding and Gillette, 1993).
Not surprisingly, in our initial experiments we found that acute application of ethanol to SCN brain slices at Zeitgeber time 16 (ZT 16, where ZT 0 = the time of lights-on and ZT 12 = the time of lights-off in the animal colony) could inhibit glutamate-induced phase delays, while ethanol inhibited glutamate-induced phase advances when co-applied at ZT 23 (Fig 1; Prosser et al., 2008). Ethanol applied by itself, in the day or night, did not alter SCN clock phase. Although the inhibition by ethanol could not be prevented by co-application of glycine or D-serine, it could be reversed by co-application of brain-derived neurotrophic factor (BDNF). These results are consistent with research in the hippocampus showing the ethanol can inhibit TrkB receptor enhancement of glutamate signaling (Kolb et al., 2005;Ron, 2004) and studies showing similar interactions between BDNF and glutamate signaling in the SCN (Kim et al., 2006;Michel et al., 2006;Mou et al., 2009).
Fig 1.

Ethanol dose-dependently inhibits in vitro glutamate-induced phase shifts. A. Shown are the 2 h means ± SEM of SCN neuronal activity from several experiments. Under control (no treatment) conditions, neuronal activity peaks near ZT 6 on the second day in vitro. Neuronal activity peaks approximately 3 h later than normal after brain slices are treated with glutamate (1mM) at ZT 16, indicating the SCN clock has been phase-delayed by 3 h. Co-application of 20mM ethanol treatment with glutamate blocks the phase delay. Glutamate treatment alone at ZT 23 advances the time of peak activity by about 3 hr, and again, this is prevented by co-treatment with 20 mM EtOH. Horizontal bars: time of lights-off in the animal colony; vertical bars: time of drug treatment; dotted line: mean time-of-peak in control experiments. B. Dose response curve for ethanol inhibition of glutamate-induced phase delays. Shown are the mean phase delays (±SEM) induced by 1 mM glutamate applied alone or co-applied with EtOH at ZT 16.
We also found that ethanol inhibition of glutamate-induced phase delays is dose-dependent, with an ED50 around 10mM. This represents a much greater ethanol sensitivity than that reported for most NMDA and GABA receptors, but is consistent with ethanol levels normally experienced during ethanol consumption by humans. This led us to run additional experiments on the neurochemistry underlying ethanol effects (McElroy et al., 2009). Ethanol inhibition of glutamate-induced phase shifts in vitro could be prevented by co-application of RO15-4513, an antagonist of a subgroup of GABAA receptors that contain [.delta] subunits and that are reported to have a high sensitivity to ethanol similar to what we had seen in our experiments (Wallner et al., 2003;Wei et al., 2004;Liang et al., 2009;Nie et al., 2011). Conversely, diazepam, which enhances GABA signaling through γ-subunit containing GABAA receptors did not mimic the inhibitory actions of ethanol (McElroy et al., 2009). Together with additional data showing expression and activity of GABAAδ subunits in the SCN (Ehlen and Paul, 2007; Ehlen and Paul, 2009), these data strongly support ethanol acting in the SCN at least in part through enhancing the tonic GABA currents thought to involve GABAAδ receptors.
B. Ethanol pharmacokinetics
Based on our exciting in vitro results, we quickly progressed to in vivo experiments in Syrian hamsters and C57BL/6J mice (Brager et al., 2011c; Ruby et al., 2009b). It had been shown previously in hamsters that chronic, free-choice ethanol drinking disrupts circadian phase-resetting responses to light pulses delivered late in the dark-phase (ZT 21; Seggio et al., 2007). However, little was known about the timing of daily ethanol self-administration and the impact of central ethanol pharmacokinetics on photic phase resetting, entrainment, and rhythm stability. We therefore explored the actions of ethanol on several critical aspects of circadian clock regulation, including 1) the pharmacokinetic profiles of ethanol and 2) the in vivo effects of acute ethanol treatments on behavioral activity patterns and photic phase resetting.
Using microdialysis, the profiles of ethanol in the extracellular fluid compartment of the SCN following acute i.p. injections in hamsters and mice were characterized. In hamsters, peak levels of ethanol (approximately 50 mM) from 2 g/kg i.p. ethanol injections, occurred within 20-40 min (Ruby et al., 2009b; Fig. 2). The half-life of this ethanol accumulation in the SCN was approximately 3 h. A similar analysis was undertaken in mice using a range of dosages (0.5 g/kg to 2.0 g/kg; Brager et al., 2011c). Peak levels of ethanol in the SCN also occurred within 20–40 min post-administration. Estimated levels in the SCN ranged from about 9 to 58 mM, and the half-lives of the ethanol ranged from 0.6 to 1.8 hr for the same doses.
Fig 2.

Pharmacokinetics of ethanol (EtOH) in the suprachiasmatic nucleus (SCN) (n = 5 hamsters) following ip injection (Inj) of EtOH (2.0 g/kg). Peak levels (estimated at 50 mM) occurred within 20–40 min of injection, and the half-life (t1/2) for clearance was 3 h. Bars represent means ± SE.
C. Ethanol effects on in vivo circadian photic phase-shifting
In our initial work in hamsters (Ruby et al., 2009b), acute i.p. injection of ethanol (2 g/kg) preceding a light pulse delivered during the late night significantly attenuated the phase-advancing action of this photic stimulus (Fig. 3). Interestingly, however, ethanol did not affect photic phase-delay shifts during the early night. This result is largely consistent with a previous study in chronically drinking hamsters where ethanol inhibited photic advances but not delays (Seggio et al., 2007). The basis of this differential effect of ethanol on phase resetting is not clear but could reflect differential influence of extra-SCN areas that prevent ethanol actions in the hamster during the early night that are absent in the late night.
Fig 3.

Left panel: representative double-plotted actograms of general locomotor activity showing EtOH attenuation of photic phase-advance responses to light pulses delivered at ZT 18.5. A and B: vehicle-injection. C and D: EtOH (2.0 g/kg) injection. Arrows denote the time of injection. Right panel: ethanol dose-dependently attenuates light-induced phase-resetting response. Bars with different letters are significantly different (p<0.05). Bars represent means ± S.E.
In extending this work, we found that in mice (as opposed to hamsters), photic phase-delays during the early night were inhibited by acute i.p. ethanol treatment (Fig. 3), but phase-advances during the late night are not (Brager et al., 2011c). Again, these results are consistent with those of Seggio et al., (2009), demonstrating attenuation of phase delays but not phase advances in mice. However, the latter result could stem, in part, from the small phase advances normally seen in mice in vivo. Notably, reverse microdialysis perfusion of the SCN with ACSF containing ethanol (50 mM estimated tissue concentration) for 1 hr also markedly inhibited photic phase-delay shifting at ZT 14. Neither ACSF nor ethanol perfusions had phase-shifting effects in the absence of a light pulse. Together with our previous in vitro data showing ethanol inhibition of glutamate-induced phase resetting in SCN brain slices (Prosser et al., 2008), inhibition of photic phase resetting by intra-SCN perfusion of ethanol in the mouse and in the hamster demonstrates that the inhibitory action of ethanol occurs directly within the SCN. However, the lack of ethanol inhibition of photic phase delays in hamsters leaves open the possibility of additional ethanol actions outside the SCN that impinge on the circadian system.
3. Ethanol effects on circadian non-photic phase resetting
Phase regulation of the SCN circadian clock involves both photic/glutamate signaling and a variety of non-photic signals. Behaviorally, these signals are usually associated with enhanced activity or arousal occurring during an animal's normal rest period (e.g., during the day for nocturnal rodents) (van Reeth and Turek, 1989;Janik and Mrosovsky, 1994;Meyer-Bernstein et al., 1997;Meyer-Bernstein and Morin, 1999). Neurochemically, these signals have been linked to increases in both serotonin and NPY input in the SCN (Cutrera et al., 1994;Janik and Mrosovsky, 1992;Biello and Mrosovsky, 1995;Dudley et al., 1998;Edgar et al., 1997;Antle et al., 2000;Vrang et al., 2003). Complicating matters, the photic and non-photic signals are generally antagonistic in their phase-resetting actions (Biello et al., 1997;Glass et al., 1992;Ralph and Mrosovsky, 1992;Selim et al., 1993; Morin and Blanchard, 1991;Prosser, 2001). Therefore, to provide a more comprehensive assessment of ethanol actions within the circadian system, we investigated the daytime effects of ethanol.
A. Ethanol enhances serotonergic phase resetting in vitro
In the SCN slice, we and others had previously shown that serotonin and serotonin agonists applied during mid-subjective day (ZT 6) induce 3 hr phase-advances of the circadian clock through activating 5HT1A and/or 5HT7 receptors (Lovenberg et al., 1993;Prosser et al., 1990,1993;Shibata et al., 1992;Sprouse et al., 2005). Other studies extended these findings to suggest involvement of 5HT5A receptors in these non-photic phase shifts (Sprouse et al., 2004). In our ethanol experiments we found that co-application of ethanol and the 5HT1A/5A/7 agonist, 8-OH-DPAT, increased the size of 8-OH-DPAT-induced phase advances by about 50%, from 3 h to about 4.5 h (Fig 4; Prosser et al., 2008). We further demonstrated that this enhancement of serotonergic phase resetting was dose-dependent, although in this case the ED50 was about 50 mM. Finally, we found that substances that inhibit glutamate/BDNF signaling in the SCN mimicked the enhancing effects of ethanol. These results are consistent with the hypothesis that the enhanced non-photic phase shifts are the result of ethanol decreasing endogenous daytime glutamate signaling in the SCN.
Fig 4.

Ethanol enhances in vitro serotonergic phase shifts. A. Shown are the 2 h means ± SEM of SCN neuronal activity after treatment with the 5-HT1A,5A,7 receptor agonist, 8-OH-DPAT (DPAT) alone at ZT 6, which induces a 3 h phase advance, and DPAT co-applied with 100mM EtOH, which induces a 4.5 h phase advance. B. Dose-response curves for DPAT when co-applied with either EtOH (circles) or the NMDA receptor agonist AP5 (triangles). Both treatments enhanced DPAT-induced phase shifts to a similar degree, which together with our other experiments, is consistent with the EtOH and AP5 effects acting through similar modes of action.
B. Ethanol inhibits serotonergic phase resetting in vivo
To further explore these effects in intact animals we first evaluated the action of acute ethanol on serotonergic phase advances in hamsters. In parallel with our in vitro experiments, we used i.p. injections of 8-OH-DPAT, as it has been employed widely in circadian studies, and induces phase shifts of similar magnitude and at the same phases as other non-photic stimuli (~1.5 h) when administered systemically (Bobrzynska et al., 1996; Tominaga et al., 1992), or in the SCN (Ehlen et al., 2001). In contrast to our in vitro results, we found that acute ethanol markedly diminished the phase-advancing action of 8-OH-DPAT (Ruby et al., 2009b; Fig. 5). The basis for these different results is unclear. It is possible that there is rapid internalization of serotonin receptors in the SCN or other circadian-related areas secondary to an ethanol-induced increase in 5-HT, resulting in a smaller serotonergic phase shift. Another possibility is that the inhibitory effects of ethanol in vivo involve pathways registered outside of the SCN, which would not occur in the deafferented SCN slice.
Fig 5.

Top panel: EtOH (2.0 g/kg) inhibition of non-photic phase-advance responses to the serotonin 5-HT1a,7 agonist (±)8-OH-DPAT (5.0 mg/kg) administered during the middle of the light phase (ZT 6; left). EtOH had no phase-resetting effect when administered before vehicle (dimethyl sulfoxide) injection at ZT 6 (right). Bars with different letters are significantly different (P < 0.05). Bars represent the means ± SE. Bottom panel: Dose-dependent EtOH inhibition of phase-advance responses to i.p. injection of (±)8-OH-DPAT delivered during the middle of the light-phase (ZT 6; bottom). Bars with different letters are significantly different (p<0.05). Bars represent means ± S.E.
Consistent with our in vivo work in hamsters, acute i.p. ethanol injections of ethanol in mice also inhibited 8-OH-DPAT phase-advance shifting. In hamsters, a dose of 2 g/kg suppressed 8-OH-DPAT shifts by 72%, while in mice this dose completely blocked shifting (Brager et al., 2011c; Fig. 5). As discussed above, the basis of this inhibition by ethanol is unclear, but this action seems contrary to observations that acute ethanol enhances serotonergic activity by increasing release and decreasing uptake of 5-HT (reviewed in Rosenwasser, 2001; LeMarquand et al., 1994). However, as suggested above, it is possible that ethanol-induced enhancement of serotonergic activity causes a rapid down-regulation of 5-HT receptors as can happen in the SCN in vitro (Prosser et al., 2006), leading to decreased 8-OH-DPAT-induced phase shifts. In our in vivo systemic trials, ethanol was administered 30 min prior to 8-OH-DPAT injection, which (according to the present pharmacokinetic analyses) potentially could have down-regulated 5-HT receptors prior to 8-OH-DPAT entry to the brain, thus attenuating its phase-advancing response.
4. Evidence of Ethanol Tolerance in the SCN circadian system
Thus far, all of the experiments described involve acute administration of ethanol, either in vivo to intact animals or in vitro to SCN brain slices. However, exclusively studying the acute effects of ethanol provides a limited view of how alcohol affects behavior and physiology. The most debilitating effects of ethanol are associated with long-term alcohol abuse, and ultimately alcohol dependency (Johnson et al., 2006;Miller and Spear, 2006;Goodman, 2008;Spanagel and Kiefer, 2008). One component of alcohol dependency is a decreased sensitivity to alcohol's effects, which leads to increased consumption (Fillmore et al., 2005;Gass and Olive, 2008;Sung et al., 2005). The strong link between ethanol tolerance and alcohol dependency let us to investigate more chronic forms of ethanol administration.
Ethanol tolerance has been subdivided into three categories based on the duration of ethanol exposure (Radcliffe et al., 2006;Vieira et al., 2008;Pohorecky and Roberts, 1992;Velazquez-Marrero et al., 2011). Acute tolerance is a form of desensitization that occurs within 5-30 min of initial ethanol exposure, and has been seen to last approximately 24 h. Rapid tolerance is seen approximately 8-24 h after initial ethanol exposure, and the amount of time it lasts has not been routinely assessed. Chronic tolerance is generally seen after multiple days or weeks of ethanol administration (or years, in the case of humans). In animal models, chronic tolerance is typically followed by a 2-4 day period of heightened responsiveness after ethanol is withdrawn, and increased ethanol preference/consumption when ethanol is again available (post-withdrawal rebound). Many experimental paradigms have been used for chronic ethanol administration, including daily ethanol injections or intra-gastric lavage, daily ethanol vapor exposure, continuous ad libitum ethanol (5-25%) drinking (either in free-choice or a forced-consumption situation), or fixed interval drinking, with ethanol available at specified times each 24 h (e.g., chronic intermittent drinking, or drinking-in-the-dark). The choice of paradigm used often depends on whether the focus of the study is on regulating the exact amount and/or timing of ethanol administered vs. allowing subjects to control their own ethanol consumption (Kliethermes et al., 2005;Logan et al., 2010;Rosenwasser et al., 2010;Iancu et al., 2013;Pisu et al., 2011;Ahmed, 2012;Damaggio and Gorman, 2013).
We have investigated all three types of tolerance using our in vitro and in vivo approaches. The various in vitro experimental protocols we use are illustrated in Fig. 6. For our acute tolerance experiments all ethanol exposure occurs in vitro in the brain slices; in the rapid and chronic tolerance experiments ethanol is initially given to the animals to drink, then tolerance is assessed in vitro.
Fig 6.

In vitro ethanol tolerance experimental paradigms. Diagrams illustrate the different experimental protocols used to assess acute tolerance (top), rapid tolerance (middle), and chronic tolerance (bottom) of in vitro phase resetting. For the latter two paradigms, EtOH is initially provided to the mice to drink, while in the first paradigm, EtOH is only applied to the brain slices for varying amounts of time immediately prior to EtOH co-application with either glutamate or the 5-HT1A,5A,7 receptor agonist, 8-OH-DPAT (DPAT). Dark bars: lights-off (in vivo), or subjective night (in vitro).
A. Acute tolerance to ethanol
In the first set of experiments we demonstrated that the SCN clock exhibits acute tolerance with respect to the ability of ethanol to inhibit glutamatergic and to enhance serotonergic phase resetting. While a 5-10 min pre-exposure of the SCN brain slices to ethanol prior to co-applying ethanol with either 8-OH-DPAT or glutamate did not prevent ethanol's normal effects, a 30 min pre-exposure to ethanol completely prevented the acute effects of ethanol, as assessed the following day (Fig 7; Prosser and Glass, 2009). Extending the pre-incubation period to 3 h did not reverse this effect, indicating that the acute tolerance does not dissipate during continued exposure to ethanol. However, this study did not assess how long acute tolerance lasts after ethanol is removed. Also, we have not investigated whether acute tolerance occurs in vivo.
Fig 7.

Acute tolerance to in vitro ethanol. Shown are the mean (SEM) phase shifts induced by glutamate and DPAT alone, or after varying lengths of pre-treatment and then co-treatment with EtOH . In both cases, a 5 min pretreatment with ethanol does not prevent ethanol modulation of glutamate or DPAT-induced phase shifts. However, ethanol pre-treatments of 30 min or longer prevent the EtOH enhancement (DPAT) and EtOH inhibition (glutamate) effects.
B. Rapid tolerance to ethanol
Next we investigated rapid tolerance using both in vivo and in vitro approaches (Lindsay et al., 2014). In both cases, mice were given no-choice 15% ethanol to drink for a single night. The following morning (for in vitro experiments) brain slices were prepared, and ethanol was co-applied with glutamate 12-18 hours later (ZT 16 or ZT 23; Fig 6). Using this protocol, we found that the sensitivity to ethanol inhibition of glutamate-induced phase shifts was decreased 10 fold by the single night of drinking (Fig 8 vs. Fig 1). Interestingly, this decreased ethanol sensitivity was not short-lived. If we delayed brain slice preparation after animals were placed back on ad libitum water, it took 3-4 days before ethanol could again inhibit glutamate-induced phase resetting with its normal effectiveness (Fig 8).
Fig 8.

Rapid tolerance to ethanol assessed in vitro. Shown on the left are the mean (SEM) phase shifts induced by 1 mM glutamate co-applied with varying concentrations of ethanol after a single night of ethanol consumption. 175-200 mM ethanol is needed to inhibit both the ZT 16 phase delays (top) and ZT 23 phase advances (bottom) induced by glutamate. Shown on the right are the phase shifts (SEM) induced by 1 mM glutamate + 20 mM ethanol applied to brain slices prepared 0-3 days after a single night of ethanol consumption. Tolerance is still evident 48 hr (ZT 16, top) and 72 hr (ZT 23, bottom) after ethanol consumption, but is gone 24 h later in each case.
We also assessed rapid tolerance in vivo. Again, mice were provided no-choice 15% ethanol for 1 night, followed by ad libitum water beginning at lights-on. The subsequent night mice were given i.p. ethanol followed by a light pulse at ZT 16. As we saw in vitro, the single night of ethanol consumption suppressed ethanol inhibition of photic phase delays (Lindsay et al., 2014).
C. Effects of chronic ethanol
Thus far, our investigation of chronic tolerance has primarily involved in vivo experiments, although we are beginning to assess the effects of chronic ethanol in vitro. As with the other aspects of our ethanol-circadian research, we have used a multi-pronged approach, and the results observed using the different protocols has been extremely useful in designing appropriate protocols and in drawing conclusions from the data.
i. Chronic ethanol pharmacokinetics
As with our in vivo experiments on acute ethanol, we felt it was important to begin these experiments by measuring ethanol pharmacokinetics during chronic consumption. Therefore, we used similar microdialysis techniques to measure ethanol levels in the SCN, with concomitant monitoring of drinking and locomotor behavior, and subcutaneous measurements of ethanol concentrations. In chronically drinking hamsters, the estimated peak level of ethanol in the SCN (~20 mM) occurred during the early dark-phase (Ruby et al., 2009a), with relatively low levels (~5 mM) at ZT 18.5 (when the phase-advancing light pulses are administered; Fig 9). Similar measurements in mice drinking 10% and 15% ethanol (Brager et al., 2010b) revealed that peak SCN ethanol levels (averaging ~10–20 mM and ~20–30 mM, respectively) also occurred predominately during the dark-phase, with smaller sporadic drinking bouts during the day. Notably, for both mice and hamsters, each ethanol drinking episode preceded a peak in SCN ethanol concentration by 20–40 min, and individual peak durations ranged from 30–60 min. Several points are evident from these experiments: first, drinking bouts are temporally matched with clustered locomotor activity bouts; second, there is a ~20-min latency between a drinking episode and increases in tissue ethanol; third, the intensity and duration of a drinking bout are proportionate to the size of the ensuing peak; and fourth, the effect of multiple drinking bouts spaced over a few hours is cumulative, resulting in a prolonged presence of ethanol in the brain and periphery.
Fig 9.
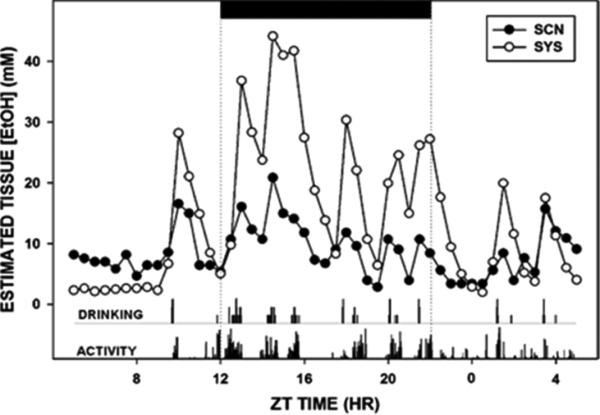
Composite 24-h pharmacokinetic profiles of SCN and subcutaneous EtOH superimposed with drinking and general locomotor activity rhythms from an individual hamster during forced 20% EtOH drinking. The black bar (top) represents the dark phase of the light-dark (LD) cycle.
ii. In vivo chronic ethanol effects
In sharp contrast to our observations of acute tolerance (in vitro) and rapid tolerance (in vivo and in vitro), we see little evidence for chronic tolerance in vivo. Beginning with our work on hamsters, we showed that chronic forced 20% ethanol consumption continued to disrupt phase-advance shifting in hamsters presented with photic stimuli late in the dark phase (Ruby et al., 2009a). The number and duration of circadian locomotor activity bouts were also affected by this ethanol treatment. Again, these results are consistent with previous studies showing that chronic ethanol consumption can affect photic phase-resetting in hamsters (Seggio et al., 2007). It is notable that this chronic ethanol consumption differentially affected multiple circadian timekeeping processes, including attenuation of photic phase-advances. However, ethanol had little apparent effect on entrainment of the locomotor activity rhythm to LD, even in hamsters maintained under very low light intensities. Similar results have been reported in other studies (Mistlberger and Nadeau, 1992). One explanation for this lack of effect is that photic phase-delays in hamsters are insensitive to ethanol (Ruby et al., 2009a;Seggio et al., 2007). Because hamsters have a short circadian period and thus entrain to 24 h LD cycles primarily through phase-delay shifts, they would theoretically have little problem entraining. Another possibility, based on the masking effect of light on activity, is that locomotor behavior is suppressed by light and stimulated by darkness, producing a seemingly entrained rhythm, irrespective of ethanol-disrupted clock function.
Similar to our results in hamsters, we and others have shown that chronic ethanol drinking (10-15%) in mice continues to inhibit photic phase delay shifts during the early night (Brager et al., 2010b; Seggio et al., 2009). Again, chronic ethanol inhibition of photic phase shifts did not prevent entrainment to the normal LD cycle or reentrainment to a shifted LD cycle, however, it disrupted the daily pattern of locomotor activity (Brager et al., 2010b; Fig. 10).
Fig 10.

A,B; Representative double-plotted actograms showing re-entrainment to a skeleton photoperiod (1 min light pulse designated by the vertical line) delivered daily at ZT 6 of the initial LD photocycle in mice chronically drinking 15% ethanol (shading) or water, respectively (n=7/group). C,D: double-plotted actograms showing the effect of chronic 15% ethanol or water, respectively on entrainment to the 1 min skeleton photoperiod delivered at ZT 11 of the initial LD photocycle (n=10/group). The horizontal black bars represent the dark phase of the initial LD cycle.
Given that ethanol inhibits light pulse-induced phase-shifting (discussed above) and alters free-running tau (Mistlberger and Nadeau, 1992; Seggio et al., 2009), it seemed reasonable to speculate that in the absence of photic masking effects, ethanol should disrupt photic entrainment. This idea is supported by the effects seen with the skeleton photocycle consisting of a daily 1 min pulse of low intensity light (25 lux), where ethanol disrupted stable photic entrainment, and in particular attenuated the robust onset of activity associated with the onset of the subjective night (Brager et al., 2010b). Thus, this work shows that chronic ethanol consumption can produce marked disturbances in circadian photic entrainment capacity and circadian locomotor activity patterns. These assessments are relevant to potential disruptive effects of alcohol in individuals exposed to shifting light-dark cycles, including rotating shift work and repeated rapid trans-meridian travel. It is noteworthy, however, that the studies provide little evidence of chronic tolerance developing in the SCN circadian clock.
iii. In vitro chronic ethanol effects
The lack of observed in vivo chronic tolerance to ethanol to inhibition of photic phase resetting is surprising in light of our data showing that both acute and rapid tolerance can develop in the SCN (Lindsay et al., 2014; Prosser and Glass, 2009). One possible explanation is that the cellular mechanisms in SCN make it resistant to chronic tolerance development. Evidence to support this possibility derives from research showing that the SCN is relatively immune to excitotoxic damage, perhaps due to strong glutamate sequestration capabilities (Bottum et al., 2010). This capacity could potentially diminish the compensatory responses to chronic ethanol. An alternative possibility is that chronic tolerance in the SCN is masked or compensated by input from other brain regions. To help differentiate between these possibilities, we have started to investigate chronic tolerance using our SCN brain slice preparation. In these experiments, we are using a restricted access paradigm, where mice have access to 15% ethanol only during a 4 h period each day, from ZT 11 to ZT 15 (Fig 6). This corresponds to the interval when our in vivo experiments indicated that mice consume the most ethanol (e.g., Fig 11). Using this paradigm, our mice consume 2.7 ± 0.3 gm/kg ethanol per day, and day-to-day variability in ethanol consumption is low. The morning following 10 days of ethanol consumption, SCN brain slices are prepared, and the ability of ethanol to inhibit glutamate-induced phase shifts is assessed. While preliminary, our data suggest that, when mice undergo this protocol for 10 days, the SCN clock subsequently shows in vitro tolerance to ethanol similar to what we previously saw with rapid tolerance – a 10-fold decrease in sensitivity to ethanol. In additional preliminary experiments, it appears to take between 7 and 9 days for this chronic tolerance to develop. Thus, our tentative conclusion is that the SCN clock does in fact develop chronic tolerance to ethanol, and that the apparent lack of tolerance observed using in vivo may be due to compensatory actions occurring outside the SCN.
Figure 11.

Representative double-plotted actograms of general circadian locomotor activity (top) and ethanol drinking (bottom) of individual mPer2 mutant (A,C) and wild-type (WT) (B,D) mice. Horizontal filled and empty bars represent the dark and light phases of the 24 hr LD cycle, respectively.
iv. Ethanol withdrawal effects
A general consequence of chronic tolerance typically is an enhanced sensitivity during ethanol withdrawal (e.g. (Gilpin and Koob, 2010;Roberts et al., 2000). In our in vivo experiments we have investigated whether the SCN clock exhibits withdrawal-associated hypersensitivity with respect to photic phase resetting. Interestingly, the results differ between hamsters and mice. Photic phase shifts in hamsters undergoing ethanol withdrawal were nearly twice the size of those exhibited by control hamsters (Ruby et al., 2009a), consistent with withdrawal-associated hypersensitivity. In contrast, photic phase shifts in mice undergoing ethanol withdrawal continued to be smaller than those seen in control mice (Brager et al., 2010b), consistent with continued ethanol inhibition of photic phase shifts 2-3 days after ethanol access ended, and 2-3 days after microdialysis measurements indicate the ethanol has been completely metabolized. Likewise, Seggio et al. (2009) observed continued inhibition of photic phase delays in mice 1 day after withdrawal from ethanol consumption. This prolonged inhibition is similar to the extended tolerance we see in vitro following rapid tolerance (i.e., 1 night of ethanol consumption), and the continued ethanol-induced hypo-locomotion during withdrawal seen by others (Logan et al., 2010, 2012).
Thus, our in vivo data are puzzling in both species: hamsters show signs of ethanol withdrawal-induced hypersensitivity while showing no signs of chronic tolerance. Conversely, mice appear to show an extended ethanol-induced inhibition well past the end of ethanol exposure. Both of these results will require more investigation to understand the cellular mechanisms involved.
Although quite preliminary, we have initiated experiments to assess the effects of ethanol withdrawal in vitro. For these experiments mice have undergone the chronic restricted-access ethanol consumption paradigm (Fig. 6), followed by 2-3 days of withdrawal. Brain slices are then prepared, and the slices treated at ZT 16 with glutamate. Our preliminary results are consistent with ethanol withdrawal-associated enhanced sensitivity: 1 uM glutamate applied to these brain slices for 10 min at ZT 16 induces a 3 h phase delay, the response normally induced by 10 uM glutamate, while in control (ethanol naïve) brain slices 1 uM glutamate does not induce a significant phase shift. Again, these effects are different from what is seen in vivo, and will require additional investigation.
5. Ethanol consumption: genetic, environmental, reward and pharmacological modulation
To fully understand the factors contributing to alcohol abuse and dependency, it is necessary to expand our perspective to assess how genetic and environmental factors modulate the processes through which ethanol acts on the brain. Moreover, with respect to our investigations on circadian/ethanol interactions, it is important to understand how they tie into ethanol's actions on reward systems. This is critical for developing treatments to prevent or reverse alcohol dependency and abuse. Again, we have taken a multi-faceted approach to this question, mostly involving in vivo experiments, utilizing genetic, environmental, behavioral, and chemical manipulations.
A. Per2 modulation of ethanol effects
Studies focusing on circadian-related influences on alcohol dependence have revealed that circadian gene mutations profoundly affect ethanol intake. Notably, mutations of circadian clock genes, including Per2 and CLOCK, which are important for stabilizing endogenous clock functions, are associated with increased alcohol intake (see Spangel and Perreau-Lenz, this volume;Spanagel et al., 2005;Ozburn et al., 2010;Sjöholm et al. 2010). Mice with mutated mPer2 have greatly potentiated ethanol intake and preference (Spanagel et al., 2005), and have an earlier onset in nocturnal wheel-running activity relative to wild-type mice (Pendergast et al., 2010). Neurochemically, the Per2 potentiation in ethanol intake is thought to arise from elevated levels of extracellular glutamate in brain areas controlling alcohol reward and craving, which is considered a key factor contributing to alcohol dependence (Dodd et al., 2000;Gass and Olive-Foster, 2007;Krystal et al., 2003;Spanagel et al., 2005). From these and other studies described below, it is likely that a normally functioning circadian timing system moderates alcohol use and dependence, and that conversely, perturbations of this system can promote alcohol intake.
We extended the findings of Spanagel et al., 2005 by exploring the behavioral circadian phenotype of the Per2 mutants as it relates to enhanced ethanol intake (Brager et al., 2011a). Our results confirmed the potentiated ethanol intake and preference in the Per2 mutants and found that it involves higher total fluid intake that does not include an increase in pure water intake (Fig. 11), The analyses also showed that the enhanced ethanol intake in Per2 mutant mice was associated, in part, with their perturbed circadian behavior: namely a ~2 h advance in the onset of nocturnal locomotor activity and corresponding increase in the duration of the nighttime activity/wakefulness period (Fig. 11). There were also more drinking bouts during the dark-phase in the Per2 mutants, and Per2 mutants exhibited more drinking episodes during the light-phase compared to wild-types, whose ethanol drinking mostly occurred near light-dark transitions. Thus, the advanced phase of behavioral activity, longer duration of nocturnal drinking, and increased daytime ethanol consumption all contributed to the increased daily ethanol intake. It will be important in future experiments to determine the extent to which elevated ethanol intake in Per2 mutant mice is associated with background strain-dependent effects on circadian behavioral and ethanol drinking rhythms.
B. Constant light-induced circadian disruptions
i. Syrian hamsters
Exposure of rodents to constant light (LL) disrupts circadian timing because it suppresses locomotor activity and can split the activity rhythm into two anti-phase components in the majority (~75%) of hamsters (de la Iglesia et al., 2000). In addition, LL induces depression-like behaviors (Fonken et al., 2009), and abolishes circadian rhythmicity in corticosterone (Claustrat et al., 2008) and melatonin secretion (Wideman and Murphy, 2009; Wurtman et al., 1963). We and others have shown that LL attenuates the circadian expression of Per gene activity within the SCN and depresses circadian activity rhythms (Sudo et al., 2003). Given these multiple circadian-related disruptions and their associations with augmented alcohol drinking, we hypothesized that LL conditions would increase the drive for ethanol reward.
In hamsters, paired comparisons between initial LD levels of activity and ethanol intake and subsequent LL levels in the group revealed that, although locomotor activity was suppressed under LL, there was little change in ethanol consumption or preference (Hammer et al., 2010; Fig. 12). This latter effect differs from that of rats, where LL decreases ethanol intake (Geller, 1971). Retrospective analysis of our data revealed marked differences between hamsters with split rhythms and those that maintained intact rhythms. Although ethanol consumption and preference were not affected by LL in either of the phenotypes, there was significantly less ethanol intake at all time-points in splitters compared to non-splitters (Fig. 12). Notably, these differences in ethanol consumption occurred under LD prior to the LL exposure, revealing the existence of an underlying predisposition for subdued ethanol intake linked to rhythm splitting. A connection between a propensity for ethanol intake and rhythm splitting under LL was also seen in a previous study in rats, where (in contrast to hamsters) selectively bred ethanol-preferring (HAD2) rats exhibited a higher degree of rhythm splitting compared to other lines of rats tested (Rosenwasser et al., 2005b). Regardless of these opposing correlations between rhythm splitting propensity in ethanol intake, it is evident that underlying genetic and/or physiological components of the SCN clock can contribute to the propensity for ethanol intake
Figure 12.

Free-choice 20% EtOH consumption (top) and EtOH preference (bottom) for hamsters under LL exhibiting non-split and split rhythms (middle and right side, respectively) and averaged for all hamsters (Group; left side). Gray bars represent the averages measured under the initial LD photocycle. White bars represent the first and last 5 days of LL. Bars represent means±SEM. For each parameter, bars with different letters are significantly different (p<0.05).
ii. Mice
We also explored the effects of LL exposure on ethanol drinking in wild-type and Per2 mutant mice (Brager et al., 2010a). As single-nucleotide polymorphisms of Per2 have been implicated in predisposition for alcohol abuse and alcoholism as discussed above, we exploited this suppressive effect of LL on Per2 and circadian activity as a physiological approach to explore the etiology of alcohol abuse and alcoholism in the male mouse. In contrast to rats and hamsters, LL exposure in wild-type mice transiently potentiated ethanol consumption and preference (Fig. 13). This effect occurred concomitantly with a severe LL-induced disruption of the circadian locomotor activity rhythm. Interestingly, unlike the wild-type mice, the Per2 mutants did not exhibit increased ethanol drinking under LL, possibly because of a ceiling effect (their ethanol drinking is normally very high) and/or because their impaired Per2 expression could not be lowered further by LL.
Figure 13.

Free-choice EtOH consumption (top) and preference (bottom) with respect to photocycle (LD; 12L:12D photocycle: LL; constant light). Constant light exposure transiently augments ethanol consumption and preference. Asterisks designate significant difference LL vs. LD (p<0.05); “a” designates significant difference wild-type vs. mPer2 mutant mice (p<0.05). Bars represent means±SEM.
C. Wheel-running as hedonic substitute
Non-photic circadian influences, including wheel-running, can be reinforcing stimuli, and brain systems involved in circadian regulation are linked to the mesocorticolimbic reward pathway (Webb et al., 2009). Ethanol and other drugs of abuse are thought to override the reward circuitry (Kelley, 2004), such that drugs are perceived as having greater subjective value than natural rewards. Moreover, evidence suggests that such agents can regulate circadian phase (Kosobud et al., 2007). Thus, the interaction of addictive drugs with the reward and circadian systems may contribute to the development of drug dependency.
Central to the maladaptation of brain and behavior to drugs is hedonic substitution, whereby ethanol or drugs are chosen by addicted humans or animals in lieu of natural rewards. Notably, the idea of hedonic substitution implies that natural rewards can also replace drug rewards. This is supported by recent evidence that wheel-running decreases cocaine self-administration in cocaine-dependent animals (Smith et al., 2008), and that environmental enrichment reduces heroin reward (El Rawas et al., 2009). We explored the possibility that rewarding manipulations that can modulate circadian timing can decrease ethanol intake in hamsters (Hammer et al., 2010). First, access to a running wheel (a non-photic phase resetting signal and a rewarding stimulus) decreased ethanol drinking, while conversely, access to ethanol decreased wheel-running (Figs. 14). These results are consistent with previous studies where running wheel access decreased ethanol intake in other high alcohol-preferring species including the C57BL/6J mouse (Ehringer et al., 2009) and the alcohol preferring (P) (but not the non-alcohol perfering [NP]) rat (McMillan et al., 1995). These observations support the hypothesis that the rewarding effects of exercise may substitute for the hedonic effects of drug and alcohol abuse. An alternative explanation relates to the proposed role of central ghrelin signaling in modulating alcohol reward. Stimulation of ghrelin receptors in the ventral tegmental area (VTA) is required for reward stimulation by ethanol (Jerlhag et al., 2009). Vigorous exercise markedly suppresses blood levels of the acylated (active) form of ghrelin that crosses the blood-brain barrier (Broom et al., 2007), and this suppression of ghrelin could decrease the incentive value of ethanol. A third possibility is that animals running on a wheel for much of the night (averaging 11–12 km) would have less time and/or inclination to drink. Regardless of the mechanism behind the wheel-running effect, the present data support exercise as a natural substitute for the hedonic aspects of alcohol.
Figure 14.

Wheel running suppresses EtOH consumption. Top panel:Top graph, mean daily ethanol consumption for all animals (dashed line; these animals were considered statistically as a single group, as they were not randomly separated into running and non-running groups until the start of Phase II), runners (solid line), and non-runners (dotted line). Phase I: ethanol acclimation period (wheels locked). Phase II (shaded): wheels unlocked for experimental group and locked for controls. Phase III: all wheels locked. Bottom graph, ethanol consumption and preference for runners and non-runners averaged over each phase. Bars represent means±SEM. Among all groups and phases, bars with different letters are significantly different (p<0.05). Bottom panel:Top graph, daily distance run by animals given free-choice between 20% ethanol and water (solid line) or water only (dotted line). Shaded area represents wheel acclimation period; EtOH introduction occurred at the beginning of the non-shaded region. Bottom graph, distance run by drinkers (EtOH) and non-drinkers (water) during the initial 3 wks of EtOH drinking. Bars represent means±SEM. Bars with different letters are significantly different (p<0.05).
It should be noted that the results discussed above contrast somewhat with results from a study in C57BL/6J mice, showing ethanol consumption was largely unaffected by running wheel access, although there was an increase in wheel-running when ethanol was withdrawn, suggestive of a reward substitution effect (Ozburn et al., 2008). Also, it has been reported that wheel-running can increase ethanol intake and preference in Lewis rats (Werme et al., 2002). These discrepancies could be related to procedural differences: the mouse study employed repeated, alternating 6-day exposures to locked and unlocked wheels with no constant locked-wheel control group, while in our study hamsters had continuous access to unlocked or locked wheels for 30 days. Another difference is that the Lewis rats exhibit a uniquely high level of addiction and excessive wheel-running behavior that could possibly produce an additive, rather than substitutive reward effect.
D. Acamprosate suppression of ethanol consumption
Acamprosate has been used clinically since 1989 as a pharmacological agent to help promote abstinence in alcohol-dependent patients. In the majority of clinical trials, this drug was shown to have a statistically significant effect over placebo in controlling relapse, at least during the initial period of abstinence (Kranzler and Gage,2008; Rosner et al., 2010). In rodents, acamprosate's overall suppressive effect on ethanol intake is consistent across studies, despite wide differences in drug and/or ethanol treatment regimens.
In our study, a six-day acamprosate treatment regimen (300 mg/kg/day i.p.) was used in wild-type and Per2 mutant mice. This treatment significantly reduced overall daily ethanol intake in both mPer2 mutants and wild-types (Brager et al., 2011a; Fig. 15). Similar to previous findings, acamprosate treatment reduced mPer2 mutant ethanol intake to that of untreated wild-type mice. Our combined drinkometer and microdialysis assessments revealed that acamprosate reduced daily total drinking bouts and systemic ethanol peaks without altering the general diurnal pattern of ethanol drinking. In agreement with other studies (Spanagel et al., 2005), we also found that systemic acamprosate treatment suppressed ethanol preference in Per2 mutants, although it was still higher than in wild-type mice. This could be related to the advanced activity onset that was unaffected by acamprosate. Our observation that systemic acamprosate treatment (300 mg/kg) significantly suppressed ethanol intake and preference in wild-type mice differs somewhat from the observation that systemic acamprosate (200 mg/kg) had no effect on wild-type intake and preference levels (Spanagel et al., 2005). A separate study in mice using a drinking-in-the-dark protocol to enhance ethanol consumption showed 200 mg/kg systemic acamprosate suppressed ethanol intake (Gupta et al., 2008). Hence, discrepancies between our findings and those of Spanagel et al., 2005 may be due to different genetic backgrounds or drinking procedures.
Figure 15.

EtOH intake (left) and preference (right) in mPer2 mutant (PER2) and wild-type (WT) mice during the daily regimen of i.p. acamprosate (ACAMP; 300 mg/kg/day) or saline injection (n=4/group). Bars represent means±SEM. For each parameter, bars with different letters are significantly different (p<0.05). Asterisks denote strain difference (p<0.05).
To address the question of the brain site(s) of acamprosate action, we undertook a separate study (Brager et al., 2011b), also in wild-type and Per2 mutant mice, using intracranial constant-release acamprosate-containing microimplants (with an output of ~50 ng/day) to map brain areas responsive to the suppressive effect of acamprosate on ethanol intake and preference. Targeted brain areas represented major alcohol reward [VTA, peduculopontine tegmentum (PPT), and nucleus accumbens (NA)] and circadian-related brain areas [intergeniculate leaflet (IGL) and SCN] (Glass et al., 2010; Koob et al., 1998; Luo et al., 2009; Samson and Chappell, 2001). In this experiment, the most pronounced response to acamprosate was in the VTA and PPT, where acamprosate decreased ethanol intake by 60% and preference by 40% in wild-type mice (Fig. 16). While there is little direct neurochemical evidence for acamprosate acting in these regions, such action is plausible given that ethanol activation of the VTA stimulates dopamine release in the NAc (Larsson and Engel, 2004), which is also affected by acamprosate via modulation of VTA ACh receptors (Chau et al., 2010a,b). Also, acamprosate inhibits NMDA (glutamatergic) receptor response in mesencephalic neurons, presumably containing elements of the VTA (Allgaier et al., 2000). Acamprosate implants in the NAc of wild-type mice also significantly suppressed ethanol intake and preference, but to a lesser extent than in the other reward areas. Nevertheless, this suppression is noteworthy given previous observations that strychnine administration into the NAc of ethanol-preferring rats (to inhibit glycine receptors) completely blocked acamprosate suppression of ethanol intake (Chau et al., 2010a). Also, previous studies have shown that acamprosate administration in the NAc increases levels of accumbal extracellular dopamine (Chau et al., 2010a,b) and dopamine reuptake transporters (Cowen et al., 2005). Further, acamprosate dampens ethanol and NMDA-induced accumbal dopamine release (Cano-Cebrian et al., 2003; Olive et al., 2002; Spanagel and Weiss, 1999). Together, these results strongly implicate the NAc as a target for acamprosate (Chau et al., 2010a,b), consistent with NAc involvement in alcohol intake (Nie et al.,2011). It is also notable that the degree to which all three reward areas responded to acamprosate in Per2 mutants was less than in wild-type mice. It is possible that the enhanced drive for ethanol in Per2 mutants rendered them less susceptible to acamprosate action.
Figure 16.
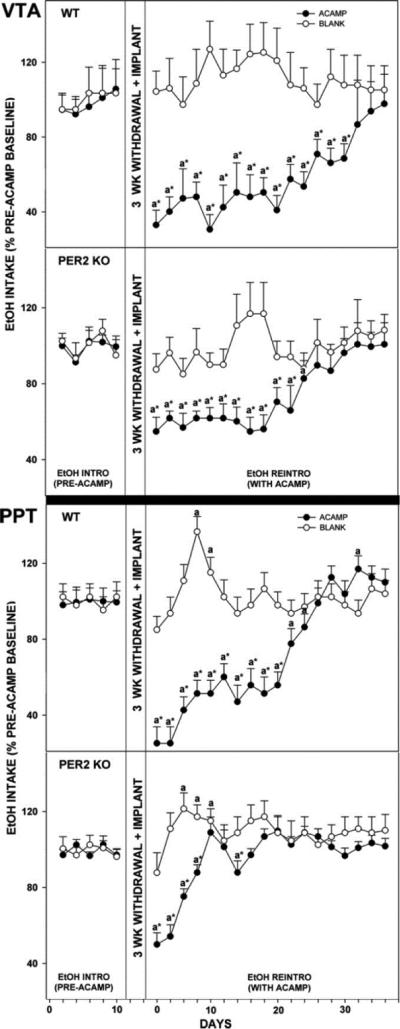
Line graphs show percent change of ethanol intake ± SE from pretreatment levels in mice receiving acamprosate (ACAMP) or no acamprosate (BLANK) from microimplants in reward areas [ventral tegmental area (VTA) andpeduculopontine tegmentum (PPT)during simulated relapse. “a” significant difference from pretreatment levels (p < 0.05). * Significant difference between treatment groups for any given day (p < 0.05). Bar graphs show means ± SEM for the time point of maximal suppression of ethanol intake by acamprosate microimplants in WT vs. mPer2 mutant (PER2 KO) mice. * Significant difference between strains (p < 0.05).
In the same study (Brager et al., 2011b), acamprosate implants were targeted to brain sites involved in regulating circadian timing. Studies have shown that the circadian system regulates ethanol seeking and consumption as reflected in the distinct daily rhythm in ethanol intake (Brager et al., 2010b; Dole and Gentry, 1984; Ruby et al., 2009a; Wasielewski and Holloway, 2001). The circadian phase of alcohol intake could be regulated by signaling from the SCN to the VTA (Luo and Aston-Jones, 2009) and/or by clock gene activity within the reward areas (Rosenwasser, 2010; Webb et al., 2009). In view of findings that 1) the SCN clock is directly disrupted by ethanol (Brager et al., 2011c; McElroy et al., 2009; Prosser et al., 2008; Ruby et al., 2009b); and 2) the SCN and IGL possess transmitter systems sensitive to acamprosate, including ACh, glutamate, and glycine (Hargraves and Fuchs, 1994; Moore and Speh, 1993; Stamp et al., 1997), these areas represent potential substrates for acamprosate's modulation of ethanol intake. Results from the present study bear out this hypothesis, as acamprosate implants targeted to the SCN and IGL inhibited ethanol intake in wild-type mice to a similar degree as acamprosate implants in the VTA and PPT. Also, unlike other targeted regions, acamprosate in the SCN suppressed ethanol intake and preference in wild-types and mPer2 mutants to a similar extent. It is notable that actogram analysis of circadian locomotor activity revealed that acamprosate suppression of ethanol intake was associated with few changes in circadian rhythm parameters, and importantly, did not perturb rhythm stability, phase angle of entrainment to the LD cycle, or the duration of nocturnal activity. This indicates that the suppression of alcohol intake by acamprosate within the SCN is not a direct consequence of altered circadian timekeeping.
6. Future directions
A. Pharmacology/receptor expression approaches
While we have made substantial progress in determining how ethanol modulates the SCN circadian clock, and how the clock in turn affects ethanol consumption, there are many questions still unanswered. It will be important to clarify the conditions under which the different forms of tolerance develop, and what brain areas are involved in modulating those effects. In particular, we believe that the timing of ethanol consumption may be a key factor regulating tolerance. We are also beginning to investigate acute ethanol-induced cellular changes that occur within the SCN, as well as the changes occurring in association with each form of tolerance. In particular, we are focusing on changes in glutamate and GABA receptors. Our initial experiments found no change in total expression or phosphorylation of the NMDA NR2B subunit (Lindsay et al., 2014) in response to rapid tolerance. However, this does not rule out changes in other NMDA subunits and/or changes in membrane localization of these receptors. Based on data showing the importance of TrkB receptors in enhancing glutamate signaling in the SCN and elsewhere (Kim et al., 2006;Michel et al., 2006;Mou et al., 2009), and ethanol's ability to disrupt these interactions (Kolb et al., 2005;Ron, 2004), we also plan to assess ethanol-induced changes in BDNF and TrkB signaling in the SCN. Finally, to fully understand the acute actions of ethanol and how these change across different time courses of ethanol consumption, it will be important to assess changes downstream from these receptors. Again, there is substantial precedence for investigating these cellular processes (Lin et al., 2006;Choi et al., 2008;Hellmann et al., 2009;Wu et al., 2011).
B. Molecular approaches
As discussed in detail by several contributors to this Special Issue, circadian clock genes participating in transcriptional, post-transcriptional, and post-translational feedback mechanisms (Ko and Takahashi, 2006) are known to play central roles in the etiology of drug abuse, including alcoholism (Bryant et al., 2009; Falcon and McClung, 2009; Perreau-Lenz and Spanagel, 2008; Rosenwasser, 2010). These genes include Period1 (Per1; Dong et al., 2011) and Period2 (Per2; Spanagel et al., 2005; Brager et al., 2011a,b), Clock (Andretic et al., 1999) and casein-kinase 1 ε/δ (CK1ε/δ; Perreau-Lenz et al., 2012). For example, Per1 mutant mice exhibit enhanced alcohol consumption under stressful conditions compared to wild-types, and Per2Brdm1 mutant mice show enhanced consumption (Brager et al., 2011a,b; Spanagel et al., 2005; Perreau-Lenz et al., 2009) and pharmacological inhibition of CK1ε/δ attenuates relapse-like ethanol intake (Perreau-Lenz et al., 2012). From a translational perspective, it has been shown that altered expression of clock genes, including Per genes in humans is linked to alcohol abuse and alcoholism (Comasco et al., 2010;Spanagel et al., 2005; Huang et al., 2010). It is apparent, therefore, that studies aimed at elucidating the mechanisms and sites for the modulatory actions of these circadian clock genes on ethanol reward offer a propitious approach for understanding the basis of drug abuse at the molecular level. Unfortunately, compensatory responses to genetic mutations and lack of regional specificity can complicate interpretation of the differences observed. However, more advanced methods of investigating genetic regulation are now available. For example, viral vectors containing short hairpin RNA (shRNA) can be used to conditionally knock down the expression of clock genes in a site-specific manner (Mukherjee et al., 2010). Microinjecting these probes into brain sites involved in ethanol reward and drive (i.e. the VTA and NAc) as well as circadian-related areas (i.e. the SCN and IGL) could provide information on which clock genes acting at what specific sites are involved in modulating various aspects of ethanol intake and dependence. Similarly, experiments utilizing conditionally regulated clock genes (e.g., tet-on/off-regulated gene expression) could clarify time-dependent roles of clock proteins in ethanol-induced effects, both in vivo and in vitro.
Another approach linking ethanol abuse to the circadian system is investigating how and where ethanol disrupts expression of microRNAs (miRNAs) that regulate the expression of mRNA and their proteins (Barbato et al., 2008). Recent studies have revealed that ethanol significantly alters miRNA expression in the brain and elsewhere (Sathayan et al., 2007; Tang et al., 2008; Pietrzykowski et al., 2008). Significantly, miRNAs likely play critical roles in SCN circadian clock photic regulation and Per gene activity (Cheng et al., 2007; Nagel et al., 2009). Clarifying potential ethanol-induced changes in miRNAs in the SCN presents an important conceptual model for understanding how ethanol directly affects circadian functioning at the molecular level.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Adinoff B, Risher-Flowers D, Dee Jong J, Ravitz B, Bone GHA, Nutt DJ, Roehrich L, Martin PR, Linnoila M. Disturbances of hypothalamic-pituitary-adrenal axis functioning during ethanol withdrawal in six men. Am J Psychiat. 1991;148:1023–1025. doi: 10.1176/ajp.148.8.1023. PMID: 1853950. [DOI] [PubMed] [Google Scholar]
- Ahmed SH. The science of making drug-addicted animals. Neuroscience. 2012;211:107–125. doi: 10.1016/j.neuroscience.2011.08.014. PMID: 21864653. [DOI] [PubMed] [Google Scholar]
- Allgaier C. Ethanol sensitivity of NMDA receptors. Neurochem Int. 2002;41:377–382. doi: 10.1016/s0197-0186(02)00046-3. PMID: 12213224. [DOI] [PubMed] [Google Scholar]
- Allgaier C, Franke H, Sobottka H, Scheibler P. Acamprosate inhibits Ca2+ influx mediated by NMDA receptors and voltage-sensitive Ca2+ channels in cultured rat mesencephalic neurones. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:440–443. doi: 10.1007/s002100000285. [DOI] [PubMed] [Google Scholar]
- Andretic R, Chaney S, Hirsh J. Requirement of Circadian Genes for Cocaine Sensitization in Drosophila. Science. 2008;285:1066–1068. doi: 10.1126/science.285.5430.1066. [DOI] [PubMed] [Google Scholar]
- Antle MC, Glass JD, Mistlberger RE. 5-HT1A autoreceptor antagonist-induced 5-HT release in the hamster suprachiasmatic nuclei: effects on circadian clock resetting. Neurosci Lett. 2000;282:97–100. doi: 10.1016/s0304-3940(00)00873-9. PMID: 10713405. [DOI] [PubMed] [Google Scholar]
- Barbato C, Giorgi C, Catalanotto C, Cogoni C. Thinking about RNA? MicroRNAs in the brain. Mamm Genome. 2008;19:541–551. doi: 10.1007/s00335-008-9129-6. [DOI] [PubMed] [Google Scholar]
- Biello SM, Golombek DA, Harrington ME. Neuropeptide Y and glutamate block each other's phase shifts in the suprachiasmatic nucleus in vitro. Neuroscience. 1997;77:1049–1057. doi: 10.1016/s0306-4522(96)00547-7. [DOI] [PubMed] [Google Scholar]
- Biello SM, Mrosovsky N. Blocking the phase-shifting effect of neuropeptide Y with light. Proc R Soc Lond B. 1995;259:179–187. doi: 10.1098/rspb.1995.0026. PMID: 7732038. [DOI] [PubMed] [Google Scholar]
- Bobrzynska KJ, Godfrey MH, Mrosovsky N. Serotonergic stimulation and nonphotic phase-shifting in hamsters. Physiol Behav. 1996;59:221–230. doi: 10.1016/0031-9384(95)02130-2. [DOI] [PubMed] [Google Scholar]
- Bottum K, Poon E, Haley B, Karmarkar S, Tischkau SA. Suprachiasmatic nucleus neurons desplay endogenous resistance to excitotoxicity. Exp Biol Med. 2010;235:237–246. doi: 10.1258/ebm.2009.009244. PMID: 20404040. [DOI] [PubMed] [Google Scholar]
- Brager AJ, Prosser RA, Glass JD. Constant light exposure in mice potentiates ethanol consumption and preference and disrupts circadian locomotor activity rhythms. Paper 7 Samuel B. Guze Symposium on Alcoholism. 2010a [Google Scholar]
- Brager AJ, Prosser RA, Glass JD. Circadian and acamprosate modulation of elevated ethanol drinking in mPer2 clock gene mutant mice. Chronobiol. International. 2011a;28:664–672. doi: 10.3109/07420528.2011.601968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager AJ, Prosser RA, Glass JD. Acamprosate-responsive brain sites for suppression of ethanol intake and preference. Am. J. Physiology. 2011b;301:R1032–R1043. doi: 10.1152/ajpregu.00179.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager AJ, Ruby CL, Prosser RA, Glass JD. Acute ethanol disrupts photic and serotonergic circadian clock phase-resetting in the mouse. Alcohol Clin Exp Res. 2011c;35:1467–1474. doi: 10.1111/j.1530-0277.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager AJ, Ruby CL, Prosser RA, Glass JD. Chronic ethanol disrupts circadian photic entrainment and impairs daily locomotor activity in the mouse. Alcohol Clin Exp Res. 2010b;34:1266–1273. doi: 10.1111/j.1530-0277.2010.01204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broom DR, Stensel DJ, Bishop NC, Burns SF, Miyashita M. Exercise-induced suppression of acylated ghrelin in humans. J Appl Physiol. 2007;102:2165–2171. doi: 10.1152/japplphysiol.00759.2006. [DOI] [PubMed] [Google Scholar]
- Brower KJ, Aldrich MS, Robinson EAR, Zucker RA, Greden JF. Insomnia, self-medication, and relapse to alcholism. Am J Psychiat. 2001;158:399–404. doi: 10.1176/appi.ajp.158.3.399. PMID: 11229980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Graham ME, Distler MG, Munoz MB, Li D, Vezina P, Sokoloff G, Palmer AA. A role for casein kinase 1 epsilon in the locomotor stimulant response to methamphetamine. Psychopharmacology (Berl) 2009;203:703–711. doi: 10.1007/s00213-008-1417-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Cebrián MJ, Zornoza-Sabina T, Guerri C, Polache A, Granero L. Local acamprosate modulates dopamine release in the rat nucleus accumbens through NMDA receptors: an in vivo microdialysis study. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:119–125. doi: 10.1007/s00210-002-0674-3. [DOI] [PubMed] [Google Scholar]
- Chau P, Lidö-Hölfödt H, Löf E, Söderpalm B, Ericson M. Glycine receptors in the nucleus accumbens involved in the ethanol intake-reducing effect of acamprosate. Alcohol Clin Exp Res. 2010a;34:39–45. doi: 10.1111/j.1530-0277.2009.01063.x. [DOI] [PubMed] [Google Scholar]
- Chau P, Stomberg R, Fagerberg A, Söderpalm B, Ericson M. Glycine receptors involved in acamprosate's modulation of accumbal dopamine levels: an in vivo microdialysis study. Alcohol Clin Exp Res. 2010b;34:32–38. doi: 10.1111/j.1530-0277.2009.01062.x. [DOI] [PubMed] [Google Scholar]
- Chen CP, Khun P, Advis JP, Sarkar DK. Chronic ethanol consumption impairs the circadian rhythm of pro-opiomelanocortin and period genes mRNA expression in the hypothalamus of the male rat. J Neurochem. 2004;88:1547–1554. doi: 10.1046/j.1471-4159.2003.02300.x. PMID: 15009656. [DOI] [PubMed] [Google Scholar]
- Cheng H-YM, Papp JW, Varlamova O, Dziema H, Russel B, Curfman JP, Nakazawa T, Shimizu K, Okamura H, Impey S, Obrietan K. microRNA modulation of circadian-clock perios and entrainment. Neuron. 2007;54:813–829. doi: 10.1016/j.neuron.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D-S, Wei W, Deitchman JK, Kharazia VN, Lesscher HMB, McMahon T, Wang D, Qi Z-H, Sieghart W, Zhang C, Shokat KM, Mody I, Messing RO. Protein kinase Cδ regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. J Neurosci. 2008;28:11890–11899. doi: 10.1523/JNEUROSCI.3156-08.2008. PMID: 19005054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CP, Gillin JC, Golshan S, Demodena A, Smith TL, Danowski S, Irwin M, Schuckit M. Increased REM sleep density at admission predicts relapse by three months in primary alcoholics with a lifetime diagnosis of secondary depression. Biol Psych. 1998;43:601–607. doi: 10.1016/s0006-3223(97)00457-5. PMID: 9564445. [DOI] [PubMed] [Google Scholar]
- Claustrat B, Valatx JL, Harthe C, Brun J. Effect of constant light on prolactin and corticosterone rhythms evaluated using a noninvasive urine sampling protocol in the rat. Horm Metab Res. 2008;40:398–403. doi: 10.1055/s-2008-1065330. [DOI] [PubMed] [Google Scholar]
- Comasco E, Nordquist N, Gokturk C, Aslund C, Hallman J, Oreland L, Nilsson KW. The clock gene PER2 and sleep problems: association with alcohol consumption among Swedish adolescents. Upsala J of Med Sci. 2010;115:41–48. doi: 10.3109/03009731003597127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen MS, Adams C, Kraehenbuehl T, Vengeliene V, Lawrence AJ. The acute anti-craving effect of acamprosate in alcohol-preferring rats is associated with modulation of the mesolimbic dopamine system. Addict Biol. 2005;10:233–242. doi: 10.1080/13556210500223132. [DOI] [PubMed] [Google Scholar]
- Cutrera RA, Kalsbeek A, Pevet P. Specific destruction of the serotonergic afferents to the suprachiasmatic nuclei prevents triazolam-induced phase advanced of hamster activity rhythms. Beh Brain Res. 1994;62:21–28. doi: 10.1016/0166-4328(94)90034-5. PMID: 7917030. [DOI] [PubMed] [Google Scholar]
- Damaggio AS, Gorman MR. Circadian phase determines effects of repeated ethanol vapor exposure and withdrawal on body temperature and activity rhythms of male mice. Alcohol Clin Expt Res. 2013:1–10. doi: 10.1111/acer.12297. PMID: 24256465. [DOI] [PubMed] [Google Scholar]
- de la Iglesia HO, Meyer J, Carpino A, Jr, Schwartz WJ. Antiphase oscillation of the left and right suprachiasmatic nuclei. Science. 2000;290:799–801. doi: 10.1126/science.290.5492.799. [DOI] [PubMed] [Google Scholar]
- Danel T, Touitou Y. Alcohol decreases the nocturnal peak of TSH in healthy volunteers. Psychopharmacology. 2003;170:213–214. doi: 10.1007/s00213-003-1532-9. PMID: 12827350. [DOI] [PubMed] [Google Scholar]
- Danel T, Vantyghem M-C, Touitou Y. Responses of the steroid circadian system to alcohol in humans: importance of the time and duration of intake. Chronobiol Int. 2006;23:1025–1034. doi: 10.1080/07420520600920742. PMID: 17050215. [DOI] [PubMed] [Google Scholar]
- Ding JM, Gillette MU. Glutamate induces light-like phase shifts in the rat SCN in brain slice. Soc Neurosci Abst. 1993;19:1815. PMID: 7903877. [Google Scholar]
- Dodd PR, Beckmann AM, Davidson MS, Wilce PA. Glutamate-mediated transmission, alcohol, and alcoholism. Neurochem Int. 2000;37:509–533. doi: 10.1016/s0197-0186(00)00061-9. [DOI] [PubMed] [Google Scholar]
- Dole VP, Gentry RT. Toward an analogue of alcoholism in mice: scale factors in the model. Proc Natl Acad Sci USA. 1984;81:3543–3546. doi: 10.1073/pnas.81.11.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Bilbao A, Laucht M, Henriksson R, Yakovleva T, Ridinger M, Desrivieres S, Clarke T-K, Lourdusamy A, Smolka MN, Cichon S, Blomeyer D, Treutlein J, Perreau-Lenz S, Witt S, Leonardi-Essmann F, Wodarz N, Zill P, Soyka M, Albrecht U, Rietschel M, Lathrop M, Bakalkin G, Spanagel R, Schumann G. Effects of the circadian rhythm gene period 1 (Per1) on psychosocial stress-induced alcohol drinking. Am J Psychiatry. 2011;168:1090–1098. doi: 10.1176/appi.ajp.2011.10111579. [DOI] [PubMed] [Google Scholar]
- Dudley TE, DiNardo LA, Glass JD. Endogenous regulation of serotonin release in the hamster suprachiasmatic nucleus. J Neurosci. 1998;18:5045–5052. doi: 10.1523/JNEUROSCI.18-13-05045.1998. PMID: 9634570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar DM, Reid MS, Dement WC. Serotonergic afferents mediate activity-dependent entrainment of the mouse circadian clock. Am J Physiol. 1997;273:R265–R269. doi: 10.1152/ajpregu.1997.273.1.R265. PMID: 9249559. [DOI] [PubMed] [Google Scholar]
- Ehlen JC, Grossman GH, Glass JD. In vivo resetting of the hamster circadian clock by 5-HT7receptors in the suprachiasmatic nucleus. J Neurosci. 2001;21:5351–5357. doi: 10.1523/JNEUROSCI.21-14-05351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlen JC, Paul KN. A role for extrasynaptic GABAA receptors in the circadian system. Soc Neurosci. 2007 Abst833.11. [Google Scholar]
- Ehlen JC, Paul KN. Regulation of light's actions in the mammalian circadian clock; role of the extrasynaptic GABAA receptor. Am.J.Physiol. 2009;296:R1606–R1612. doi: 10.1152/ajpregu.90878.2008. PMID: 19244580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehringer MA, Hoft NR, Zunhammer M. Reduced alcohol consumption in mice with access to a running wheel. Alcohol. 2009;43:443–452. doi: 10.1016/j.alcohol.2009.06.003. [DOI] [PubMed] [Google Scholar]
- El Rawas R, Thiriet N, Lardeux V, Jaber M, Solinas M. Environmental enrichment decreases the rewarding but not the activating effects of heroin. Psychopharmacology. 2009;203:561–570. doi: 10.1007/s00213-008-1402-6. [DOI] [PubMed] [Google Scholar]
- Falcon E, McClung CA. A role for the circadian genes in drug addiction. Neuropharmacology. 2009;56:91–96. doi: 10.1016/j.neuropharm.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillmore MT, Marczinski CA, Bowman AM. Acute tolerance to alcohol effects on inhibitory and activational mechanisms of behavioral control. J Stud Alcohol. 2005;66:663–672. doi: 10.15288/jsa.2005.66.663. PMID: 16331852. [DOI] [PubMed] [Google Scholar]
- Fonken LK, Finy MS, Walton JC, Weil ZM, Workman JL, Ross J, Nelson RJ. Influence of light at night on murine anxiety- and depressive-like responses. Behav Brain Res. 2009;205:349–354. doi: 10.1016/j.bbr.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Gass JT, Olive MF. Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol. 2008;75:218–265. doi: 10.1016/j.bcp.2007.06.039. PMID: 17706608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller I. Ethanol preference in the rat as a function of photoperiod. Science. 1971;173:456–459. doi: 10.1126/science.173.3995.456. [DOI] [PubMed] [Google Scholar]
- Gillette MU, Reppert SM. The hypothalamic suprachiasmatic nuclei: Circadian patterns of vasopressin secretion and neuronal activity [u]in vitro[/u]. Brain Res Bull. 1987;19:135–139. doi: 10.1016/0361-9230(87)90176-6. PMID: 3651837. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Koob GF. Effects of β-adrenoceptor antagonists on alcohol drinking by alcohol-dependent rats. Psychopharmacology. 2010;212:431–439. doi: 10.1007/s00213-010-1967-8. PMID: 20676608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Francl JM, Kaur G. On the intrinsic regulation of neuropeptide Y in the SCN circadian clock. Eur J Neurosci. 2010;31:1117–1126. doi: 10.1111/j.1460-9568.2010.07139.x. [DOI] [PubMed] [Google Scholar]
- Glass JD, Randolph WW, Ferreira SA, Rea MA, Hauser UE, Blank JL, De Vries MJ. Diurnal variation in 5-hydroxyindole-acetic acid output in the suprachiasmatic region of the siberian hamster assessed by in vivo microdialysis: evidence for nocturnal activation of serotonin release. Neuroendocrinology. 1992;56:582–590. doi: 10.1159/000126277. PMID: 1282219. [DOI] [PubMed] [Google Scholar]
- Goodman A. Neurobiology of addiction. An integrative review. Biochem Pharmacol. 2008;75:266–322. doi: 10.1016/j.bcp.2007.07.030. PMID: 17764663. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Browning MD. Protein kinase C activation induces tyrosine phosphorylation of the NR2A and NR2B subunits of the NMDA receptor. J Neurochem. 2001;76:737–744. doi: 10.1046/j.1471-4159.2001.00034.x. PMID: 11158244. [DOI] [PubMed] [Google Scholar]
- Gupta T, Syed YM, Revis AA, Miller SA, Martinez M, Cohn KA, Demeyer MR, Patel KY, Brzezinska WJ, Rhodes JS. Acute effects of acamprosate and MPEP on ethanol drinking-in-the-dark in male C57BL/6J mice. Alcohol Clin Exp Res. 2008;32:1992–1998. doi: 10.1111/j.1530-0277.2008.00787.x. [DOI] [PubMed] [Google Scholar]
- Hammer SB, Ruby CL, Brager AJ, Prosser RA, Glass JD. Environmental modulation of alcohol intake in hamsters: Effects of wheel-running and constant light exposure. Alcoholism: Clinical and Experimental Res. 2010;34:1651–1658. doi: 10.1111/j.1530-0277.2010.01251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartgraves MD, Fuchs JL. NMDA receptor binding in rodent suprachiasmatic nucleus. Brain Res. 1994;640:113–118. doi: 10.1016/0006-8993(94)91863-5. [DOI] [PubMed] [Google Scholar]
- Hellmann J, Rommelspacher H, Wernicke C. Long-term ethanol exposure impairs neuronal differentation of human neuroblastoma cells involving neurotrophin-mediated intracellular signaling and in particular protein kinase C. Alcohol Clin Expt Res. 2009;33:538–550. doi: 10.1111/j.1530-0277.2008.00867.x. [DOI] [PubMed] [Google Scholar]
- Hoffman PL. NMDA receptors in alcoholism. Int Rev Neurobiol. 2003;56:35–82. doi: 10.1016/s0074-7742(03)56002-0. PMID: 14696310. [DOI] [PubMed] [Google Scholar]
- Holloway FA, Miller JM, King DA, Bedingfield JB. Delayed ethanol effects on physiological and behavioral indices in the rat. Alcohol. 1993;10:511–519. doi: 10.1016/0741-8329(93)90075-y. PMID: 8123210. [DOI] [PubMed] [Google Scholar]
- Huang MC, Ho CW, Chen CH, Liu SC, Chen CC, Leu SJ. Reduced expression of circadian clock genes in male alcoholic patients. Alcohol Clin Exp Res. 2010;34:1899–1904. doi: 10.1111/j.1530-0277.2010.01278.x. [DOI] [PubMed] [Google Scholar]
- Iancu OD, Oberbeck D, Darakjian P, Metten P, McWeeney S, Crabbe JC, Hitzemann R. Selection for drinking in the dark alters brain gene coexpression networks. Alcohol Clin Expt Res. 2013;37:1295–1303. doi: 10.1111/acer.12100. PMID: 23550792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janik D, Mrosovsky N. Gene expression in the geniculate induced by a nonphotic circadian phase shifting stimulus. NeuroReport. 1992;3:575–578. doi: 10.1097/00001756-199207000-00007. PMID: 7922565. [DOI] [PubMed] [Google Scholar]
- Janik D, Mrosovsky N. Intergeniculate leaflet lesions and behaviorally-induced shifts of circadian rhythms. Brain Res. 1994;651:174–182. doi: 10.1016/0006-8993(94)90695-5. PMID: 1421110. [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu SL, Salome N, Heilig M, Moechars D, Datta R, Perrissoud D, Dickson SL, Engel JA. Requirement of central ghrelin signaling for alcohol reward. Proc Nat Acad Sci. 2009;106:11318–11323. doi: 10.1073/pnas.0812809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Koob GF, Schuckit MA, Mason BJ, Ait-Daoud N. Understanding and treating alcohol dependence. Alcohol Clin Expt Res. 2006;30:567–584. doi: 10.1111/j.1530-0277.2005.00066.x. PMID: 16499499. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Kim YI, Choi H-J, Colwell CS. Brain-derived neurotrophic factor regulation of N-methyl-D-aspartate receptor-mediated synaptic currents in suprachiasmatic nucleus neurons. J Neurosci Res. 2006;84:1512–1520. doi: 10.1002/jnr.21063. PMID: 16983663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliethermes CL, Cronise K, Crabbe JC. Home cage activity and ingestive behaviors in mice following chronic ethanol vapor inhalation. Physiol Behav. 2005;85:479–488. doi: 10.1016/j.physbeh.2005.05.009. PMID: 16005034. [DOI] [PubMed] [Google Scholar]
- Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Human Mol Genet. 2006;15:R271–R277. doi: 10.1093/hmg/ddl207. Spec No 2. [DOI] [PubMed] [Google Scholar]
- Kolb JE, Trettel J, Levine ES. BDNF enhancement of postsynaptic NMDA receptors is blocked by ethanol. Synapse. 2005;55:52–57. doi: 10.1002/syn.20090. PMID: 15515007. [DOI] [PubMed] [Google Scholar]
- Koob GF, Roberts AJ, Schulteis G, Parson LH, Heyser CJ, Hyytiä P, Merlo-Pich E, Weiss F. Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- Kosobud AEK, Gillman AG, Leffel JK, Pecoraro NC, Rebec GV, Timberlake W. Drugs of abuse can entrain circadian rhythms. ScientificWorldJournal. 2007;7:203–212. doi: 10.1100/tsw.2007.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzler HR, Gage A. Acamprosate efficacy in alcohol-dependent partients: summary of results from three pivotal trials. Am J Addict. 2008;17:70–76. doi: 10.1080/10550490701756120. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC. N-methyld-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther. 2003;99:79–94. doi: 10.1016/s0163-7258(03)00054-8. [DOI] [PubMed] [Google Scholar]
- Kuhlwein E, Hauger RL, Irwin MR. Abnormal nocturnal melatonin secretion and disordered sleep in abstinent alcoholics. Biol Psych. 2003;54:1437–1443. doi: 10.1016/s0006-3223(03)00005-2. PMID: 14675809. [DOI] [PubMed] [Google Scholar]
- Landolt HP, Gillin JC. Sleep abnormalities during abstinence in alcohol-dependent patients: aetiology and management. CNS Drugs. 2001;15:413–425. doi: 10.2165/00023210-200115050-00006. [DOI] [PubMed] [Google Scholar]
- Larsson A, Engel JA. Neurochemical and behavioral studies on ethanol and nicotine interactions. Neurosci Biobehav Rev. 2004;27:713–20. doi: 10.1016/j.neubiorev.2003.11.010. [DOI] [PubMed] [Google Scholar]
- LeMarquand D, Pihl RO, Benkelfat C. Serotonin and alcohol intake, abuse and dependence: Findings of animal studies. Biol Psychiat. 1994;36:395–421. doi: 10.1016/0006-3223(94)91215-7. [DOI] [PubMed] [Google Scholar]
- Liang J, Spigelman I, Olsen RW. Tolerance to sedative/hypnotic actions of GABAergic drugs correlates with tolerance to potentiation of extrasynaptic tonic currents of alcohol-dependent rats. J Neurophysiol. 2009;102:224–233. doi: 10.1152/jn.90484.2008. PMID: 19420124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HH, Chang S-J, Shie H-J, Lai CC. Ethanol inhibition of NMDA-induced responses and acute tolerance to the inhibition in rat rostral ventrolateral medulla in vivo: involvement of cAMP-dependent protein kinases. Neuropharmacology. 2006;51:747–755. doi: 10.1016/j.neuropharm.2006.05.018. PMID: 16806304. [DOI] [PubMed] [Google Scholar]
- Lindsay JH, Glass JD, Amicarelli M, Prosser RA. The mammalian circadian clock in the suprachiasmatic nucleus exhibits rapid tolerance to ethanol in vivo and in vitro. Alcohol Clin Expt Res. 2014;38:760–769. doi: 10.1111/acer.12303. PMID: 24512529. [DOI] [PubMed] [Google Scholar]
- Logan RW, McCulley WDI, Seggio JA, Rosenwasser AM. Effects of withdrawal from chronic intermittent ethanol vapor on the level and circadian periodicity of running-wheel activity in C57BL/6J and C3H/HeJ mice. Alcohol Clin Expt Res. 2012;36:467–476. doi: 10.1111/j.1530-0277.2011.01634.x. PMID: 24512529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan RW, Seggio JA, Robinson SL, Richard GR, Rosenwasser AM. Circadian wheel-running activity during withdrawal from chronic intermittent ethanol exposure in mice. Alcohol. 2010;44:239–244. doi: 10.1016/j.alcohol.2010.02.011. PMID: 20682191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, Foye PE, Racke M, Slone AL, Siegel BW, Danielson PE, Sutcliffe JG, Erlander MG. A novel adenylate cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms. Neuron. 1993;11:449–458. doi: 10.1016/0896-6273(93)90149-l. PMID: 8398139. [DOI] [PubMed] [Google Scholar]
- Luo AH, Aston-Jones G. Circuit projection from suprachiasmatic nucleus to ventral tegmental area: a novel circadian output pathway. Eur J Neurosci. 2009;29:748–760. doi: 10.1111/j.1460-9568.2008.06606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy B, Zakaria A, Glass JD, Prosser RA. Ethanol modulates mammalian circadian clock phase resetting through extrasynaptic GABA receptor activation. Neuroscience. 2009;164:842–848. doi: 10.1016/j.neuroscience.2009.08.020. PMID: 19695310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan DE, McClure GYH, Hardwick WC. Effects of access to a running wheel on food, water and ethanol intake in rats bred to accept ethanol. Drug Alcohol Dependence. 1995;40:1–7. doi: 10.1016/0376-8716(95)01162-5. [DOI] [PubMed] [Google Scholar]
- Meyer-Bernstein EL, Blanchard JH, Morin LP. The serotonergic projection from the median raphe nucleus to the suprachiasmatic nucleus modulates activity phase onset, but not other circadian rhythm parameters. Brain Res. 1997;755:112–120. doi: 10.1016/s0006-8993(97)00111-x. PMID: 9163546. [DOI] [PubMed] [Google Scholar]
- Meyer-Bernstein EL, Morin LP. Electrical stimulation of the median or dorsal raphe nuclei reduces light-induced fos protein in the suprachiasmatic nucleus and causes circadian activity rhythm phase shifts. Neuroscience. 1999;92:267–279. doi: 10.1016/s0306-4522(98)00733-7. PMID: 10392849. [DOI] [PubMed] [Google Scholar]
- Michel S, Clark JP, Ding JM, Colwell CS. Brain-derived neurotrophic factor and neurotrophin receptors modulate glutamate-induced phase shifts of the suprachiasmatic nucleus. Eur J Neurosci. 2006;24:1109–1116. doi: 10.1111/j.1460-9568.2006.04972.x. PMID: 16930436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, Spear LP. The alcoholism generator. Alcohol Clin Expt Res. 2006;30:1466–1469. doi: 10.1111/j.1530-0277.2006.00177.x. PMID: 16930208. [DOI] [PubMed] [Google Scholar]
- Mistlberger RE, Nadeau J. Ethanol and circadian rhythms in the Syrian hamster: effects on entrained phase, reentrainment rate, and period. Pharmacol Biochem Beh. 1992;43:159–165. doi: 10.1016/0091-3057(92)90652-v. PMID: 1409799. [DOI] [PubMed] [Google Scholar]
- Moore RY, Speh JC. GABA is the principal neurotransmitter of the circadian system. Neurosci Lett. 1993;150:112–116. doi: 10.1016/0304-3940(93)90120-a. [DOI] [PubMed] [Google Scholar]
- Morin LP, Blanchard J. Depletion of brain serotonin by 5,7-DHT modifies hamster circadian rhythm response to light. Brain Res. 1991;566:173–185. doi: 10.1016/0006-8993(91)91696-x. PMID: 1814534. [DOI] [PubMed] [Google Scholar]
- Mou X, Peterson CB, Prosser RA. Tissue-type plasminogen activator-plasmin-BDNF modulate glutamate-induced phase-shifts of the mouse suprachiasmatic circadian clock in vitro. Eur J Neurosci. 2009;30:1451–1460. doi: 10.1111/j.1460-9568.2009.06946.x. PMID: 19811533. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Coque L, Cao J-L, Kumar J, Chakravarty S, Asaithamby A, Graham A, Gordon E, Enwright JF, DiLeone RJ, Birnbaum SG, Cooper DC, McClung CA. Knockdown of Clock in the ventral tegmental area through RNA interference results in a mixed state of mania and depression-like behavior. Biol Psychiatry. 2010;68:503–511. doi: 10.1016/j.biopsych.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel R, Clijsters L, Agami R. The miRNA-192/194 cluster regulates the Period gene family and the circadian clock. FEBS J. 2009;296:5447–5455. doi: 10.1111/j.1742-4658.2009.07229.x. [DOI] [PubMed] [Google Scholar]
- Newman GC, Hospod FE, Patlak CS. Brain slice glucose utilization. J Neurochem. 1988;51:1783–1796. doi: 10.1111/j.1471-4159.1988.tb01160.x. PMID: 3183659. [DOI] [PubMed] [Google Scholar]
- Newman GC, Hospod FE, Patlak CS, Moore RY. Analysis of in vitro glucose utilization in a circadian pacemaker model. J Neurosci. 1992;12:2015–2021. doi: 10.1523/JNEUROSCI.12-06-02015.1992. PMID: 1607926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H, Rewal M, Gill TM, Ron D, Janak PH. Extrasynaptic δ-containing GABAA receptors in the nucleus accumbens dorsomedial shell contribute to alcohol intake. PNAS. 2011;108:4459–4464. doi: 10.1073/pnas.1016156108. PMID: 21368141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF, Nannini MA, Ou CJ, Koenig HN, Hodge CW. Effects of acute acamprosate and homotaurine on ethanol intake and ethanol-stimulated mesolimbic dopamine release. Eur J Pharmacol. 2002;437:55–61. doi: 10.1016/s0014-2999(02)01272-4. [DOI] [PubMed] [Google Scholar]
- Ozburn AR, Gordon EA, McClung CA. ClockΔ19 mutants exhibit increased ethanol preference and consumption. Alcohol Clin Exp Res. 2010;34(6):39A. [Google Scholar]
- Ozburn AR, Harris RA, Blednov YA. Wheel running, voluntary ethanol consumption, and hedonic substitution. Alcohol. 2008;42:417–424. doi: 10.1016/j.alcohol.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreau-Lenz S, Spanagel R. The effects of drugs of abuse on clock genes. Drug News Perspect. 2008;21:211–217. doi: 10.1358/dnp.2008.21.4.1213350. [DOI] [PubMed] [Google Scholar]
- Perreau-Lenz S, Zghoul T, de Fonseca FR, Spanagel R, Bilbao A. Circadian regulation of central ethanol sensitivity by the mPer2 gene. Addict Biol. 2009;14:253–259. doi: 10.1111/j.1369-1600.2009.00165.x. [DOI] [PubMed] [Google Scholar]
- Perreau-Lenz S, Vengeliene V, Noori HR, Merlo-Pich EV, Corsi MA, Corti C, Spanagel R. Inhibition of the casein-kinase-1-ε/δ/ prevents relapse-like alcohol drinking. 2012;37:2121–2131. doi: 10.1038/npp.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast JS, Friday RC, Yamazaki S. Distinct functions of Period2 and Period3 in the mouse circadian system revealed by in vitro analysis. PLoS ONE. 2010;5:e8552. doi: 10.1371/journal.pone.0008552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, Siegelmann HT, Triestman SN. Posttranslational regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59:274–287. doi: 10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisu MG, Mostallino MC, Dore R, Maciocco E, Secci PP, Serra M. Effects of voluntary ethanol consumption on emotional state and stress responsiveness in socially isolated rats. Eur Neuropsychopharm. 2011;21:414–425. doi: 10.1016/j.euroneuro.2010.07.006. PMID: 21067904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohorecky LA, Roberts P. Daily dose of ethanol and the development and decay of acute and chronic tolerance and physical dependence in rats. Pharmacol Biochem Beh. 1992;42:831–842. doi: 10.1016/0091-3057(92)90037-g. PMID: 1513866. [DOI] [PubMed] [Google Scholar]
- Prosser RA. Glutamate blocks serotonergic phase advances of the mammalian circadian pacemaker through AMPA and NMDA receptors. J Neurosci. 2001;21:7815–7822. doi: 10.1523/JNEUROSCI.21-19-07815.2001. PMID: 11567072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RA. Serotonin phase-shifts the mouse suprachiasmatic circadian clock in vitro. Brain Res. 2003;966:110–115. doi: 10.1016/s0006-8993(02)04206-3. PMID: 12646314. [DOI] [PubMed] [Google Scholar]
- Prosser RA, Dean RR, Edgar DM, Heller HC, Miller JD. Serotonin and the mammalian circadian system: I. In vitro phase shifts by serotonergic agonists and antagonists. J Biol Rhythms. 1993;8:1–16. doi: 10.1177/074873049300800101. PMID: 8490207. [DOI] [PubMed] [Google Scholar]
- Prosser RA, Glass JD. The mammalian circadian clock exhibits acute tolerance to ethanol. Alcohol Clin Exp Res. 2009;33:2088–2093. doi: 10.1111/j.1530-0277.2009.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RA, Lee H-M, Wehner A. Serotonergic pre-treatments block in vitro serotonergic phase shifts of the mouse suprachiasmatic nucleus circadian clock. Neuroscience. 2006;142:547–555. doi: 10.1016/j.neuroscience.2006.06.014. PMID: 16876330. [DOI] [PubMed] [Google Scholar]
- Prosser RA, Mangrum CA, Glass DJ. Acute ethanol modulates glutamatergic and serotonergic phase shifts of the mouse circadian clock in vitro. Neuroscience. 2008;152:837–848. doi: 10.1016/j.neuroscience.2007.12.049. PMID: 18313227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RA, Miller JD, Heller HC. A serotonin agonist phase-shifts the circadian clock in the suprachiasmatic nucleus in vitro. Brain Res. 1990;534:336–339. doi: 10.1016/0006-8993(90)90153-3. [DOI] [PubMed] [Google Scholar]
- Radcliffe RA, Floyd KL, Lee MJ. Rapid ethanol tolerance mediated by adaptations in acute tolerance in inbred mouse strains. Pharmacol Biochem Beh. 2006;84:524–534. doi: 10.1016/j.pbb.2006.06.018. PMID: 16899285. [DOI] [PubMed] [Google Scholar]
- Rajakrishnan V, Subramanian P, Viswanathan P, Menon VP. Effect of chronic ethanol ingestion on biochemical circadian rhythms in Wistar rats. Alcohol. 1999;18:147–152. doi: 10.1016/s0741-8329(98)00077-9. PMID: 10456565. [DOI] [PubMed] [Google Scholar]
- Ralph MR, Mrosovsky N. Behavioral inhibition of circadian responses to light. J Biol Rhythms. 1992;7:353–359. doi: 10.1177/074873049200700408. PMID: 1286206. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF. Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. PMID: 10788758. [DOI] [PubMed] [Google Scholar]
- Roehrs T, Petrucelli N, Roth T. Sleep restriction, ethanol effects and time of day. Human Psychoparm. 1996;11:199–204. [Google Scholar]
- Roehrs T, Roth T. Sleep, sleepiness, sleep disorders and alcohol use and abuse. Sleep Med Rev. 2001;5:287–297. doi: 10.1053/smrv.2001.0162. PMID: 12530993. [DOI] [PubMed] [Google Scholar]
- Rojdmark S, Wikner J, Adner N, Andersson DEH, Wetterberg L. Inhibition of melatonin secretion by ethanol in man. Metab Clin Exp. 1993;42:1047–1051. doi: 10.1016/0026-0495(93)90021-f. PMID: 8345809. [DOI] [PubMed] [Google Scholar]
- Ron D. Signaling cascades regulating NMDA receptor sensitivity to ethanol. The Neuroscientist. 2004;10:325–336. doi: 10.1177/1073858404263516. PMID: 15271260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwasser AM. Alcohol, antidepressants, and circadian rhythms. Alcohol Res Health. 2001;25:126–135. [PMC free article] [PubMed] [Google Scholar]
- Rosenwasser AM. Effects of ethanol and ethanol preference on the mammalian circadian pacemaker: behavioral characteristics. Alcohol Clin Expt Res. 2004;28S:60A. [Google Scholar]
- Rosenwasser AM. Circadian clock genes: non-circadian roles in sleep, addiction, and psychiatric disorders? Neurosci Biobehav Rev. 2010;38:1249–1255. doi: 10.1016/j.neubiorev.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Rosenwasser AM, Clark JW, Fixaris MC, Belanger GV, Foster JA. Effects of repeated light-dark phase shifts on voluntary ethanol and water intake in male and female Fischer and Lewis rats. Alcohol. 2010;44:229–237. doi: 10.1016/j.alcohol.2010.03.002. PMID: 20488643. [DOI] [PubMed] [Google Scholar]
- Rosenwasser AM, Fecteau ME, Logan RW. Effects of ethanol intake and ethanol withdrawal on free-running circadian activity rhythms in rats. Physiol Behav. 2005a;84:537–542. doi: 10.1016/j.physbeh.2005.01.016. PMID: 15811388. [DOI] [PubMed] [Google Scholar]
- Rosenwasser AM, Fecteau ME, Logan RW, Reed JD, Cotter SJ, Seggio JA. Circadian activity rhythms in selectively bred ethanol-preferring and nonpreferring rats. Alcohol. 2005b;36:69–81. doi: 10.1016/j.alcohol.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Rosenwasser AM, Logan RW, Fecteau ME. Chronic ethanol intake alters circadian period-responses to brief light pulses in rats. Chronobiol Int. 2005c;22:227–236. doi: 10.1081/cbi-200053496. PMID: 16021840. [DOI] [PubMed] [Google Scholar]
- Rösner S, Hackl-Herrwerth A, Leucht S, Lehert P, Vecchi S, Soyka M. Acamprosate for alcohol dependence. Cochrane Database Syst Rev. 2010;8:CD004332. doi: 10.1002/14651858.CD004332.pub2. [DOI] [PubMed] [Google Scholar]
- Ruby CL, Brager AJ, DePaul MA, Prosser RA, Glass JD. Chronic ethanol disrupts circadian behavior and photic phase-resetting in the hamster. Am J Physiol. 2009a;297:R729–R737. doi: 10.1152/ajpregu.00268.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby CL, Prosser RA, DePaul MA, Roberts RJ, Glass JD. Acute ethanol impairs photic and nonphotic circadian phase resetting in the Syrian hamster. Am J Physiol. 2009b;296:R411–R418. doi: 10.1152/ajpregu.90782.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson HH, Chappell A. Injected muscimol in pedunculopontine tegmental nucleus alters ethanol self-administration. Alcohol. 2001;23:41–8. doi: 10.1016/s0741-8329(00)00122-1. [DOI] [PubMed] [Google Scholar]
- Sathyan P, Golden HB, Miranda RC. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of fetal cerebral cortical neuroepithelium. J Neurosci. 2007;27:8546–8557. doi: 10.1523/JNEUROSCI.1269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M, Frey R, Pichler P, Ropke H, Anderer P, Saletu B, Rudas S. Sleep quality during alcohol withdrawal with bright light therapy. Prog Neuropsychopharmacol Bio Psychiatry. 1997;21:965–977. doi: 10.1016/s0278-5846(97)00092-4. [DOI] [PubMed] [Google Scholar]
- Seggio JA, Fixaris MC, Reed JD, Logan RW, Rosenwasser AM. Chronic ethanol intake alters circadian phase shifting and free-running period in mice. J Biol Rhythms. 2009;24:304–312. doi: 10.1177/0748730409338449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seggio JA, Logan RW, Rosenwasser AM. Chronic ethanol intake modulates photic and non-photic circadian phase responses in the Syrian hamster. Pharmacol Biochem Beh. 2007;87:297–305. doi: 10.1016/j.pbb.2007.05.001. PMID: 17544066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selim M, Glass JD, Hauser UE, Rea MA. Serotonergic inhibition of light-induced fos protein expression and extracellular glutamate in the suprachiasmatic nuclei. Brain Res. 1993;621:181–188. doi: 10.1016/0006-8993(93)90105-v. PMID: 7902183. [DOI] [PubMed] [Google Scholar]
- Shibata S, Tsuneyoshi A, Hamada T, Tominaga K, Watanabe S. Phase-shifting effect of 8-OH-DPAT, a serotonin1A receptor agonist, on the circadian rhythm of firing rate in the rat suprachiasmatic nuclei in vitro. Brain Res. 1992;582:353–356. doi: 10.1016/0006-8993(92)90156-4. [DOI] [PubMed] [Google Scholar]
- Sjöholm LK, Kovanen L, Sarrikoski ST, Schalling M, Lavebratt C, Partonen T. CLOCK is suggested to associate with comorbid alcohol use and depressive disorders. J Circadian Rhythms. 2010;8:1–9. doi: 10.1186/1740-3391-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Schmidt KT, Iordanou JC, Mustroph ML. Aerobic exercise decreases the positive-reinforcing effects of cocaine. Drug Alcohol Depend. 2008;98:129–135. doi: 10.1016/j.drugalcdep.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Kiefer F. Drugs for relapse prevention of alcoholism: ten years of progress. TIPS. 2008;29:109–115. doi: 10.1016/j.tips.2007.12.005. PMID: 18262663. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, Lascorz J, Depner M, Holzberg D, Soyka M, Schreiber S, Matsuda F, Lathrop M, Schumann G, Albrecht U. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nature Med. 2005;11:35–42. doi: 10.1038/nm1163. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci. 1999;22:521–527. doi: 10.1016/s0166-2236(99)01447-2. [DOI] [PubMed] [Google Scholar]
- Sprouse J, Li X, Stock J, McNeish J, Reynolds L. Circadian rhythm phenotype of 5-HT7 receptor knockout mice: 5-HT and 8-OH-DPAT-induced phase advances of SCN neuronal firing. J.Biol.Rhythms. 2005;20:122–131. doi: 10.1177/0748730404273432. PMID: 15834109. [DOI] [PubMed] [Google Scholar]
- Sprouse J, Reynolds L, Braselton J, Schmidt A. Serotonin-induced phase advances of SCN neuronal firing in vitro: a possible role for 5-HT5A receptors? Synapse. 2004;54:111–118. doi: 10.1002/syn.20070. PMID: 15352136. [DOI] [PubMed] [Google Scholar]
- Stamp JA, Piggins HD, Rusak B, Semba K. Distribution of ionotropic glutamate receptor subunit immunoreactivity in the suprachiasmatic nucleus and intergeniculate leaflet of the hamster. Brain Res. 1997;756:215–224. doi: 10.1016/s0006-8993(97)00199-6. [DOI] [PubMed] [Google Scholar]
- Sudo M, Sasahara K, Moriya T, Akiyama M, Hamada T, Shibata S. Constant light housing attenuates circadian rhythms of mPer2 mRNA and mPer2 protein expression in the suprachiasmatic nucleus of mice. Neuroscience. 2003;121:493–499. doi: 10.1016/s0306-4522(03)00457-3. [DOI] [PubMed] [Google Scholar]
- Sung JY, Park SM, Lee C-H, Um JW, Lee HJ, Kim J, Oh YJ, Lee S-T, Paik SR, Chung KC. Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J Biol Chem. 2005;280:25216–25224. doi: 10.1074/jbc.M503341200. PMID: 15863497. [DOI] [PubMed] [Google Scholar]
- Tang Y, Banan A, Forsyth CB, Fields JZ, Lau CK, Zhang LJ, Keshavarzian A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin Expt Res. 2008;32:355–364. doi: 10.1111/j.1530-0277.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- Tominaga K, Shibata S, Ueki S, Watanabe S. Effects of 5-HT1A receptor agonists on the circadian rhythm of wheel-running activity in hamsters. Europ J Pharm. 1992;214:79–84. doi: 10.1016/0014-2999(92)90099-p. [DOI] [PubMed] [Google Scholar]
- van Reeth O, Turek FW. Stimulated activity mediates phase shifts in the hamster circadian clock induced by dark pulses or benzodiazepines. Nature. 1989;339:49–51. doi: 10.1038/339049a0. PMID: 2654641. [DOI] [PubMed] [Google Scholar]
- Velazquez-Marrero C, Wynne P, Bernardo A, Palacio S, Martin G, Treistman SN. The relationship between duration of initial alcohol exposure and persistence of molecular tolerance is markedly nonlinear. J Neurosci. 2011;31:2436–2446. doi: 10.1523/JNEUROSCI.5429-10.2011. PMID: 21325511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira E, Nilsson EC, Nerstedt A, Ormestad M, Long YC, Garcia-Roves PM, Zierath JR, Mahlapuu M. Relationship between AMPK and the transcriptional balance of clock-related genes in skeletal muscle. Am J Physiol. 2008;295:E1032–E1037. doi: 10.1152/ajpendo.90510.2008. [DOI] [PubMed] [Google Scholar]
- Vrang N, Mrosovsky N, Mikkelsen JD. Afferent projections to the hamster intergeniculate leaflet demonstrated by retrograde and anterograde tracing. Brain Res Bull. 2003;59:267–288. doi: 10.1016/s0361-9230(02)00875-4. [DOI] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW. Ethanol enhances α4β3δ and α6β3δ γ-aminobutyric acid type A receptors at low concentrations known to affect humans. PNAS. 2003;100:15218–15223. doi: 10.1073/pnas.2435171100. PMID: 14625373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasielewski JA, Holloway FA. Alcohol's interactions with circadian rhythms. Alcohol Res Health. 2001;25:94–100. [PMC free article] [PubMed] [Google Scholar]
- Webb IC, Baltazar RM, Wang X, Pitchers KK, Coolen LM, Lehman MN. Diurnal variations in natural and drug reward, mesolimbic tyrosine hydroxylase, and clock gene expression in the male rat. J Biol Rhythms. 2009;24:465–76. doi: 10.1177/0748730409346657. [DOI] [PubMed] [Google Scholar]
- Wei W, Coutinho Faria L, Mody I. Low ethanol concentrations selectively augment the tonic inhibition mediated by δ subunit-containing GABAA receptors in hippocampal neurons. J Neurosci. 2004;24:8379–8382. doi: 10.1523/JNEUROSCI.2040-04.2004. PMID: 15385620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werme M, Lindholm S, Thoren P, Franck J, Brene S. Running increases ethanol preference. Behav Brain Res. 2002;133:301–308. doi: 10.1016/s0166-4328(02)00027-x. [DOI] [PubMed] [Google Scholar]
- Wideman CH, Murphy HM. Constant light induces alterations in melatonin levels, food intake, feed efficiency, visceral adiposity, and circadian rhythms in rats. Nutritional Neurosci. 2009;12:233–240. doi: 10.1179/147683009X423436. [DOI] [PubMed] [Google Scholar]
- Wu PH, Coultrap SJ, Browning MD, Proctor WR. Functional adaptation of the N-methyl-D-aspartate receptor to inhibition by ethanol is modulated by striatal-enriched protein tyrosine phosphatase and p38 mitogen-activated protein kinase. Molec Pharmacol. 2011;80:529–537. doi: 10.1124/mol.110.068643. PMID: 21680777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtman RJ, Axelrod J, Phillips LS. Melatonin synthesis in the pineal gland: Control by light. Science. 1963;142:1071–1073. doi: 10.1126/science.142.3595.1071. [DOI] [PubMed] [Google Scholar]