

Abstract
In mammals, β-carotene-15,15′-oxygenase (BCO1) is the main enzyme that cleaves β-carotene, the most abundant vitamin A precursor, to generate retinoids (vitamin A derivatives), both in adult and developing tissues. We previously reported that, in addition to this function, BCO1 can also influence the synthesis of retinyl esters, the storage form of retinoids, in the mouse embryo at mid-gestation. Indeed, lack of embryonic BCO1 impaired both lecithin-dependent and acyl CoA-dependent retinol esterification, mediated by lecithin:retinol acyltransferase (LRAT) and acyl CoA:retinol acyltransferase (ARAT), respectively. Furthermore, embryonic BCO1 also influenced the ester pools of cholesterol and diacylglycerol. In this report, we gained novel insights into this alternative function of BCO1 by investigating whether BCO1 influenced embryonic retinoid and lipid metabolism in a tissue-dependent manner. To this end, livers and brains from wild-type and BCO1−/− embryos at mid-gestation were analyzed for retinoid and lipid content, as well as gene expression levels. We also asked whether or not the role of BCO1 as a regulator of lecithin- and acyl CoA-dependent retinol esterification was exclusively restricted to the developing tissues. Thus, a survey of retinol and retinyl ester levels in adult tissues of wild-type, BCO1−/−, LRAT−/− and LRAT−/−BCO1−/− mice was performed. We showed that the absence of BCO1 affects embryonic retinoid and lipid homeostasis in a tissue-specific manner and that retinyl ester formation is also influenced by BCO1 in a few adult tissues (pancreas, lung, heart and adipose) in a sex- dependent manner.
Keywords: β-carotene 15, 15′ oxygenase (BCO1 or CMOI or BCMO1), embryonic organs, adult organs, retinoid metabolism, β-carotene metabolism
INTRODUCTION
In mammalian tissues, the enzyme β-carotene-15,15′-oxygenase (BCO1) cleaves β-carotene, the most abundant vitamin A precursor, to generate retinoids (vitamin A and its derivatives) which are needed to sustain critical biological functions, including embryonic development [1, 2]. BCO1 cleaves β-carotene at the 15,15′ double bond, yielding two molecules of retinaldehyde, which can then be oxidized to retinoic acid, the transcriptionally active form of vitamin A [3], or reduced to retinol (vitamin A alcohol) [4]. Retinol can form retinyl esters for storage in tissues upon esterification by lecithin:retinol acyltransferase (LRAT) which uses lecithin as a fatty acyl donor [5]. At least in the intestine, an unidentified acyl CoA:retinol acyltransferase (ARAT), catalyzing the fatty acyl CoA-dependent esterification of retinol, also exists and may work in conjunction with LRAT to synthesize retinyl esters [6–8]. The liver is the major storage site of vitamin A, from which retinol is recovered and secreted into the circulation bound to retinol-binding protein (RBP) to be delivered to all peripheral organs [9, 10]. However, retinyl esters can accumulate to various degrees in a number of other sites within the body, including the lung and adipose tissue [5]. β-carotene can also be cleaved asymmetrically by a second enzyme, β-carotene-9′,10′-oxygenase (BCO2), generating one β-ionone ring and β-apo 10′ carotenal [11]. The latter can be converted into one molecule of retinaldehyde by a chain shortening mechanism that seems to be mediated by BCO1 [11]. This enzyme, however, does not likely play a major role in the generation of retinoids in vivo, but it rather seems to protect the cell, specifically the mitochondria where it is localized [12], from the toxic effects of carotenoid accumulation [13].
The importance of BCO1 in supporting vitamin A-dependent functions via cleavage of the dietary provitamin A carotenoid, β-carotene, has been shown in adult and developing tissues [11, 14, 15]. By using mice lacking BCO1, our laboratory demonstrated that the mammalian embryo obtains β-carotene from the maternal bloodstream upon placental uptake and consequent transfer to the developing tissues, where the provitamin A is metabolized via BCO1 to support normal development [1, 14]. These studies also provided evidence that BCO1 deficiency reduced embryonic retinyl ester levels, as well as LRAT mRNA expression and activity [14]. Our data also indicated that an ARAT activity may exist in the developing tissues and that such activity was also impaired in embryos lacking BCO1 [16]. Further analysis of whole-embryo lipid content also suggested that the BCO1 genotype impacts on cholesteryl ester, triacylglycerol and even phosphatidylcholine (PC) and phosphatidylethanolamine (PE) synthesis. Indeed, in embryos knockout for the cleavage enzyme, the above-mentioned lipid classes were significantly reduced compared to wild-type embryos, and so were the mRNA levels of LCAT (lecitin:cholesterol acyltransferase), ACAT1 (acyl CoA:cholesterol acyltransferase 1) and DGAT2 (acyl CoA:diacylglycerol acyltransferase 2) genes encoding the key enzymes catalyzing cholesteryl ester and triacylglycerol synthesis [16].
Whereas the tissue-specific expression of BCO1 in adult mammalian tissues has been extensively reported [17–19], a thorough analysis of the tissue BCO1 expression pattern throughout embryogenesis is still lacking. At mid-gestation (14.5 days post coitum, dpc), the developmental window that was the focus of our initial findings, the majority of the organogenesis is completed, and the embryo can be considered “metabolically” active and capable of regulating its own retinoid and carotenoid metabolism [1, 14, 16]. We were therefore interested in understanding whether the BCO1 genotype would impact retinoid and lipid metabolism in the embryo in a tissue-dependent manner. To this end, we used wild-type and BCO1 knockout mice (BCO1−/−) to study whether, in the absence of BCO1, retinoid and lipid content or composition was reduced in the embryonic liver and brain. We chose to analyze the liver, as it is a major site of retinoid metabolism [5, 20], and brain, as this organ has a very high demand of lipids during embryonic development [21, 22]. Furthermore, ablation of BCO1 in the background of a mouse model that is susceptible to developing vitamin A deficiency (RBP−/− mice; [23]) resulted in a severe brain malformation (exencephaly) [14]. The failure to improve this developmental defect upon β-carotene supplementation of dams bearing BCO1+/-RBP−/− embryos, suggested to us that this phenotype was not vitamin A- but lipid-dependent [14, 16]. We were also interested in understanding whether the impact of the BCO1 genotype on retinyl ester formation, both lecithin- and acyl CoA-dependent, was restricted exclusively to the developing tissues. To this end, we compared retinol and retinyl ester levels in various tissues of wild-type vs. BCO1−/− adult females and LRAT−/− vs. LRAT−/− BCO1−/− adult male and female mice. Here we report that the BCO1 genotype impacts embryonic retinoid and lipid homeostasis in a tissue-specific manner and that retinyl ester formation is also influenced by BCO1 in a few adult tissues (pancreas, lung, heart and adipose) in a sex-dependent manner.
MATERIALS AND METHODS
Animals
Wild-type (WT), BCO1 knockout mice (BCO1−/−; [24]), LRAT knockout mice (LRAT−/−; [25]), and mice lacking both LRAT and BCO1 (LRAT−/−BCO1−/−; [16]) were used in these studies. All mice have a mixed genetic background C57BL/6 X 129sv. Mice were maintained on a standard nutritionally complete vitamin A-sufficient chow diet (vitamin A, 18 IU/g diet; carotenoids 1.2 ppm) throughout life and gestation. Food and water were provided ad libitum and the animals were housed in the facility under regular dark-light cycle (light on 7am to 7pm) without challenging time difference. For the experiments conducted on the embryonic organs, WT (n= 4) and BCO1−/− (n= 4) female mice were mated with matching genotype males at three months of age. At the time of vaginal plug detection (set as 0.5 dpc, the onset of gestation), females were separated from males and maintained on the same diet until the day of sacrifice (14.5 dpc). Dissected embryonic tissues were immediately frozen and kept at −80°C for further use. For the experiments conducted on the adult organs, groups of WT, LRAT−/−, BCO1−/− and LRAT−/−BCO1−/− male and female (n = 3 mice/group) mice were sacrificed at 55 and 110 days of age, respectively. Dissected organs were immediately frozen and kept at −80°C for further use. The adult mice used in this study were not dark-adapted prior to sacrifice. All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Rutgers University Institutional Committee on Animal Care.
Reagents
All solvents for analytical determinations were HPLC grade. Tetrahydrofuran, 2,2,4-trimethylpentane, toluene, isooctane and isopropanol were obtained from J.T. Baker (Avantor Performance Materials Inc., Center Valley, PA); acetone and water were from EMD Chemicals, Inc. (Gibbstown, NJ). Stigmastanol was purchased from Sigma (St. Louis, MO); C14:0 ceramide, di-C20:1 PC, and di-C14:0 PE standards were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL); trinonanoin, was purchased from Nu-Chek Prep, Inc. (Elysian, MN). Acetonitrile, methylene chloride, and methanol were from Fisher Scientific (Pittsburgh, PA); retinol, retinyl palmitate, and retinyl acetate were purchased from Sigma (St. Louis, MO).
Retinol and retinyl ester measurements by HPLC
Adult and embryo tissue retinol and retinyl ester levels were analyzed by HPLC ([26]). Briefly, serum (100–200 ul) and tissues (100–200 mg) were extracted with hexane followed by loading on HPLC. Retinol and four major retinyl ester peaks (retinyl palmitate, oleate, linoleate and stearate) were separated on a 4.6 × 250 mm Denali C18 column (Grace, Deerfield, IL) preceded by a C18 guard column (PerkinElmer, Waltham, MA) using acetonitrile, methanol, and methylene chloride (70:15:15, v/v) as the mobile phase flowing at 1.8 ml/min. Quantitative analysis was performed by comparing retention times and spectral data of experimental compounds with those of authentic standards. Retinyl acetate was added as an internal standard. Detection limits were as follows: serum < 0.1 ng/dl and tissues <1 ng/g.
Lipid analysis by LC/MS
The analyses were performed as we previously reported [16]. Lipids were extracted (Folch method) from individual embryo liver and brain and were evaporated to dryness under a nitrogen stream and re-dissolved in isooctane/tetrahydrofuran (9:1, v/v). Total TAG and phospholipids were measured in a single chromatographic run without pre-separation, using a Dionex UltiMate 3000 LC system coupled to an Applied Biosystems 4000 QTrap mass spectrometer with a Turbo V electrospray ionization source (ABSciex, Foster City, CA) (ref). Free cholesterol, cholesteryl esters, and ceramides were measured in a single run using LC/MS with the normal phase chromatographic method described above. Quantitative analyses of total TAG and phospholipids were performed by integrating the total peak for all the compounds of those class combined. Mass amount of free cholesterol, and cholesteryl esters were measured as previously described (ref). Ceramides elute at about 12.5 min and were measured with a scan for precursors of m/z 264. The electrode voltage was 900V, the collision gas setting was 2, the declustering potential was 20, the collision energy was 32, the entrance potential was 10, and the collision cell exit potential was 9. A standard curve containing 0.25–30 ng/injection ceramide (18:1-14:0 and 18:1-17:0) was used, and samples were spiked with an internal standard mixture containing stigmastanol, 18:1-17:0 or 18:1-14:0 ceramide, and cholesteryl nonanoate.
Quantitative real-time PCR (QPCR)
Total RNA from individual embryo liver and brain RNA was isolated using the Stratagene absolutely RNA kit (Agilent Tech., CA) according to the manufacturer’s instructions. A detailed method has been described previously [14, 27]. Briefly, RNA concentrations were measured followed by DNase I treatment (Roche Diagnostics, IN). RNA was reverse transcribed to cDNA and used for QPCR. For the QPCR experiments, 300 nmol/l of each primer (final concentration), together with an amount of cDNA for each sample equivalent to 10–50 ng of the total RNA input in a final volume of 15 ul was used. Primer sequences were as follows: BCO1, Fwd 5′ GAGCAAGTACAACCATTGGT 3′, Rev 5′ AACTCAGACACCACGATTC 3′; BCO2, Fwd 5′ AGGACCAGGGCTGTATTGTG 3′, Rev 5′ CGCTGGCTGAAGAATAGGAC 3′; LRAT, Fwd 5′ GCAGTTGGGACTGACTCCAT 3′, Rev 5′ CAGATTGCAGGAAGGGTCAT 3′; ACAT1, Fwd 5′ AGCCCAGAGGCTCAATGTTA 3′, Rev 5′ GGCTAGCACAACCACACTGA 3′; ACAT2, Fwd 5′ TGTCACAGAACAGGGCAGAG 3′, Rev 5′ TGACAGTTCCTGTCCCATCA 3′; LCAT, Fwd 5′ GGATATGTGCGGGATGAGAC 3′, Rev 5′ AGTGGAGCACATGCAGACAG 3′; DGAT1, Fwd 5′ GCTTCTGCAGTTTGGAGACC 3′, Rev 5′ CTCATGGAAGAAGGCTGAGG 3′; DGAT2, Fwd 5′ ACGCAGTCACCCTGAAG AAC 3′, Rev 5′ AGGGGGCGAAACCAATATAC 3′; SPT1, Fwd 5′ AGGGTTCTATGGCACATTTGATG 3′, Rev 5′ TGGCTTCTTCGGTCTTCATAAAC 3′; SPT2, Fwd 5′ CAAAGAGCTTCGGTGCTTCAG 3′, Rev 5′ GAATGTGTGCGCAGGTAGTCTATC 3′; Actin, Fwd 5′ AACACAGTGCTGTCTGGTGG 3′; Rev 5′ GAAAGGGTGTAAAACGCAGC 3′. Reactions were run in the Light Cycler 480 machine (Roche Diagnostics, IN). All samples were run in triplicate with NRTC and the non-template controls. The expression of each gene relative to the calibrator was calculated using 2(-ΔΔCt). All groups were expressed as fold change of the control group.
Statistical analyses
Normality of variables was assessed by the Shapiro–Wilk test. When the data were not normally distributed, values were logarithmically transformed prior to statistical analysis (embryonic liver retinyl esters). Normally distributed values were statistically analyzed by Students’ t-test for the comparison. Not normally distributed values were analyzed by Mann–Whitney test for the comparison. A p-value of <0.05 was used to establish statistical significance. Analyses were performed by means of SPSS (SPSS Inc., Chicago, IL, USA).
RESULTS
Embryonic expression of BCO1 and BCO2 at mid-gestation
To confirm expression of BCO1 and BCO2 in the embryonic liver and brain, mRNA of the above-mentioned organs from wild-type embryos at 14.5 dpc were analyzed by QPCR. Interestingly, BCO1 mRNA levels were negligible in embryonic brain compared to the liver (average brain CT value = 30.9 vs. average liver CT value = 25.9) (Figure 1). BCO2 mRNA expression was similar but also extremely low in both organs (average brain CT value = 29.7 vs. average liver CT value = 30) (Figure 1).
Figure 1. BCO1 and BCO2 mRNA levels in wild type embryonic organs.

mRNA levels were measured by QPCR in wild-type liver (E-liver) and brain (E-brain) from embryos at 14.5 dpc. E-Values are shown as means ± SD using the 2(−ΔΔCt) method, and are expressed as fold change from liver mRNA level, set as a calibrator at 1; n = 4 samples/group. Average CT values areas are as follows: for BCO1, liver = 25.9, brain = 30.9; for BCO2, liver = 30.0, brain = 29.7. BCO1, β-carotene-15,15′-oxygenase; BCO2, β-carotene-9′,10′-oxygenase.
Retinoid levels in the developing liver and brain at mid-gestation
To assess the tissue-specificity of BCO1 deficiency on embryonic retinoid metabolism, retinol and retinyl ester levels were measured by reverse phase HPLC in livers and brains from wild-type and BCO1−/− embryos at 14.5 dpc. Deficiency of BCO1 in the liver did not significantly affect retinol or retinyl ester concentrations compared to the wild-type group, even though retinyl ester levels tended to be lower in the absence of BCO1 (Figure 2). Retinyl esters were undetectable in the brains of both wild-type and BCO1−/− embryos. Moreover, no significant difference was observed in the concentration of retinol in this organ (Figure 2).
Figure 2. Retinol and retinyl ester levels in embryonic liver and brain.
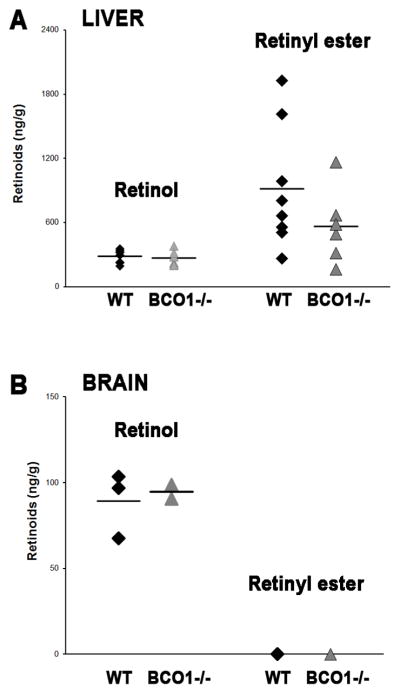
Retinoid levels were analyzed by reverse phase HPLC in liver (A; n = 6 – 8 samples/group) and brain (B; n = 4 samples/group) from embryos at 14.5 dpc. One to three embryos/dam from each genotype were used. Individual measurements (raw data) are shown and the mean values are indicated as a horizontal line. Detection limits of retinol and retinyl ester were as follows: serum, 0.1 ng/dl and tissues, 1 ng/g.
Lipid content of the developing liver and brain at mid-gestation
To assess the tissue-specificity of BCO1 deficiency on embryonic lipid metabolism, total cholesterol, cholesteryl esters, triacylglycerols, phosphatidylinositol (PI), PE, PC, phosphatidylserine (PS) and ceramides were measured by LC/MS analysis in the livers and brains (whole heads) of wild-type and BCO1−/− embryos at mid-gestation. As shown in Table 1, total hepatic cholesteryl ester, PI and PE levels were significantly reduced in the BCO1−/− embryos compared to wild-type. The brains of the BCO1-deficient embryos had reduced levels of cholesteryl esters, triacylglycerols, PI, and ceramides compared to wild-type. Total brain PE levels, however, were significantly greater in the absence of BCO1.
Table 1.
Lipid LC/MS analysis of liver and brain from wild-type and BCO1−/− embryos at 14.5 dpc
| Liver | Brain | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Wild type | BCO1−/− | % changes | Wild type | BCO1−/− | % changes | |
|
|
|
|||||
| μg/mg protein | μg/mg protein | |||||
| Cholesterol | 22.0 ± 11.3 | 19.3 ± 5.8 | NA | 28.9 ± 2.1 | 26.6 ± 1.2 | NA |
| Cholesteryl esters | 6.1 ± 1.8 | 2.5 ± 1.0* | -59.0 | 21.3 ± 2.2 | 16.4 ± 0.9* | −23.0 |
| Total TAGs | 42.8 ± 14.9 | 46.3 ± 11.1 | NA | 12.3 ± 2.5 | 7.6 ± 1.2* | −38.2 |
| Total PI | 16.0 ± 1.9 | 11.7 ± 1.8* | -26.9 | 16.8 ± 0.7 | 9.2 ± 3.1* | −45.2 |
| Total PS | 24.6 ± 2.7 | 21.0 ± 3.1 | NA | 35.9 ± 2.1 | 30.9 ± 1.7 | NA |
| Total PE | 32.0 ± 4.9 | 21.6 ± 2.2* | -32.5 | 36.8 ± 2.9 | 51.9 ± 5.2* | 41.0 |
| Total PC | 63.2 ± 18.4 | 61.3 ± 12.8 | NA | 112.4 ± 11.0 | 109.2 ± 14.9 | NA |
| Ceramides | 0.8 ± 0.2 | 0.6 ± 0.1 | NA | 3.1 ± 0.3 | 2.8 ± 0.2* | −9.7 |
Liver and brain (whole head) from wild-type and BCO1 knockout (BCO1−/−) embryos at 14.5 dpc were analyzed by LC-MS as described in the Materials and Methods. Values are expressed as mean ± SD. Statistical analysis was performed as described in Materials and Methods; n= 5 – 6 samples/group
, p < 0.05 vs. the corresponding wild-type organ. Percent changes of the corresponding wild type value (% changes) are shown only when the difference between the two genotypes is statistically significant. In the case of non statistically significant differences, the % change is not shown and indicated as NA = not applicable. Triacylglycerols (TAGs), phosphatidylinositol (PI), phosphatidylserine (PS), phosphatidylethanolamine (PE), phosphatidylcholine (PC).
mRNA expression levels of the key enzymes involved in retinyl esters, cholesteryl esters, triacylglycerols and ceramides synthesis
To establish whether any particular organ was responsible for the transcriptional changes previously reported upon analysis of the whole embryo [16], QPCR analysis was conducted on E14.5 livers and brains of BCO1−/− and wild-type embryos. In addition, mRNA levels of serine palmitoyl transferase 1 and 2 (SPT1 and SPT2), were also measured. These enzymes catalyze the acyltransferase reaction that condenses serine and palmitoyl-CoA to form 3-keto sphinganine which constitutes the first step of the complex biosynthetic pathway that enables de novo ceramides production [28]. As shown in Figure 3A, only LRAT mRNA levels were significantly reduced in the BCO1−/− embryonic livers compared to wild-type, with no significant changes in the transcription of any other tested genes (Figure 3A). In agreement with the undetectable retinyl ester levels in the embryonic brain, LRAT mRNA was also undetectable in this organ (Figure 3B). Furthermore, only ACAT2 showed a significant transcriptional upregulation in the BCO1−/− embryonic brains compared to wild-type (Figure 3B).
Figure 3. LRAT, ACAT1, ACAT2, LCAT, DGAT1, DGAT2, SPT1, and SPT2 mRNA levels in embryonic liver and brain.

mRNA levels were measured by QPCR in wild-type (WT) and BCO1 knockout (BCO1−/−) embryo liver (A) and brain (B) at 14.5 dpc. Data are shown as mean ± SD using 2(-ΔΔCt) method, and are expressed as fold change from the WT organ, set as a calibrator at 1; n = 4 samples/group. One embryo/dam from each genotype was used. Statistical analysis performed as described in Materials and Methods. *, p < 0.05 vs. the calibrator (wild-type liver (A) and wild-type brain (B)). LRAT, lecithin:retinol acyltransferase; ACAT1, acyl CoA:cholesterol acyltransferase 1; ACAT2, acyl CoA:cholesterol acyltransferase 2; LCAT, lecithin:cholesterol acyltransferase; DGAT1, acyl CoA:diacylglycerol acyltransferase 1; DGAT2, acyl CoA:diacylglycerol acyltransferase 2; SPT1, serine palmitoyl transferase 1; and SPT2, serine palmitoyl transferase 2.
All together these data suggest that the BCO1 genotype impacts retinoid and lipid homeostasis in the embryo in a tissue-specific manner.
Survey of retinoid levels in various organs of adult mice
We next asked whether or not the influence of BCO1 on retinoid homeostasis was a function exclusively restricted to the developing organs. To this end, we set out to measure retinol and retinyl ester levels by HPLC in adult mouse organs. An initial survey performed on the tissue of BCO1−/− and wild-type female mice revealed a statistically significant decrease in retinol content in the heart, pancreas, spleen, eye and adipose tissue of the knockout females. Retinol concentrations were greater in the lungs of the BCO1−/− females compared to wild-type (Figure 4A). Kidney and spleen retinyl ester levels were also greater in the knockout mice compared to wild-type (Figure 4B). We next used mice on the LRAT−/− background to assess the effect of BCO1 on the acylCoA-dependent retinol esterification, which is otherwise difficult to detect in the wild-type mice, given the large contribution of LRAT to the formation of retinyl ester stores. We compared adult tissue retinoid levels in LRAT−/− mice vs. those of mice lacking both LRAT and BCO1 (LRAT−/−BCO1−/−). In the case of the age-matched males, we observed a significant reduction in the retinol concentration in the heart and pancreas (Figure 5A), as well as a significant decrease in the heart, pancreas and lung retinyl ester levels in the LRAT−/−BCO1−/− compared to LRAT−/− mice (Figure 5B). In contrast to males, retinol levels were significantly greater in the adipose tissue (Figure 5C), and retinyl ester concentrations were elevated in the adipose and heart of the LRAT−/−BCO1−/− vs. LRAT−/− age-matched female mice (Figure 5D). As previously reported [25, 29, 30], retinyl esters were non detectable in the liver in the absence of LRAT, regardless of the sex (Figure 5B and 5D).
Figure 4. Retinol and retinyl ester levels in various adult mouse organs from wild-type and BCO1−/− female mice.

Retinol (A) and retinyl ester (B) levels were analyzed by reverse phase HPLC in tissues from age-matched mice. Statistical analysis performed as described in Materials and Methods; n = 3 mice/group. *, p <0.05 indicated on the BCO1−/− tissue with significant difference vs. wild-type. Detection limits of retinol and retinyl ester were as follows: serum, 0.1 ng/dl and tissues, 1 ng/g. S, serum; Lu, lung; Kid, kidney; Mus, muscle; H, heart; MG, mammary gland; Sp, spleen; Ey, eye; Pan, pancreas; Liv, liver; Adi, visceral (gonadal) adipose tissue.
Figure 5. Retinol and retinyl ester levels in adult mouse organs from LRAT−/− and LRAT−/−BCO1−/− mice.

Retinol and retinyl ester levels were analyzed in male (A – B) and female (C – D) mice by reverse phase HPLC in tissues from sex- and age-matched mice. Animals were fasted overnight before collection of the tissues at the time of sacrifice. Statistical analysis performed as described in Materials and Methods; n = 3 mice/group. *, p <0.05 indicated on the LRAT−/− BCO1−/− tissue with significant difference vs. LRAT−/−. Detection limits of retinol and retinyl ester were as follows: serum, 0.1 ng/dl and tissues, 1 ng/g. S, serum; Lu, lung; Kid, kidney; Mus, muscle; H, heart; Muc, intestinal mucosa; Sp, spleen; Ey, eye; Pan, pancreas; Liv, liver; Adi, visceral (gonadal) adipose tissue.
Overall these results imply that the BCO1 genotype impacts retinoid metabolism not only in developing tissues but also in a few adult tissues in a sex-dependent manner.
DISCUSSION
Over the past fifteen years, a number of seminal discoveries in the field of carotenoid metabolism have significantly expanded our knowledge of the function of β-carotene as a vitamin A precursor in adult and developing tissues [14, 24, 31, 32], the role of the symmetric vs. the asymmetric cleavage pathway in the generation of retinoids [11], the biological actions of apocarotenoids [33, 34], the interplay and the specific functions of BCO1 and BCO2 [11, 12]. Furthermore, a number of laboratories have provided evidence that BCO1 can be placed at a critical intersection of carotenoid, retinoid and lipid metabolism in various tissues [15, 16, 31, 35, 36]. Our laboratory first proposed that BCO1, in addition to its main function as an enzyme that metabolizes β-carotene and apocarotenoids, can influence ester formation from various substrates, such as retinol, cholesterol and diacylglycerols in the mouse embryo at mid-gestation [16]. The mechanisms underlying this alternative function of BCO1 are still not understood. Our previously published data suggest two non-mutually exclusive possibilities. On the one hand, BCO1 may influence the transcription of certain genes, such as LRAT, LCAT, ACAT1 and DGAT2, thus ultimately affecting the enzymatic activity of the corresponding proteins and the levels of their specific reaction products, i.e. retinyl esters, cholesteryl esters and triacylglycerols. On the other hand, BCO1 may be more directly involved in these reactions itself, facilitating for example intracellular lipid trafficking, and thus affecting the tissue concentrations of the above-mentioned lipid products [16].
Given that β-carotene is predominantly metabolized in the liver and that the developing brain has a high demand for lipids, we set out to assess the contribution of BCO1 expressed in liver and brain to the formation of retinyl esters and various lipid classes during embryonic development. As mentioned earlier, data on the tissue expression pattern of the two β-carotene cleavage enzymes during mammalian embryogenesis are lacking. Thus, this is the first report on BCO1 and BCO2 mRNA levels in the above-mentioned organs of the wild-type mouse embryo at mid-gestation. Surprisingly, embryonic liver only showed well-detectable levels of BCO1 mRNA, with BCO2 levels being negligible. This is in contrast to both the mouse and human adult liver, where expression of both genes has been reported with a cell-type-specific distribution [17, 18, 32]. Perhaps, at this developmental stage, BCO2 is not expressed in the liver or its expression is restricted to a subset of specific cell types within this organ. In the brain, both enzymes were expressed at extremely low levels. This finding confirms the literature on adult human tissues [17, 18], but is in contrast with a report of BCO2, but not BCO1, mRNA expression in the adult mouse brain [37]. We cannot exclude that expression restricted to specific cell types could also be the reason for the extremely low brain transcription levels of BCO1 and BCO2 reported here. Further studies are needed to address this question.
The significant reduction in hepatic LRAT mRNA levels observed in the BCO1-deficient embryos suggest that the developing liver may be one of the organs that contribute to the previously reported retinyl ester difference in the whole embryo [16]. Hepatic retinyl ester levels assessed by HPLC, however, were not significantly different between wild-type and BCO1−/− embryos, likely due to the large variability of our measures. The low tissue weight of the embryonic liver samples (average 15 mg) could account for this discrepancy. Further measurements by LC-MS analysis should be performed to obtain a more reliable estimate of the retinoid concentration in this developing organ. We previously reported that retinoic acid levels were similar in wild-type and BCO1−/− embryos from dams at mid-gestation maintained on a vitamin A-sufficient diet (as in the current study) [14]. Thus, it is unlikely that BCO1-deficiency might affect expression of LRAT, a well-known target for retinoic acid action [38], via retinoic acid signaling. However, only direct measurements of retinoic acid in the individual embryonic organs can unequivocally rule out the above-mentioned hypothesis. In the embryonic brain, both LRAT mRNA and retinyl ester levels were undetectable, even in the wild-type embryo. This is in contrast to the adult mouse brain where low but detectable levels of retinyl esters have been reported [29, 30]. Perhaps, LRAT gene expression is turned on at later stages of gestation or after birth. This remains to be established. In the future, it could be important to evaluate the contribution to retinyl ester formation of other organs, such as the lungs, that could serve as retinoid storage sites for the mid-gestation embryo [39].
Various lipid classes were affected by the lack of BCO1 in both the embryonic liver and brain, but with a different pattern. Only cholesteryl esters, PI and PE were reduced in the knockout embryonic liver by 59, 27 and 32%, respectively. In the BCO1−/− brain, cholesteryl esters, triacylglycerols, PI and ceramides were reduced by 23, 38, 45 and 10%, respectively. In contrast, in this same organ, PE levels were increased by 40%. It is of note that in our previous study [16], with the exception of cholesteryl esters, all the other lipid species analyzed were similar between wild-type and BCO1−/− embryos when total levels of such lipids were compared. Nevertheless, differences became evident when the fatty acid moieties of the various lipid classes were analyzed [16]. In the current study, only total levels of the lipid species were measured in the two developing organs, and yet statistically significant differences were observed between the two genotypes. Based on the percent changes between the two genotypes, we could infer that the embryonic liver may be the major contributor to the cholesteryl ester reduction previously reported in the whole embryo [16], whereas the brain could mainly account for the reduced embryonic triacylglycerol levels [16]. In the case of the whole embryo analysis, PI and PS did not show dramatic differences between the wild-type and BCO1−/− strains [16]. In contrast, significant reductions in certain species of PC and PE with higher degrees of unsaturation were observed in embryos lacking BCO1 [16]. In this report, we showed that total PI was significantly reduced in both the embryonic liver and brain, with no changes in PS and PC content. It is likely that the current organ-specific analysis unmasked previously un-noticed differences. On the other hand, other embryonic organs may account for the significant changes in PC levels detected in the whole embryo [16]. Furthermore, the opposite effects of the lack of BCO1 on the PE content of the liver and brain further suggests a tissue-specific function of the provitamin A cleavage enzyme in regulating embryonic lipid metabolism, and implies that BCO1 may exert this action in other embryonic organs too. Finally, whereas ceramide levels only showed a trend towards a reduction in the whole BCO1−/− embryos compared to wild-type (data not shown), this difference became statistically significant when ceramide concentrations were measured in the embryonic brain (head).
The transcription of the genes controlling the synthesis of the above-mentioned lipid classes, however, was similar between wild-type and BCO1 knockout embryos, in both organs, with the exception of an increase in brain mRNA levels of ACAT2, one of the genes controlling cholesteryl ester synthesis [40]. This seems to be a paradox, given the significantly lower cholesteryl ester levels in the BCO1−/− brains. Perhaps, it is an ineffective attempt to counteract the reduction of this lipid species in the brain. Overall, our results do not seem to favor the hypothesis that BCO1 affects retinoid and lipid homeostasis in the embryonic liver and brain by somehow regulating gene transcription. Also, it is puzzling that the brain lipid composition of the knockout embryos was so dramatically affected by the BCO1 deficiency, given that BCO1 is barely expressed in mouse brain at this stage of gestation. More investigations are needed to assess brain BCO1 activity levels, expression levels at earlier stages of embryogenesis, as well as cell-specific expression.
Is the role of BCO1 as a regulator of retinoid homeostasis exclusively restricted to the developing tissues (i.e. embryo [14, 16] and yolk sac [41])? To begin addressing this question we performed an initial HPLC survey on adult tissues from BCO1−/− and wild-type female mice. Interestingly, BCO1 influenced retinyl ester levels in a tissue-dependent manner (only in spleen and pancreas) and, in contrast to what we previously reported for the whole embryo [14, 16], they were increased in comparison to the wild-type. Overall, we took these findings as an indication that lecithin-dependent retinol esterification may be affected by BCO1 in adult mouse tissues. We next used mice on the LRAT−/− background to assess the effect of the lack of BCO1 on the acylCoA-dependent retinol esterification that could also take place in tissues and that we showed also being influenced by the absence of BCO1 in the whole embryo [16]. Our data confirm the current understanding of the absence of an ARAT activity in the liver. While the older literature based on in vitro analyses suggested that an ARAT activity was involved in hepatic retinyl ester formation [6], subsequent in vivo studies from independent groups convincingly disputed this notion [7, 8, 30]. Interestingly, in a few adult tissues the absence of BCO1 affected retinyl ester levels in a tissue- and sex-dependent manner, resulting in reduced retinoid stores in the LRAT−/−BCO1−/− males (pancreas, lung and heart), and increased retinoid stores in the LRAT−/−BCO1−/− females (heart and adipose). Perhaps these tissue differences in retinyl ester levels reflect differences in an intestinal ARAT activity and their consequent effect on tissue uptake of chylomicron retinyl esters. Interestingly, the heart, one of the organs where such changes in retinoid stores were detected, is highly dependent on dietary vitamin A (retinly esters in chylomicrons) as a source of retinoids [42]. However, that different organs were affected in males vs. females, with opposite changes in retinyl esters levels, could also suggest differences in cellularity within these tissues, accompanied by sex-dependent effects. Notably, retinoid and carotenoid metabolisms are influenced by sex hormones [43, 44]. We would like to point out that the sex of the embryos was not determined in our previous studies.
Pancreas displayed the greatest reduction in retinyl ester levels in the LRAT−/−BCO1−/− males. In agreement with a recent report from the Gudas group showing that vitamin A is required for both the maintenance of pancreatic β-cell and α-cell mass and for glucose-stimulated insulin secretion in adult LRAT−/− male mice [45], our data suggest that BCO1 may also play a role in maintaining pancreatic functions in adult life, possibly impacting retinoid homeostasis in this organ. In the double knockout females, adipose tissue displayed the greatest increase in retinyl ester stores. Ziouzenkova and colleagues showed how the transcription of aldehyde dehydrogenase-1A1 (ALDH1A1), the enzyme that catalyzes the conversion of retinaldehyde to retinoic acid, is differentially regulated by estrogen and androgen in visceral fat, establishing a female-specific mechanism whereby ALDH1A1 regulates adipogenesis, abdominal fat formation, glucose tolerance, and suppression of thermogenesis in adipocytes [46, 47]. Our data call for further studies to address whether a similar female-specific mechanism may also involve BCO1 as a potential target gene in the adipose tissue.
Interestingly, retinyl ester stores in the heart were affected by BCO1- and LRAT-deficiency in both males and females, although in an opposite way. BCO1 expression has been reported in all the above-mentioned tissues in mice and humans, with the exception of the adult heart, where it seems not to be expressed [15, 18]. Thus, it is puzzling that the absence of this enzyme results in altered retinoid metabolism in this organ. It is relevant, however, that Blaner and colleagues recently reported alterations in heart retinoid and lipid metabolism as well as compromised heart function, in BCO1−/− male mice [15]. The reasons for this apparent inconsistency are currently not known. BCO1-dependent alterations in lipid metabolism have been reported in the adult liver, adipose tissue and heart [15, 24, 31, 32, 48], but it remains to be established whether the β-carotene cleavage enzyme may be able to influence lipid metabolism in other adult tissues as well. Our data also suggest that the absence of BCO1 may influence retinol levels in a tissue- and sex-dependent manner. The mechanisms underlying this effect are currently unknown.
In conclusion, this study provides novel insights into the tissue- and sex-specificity of the previously reported alternative function that BCO1 may exert by regulating ester formation from different substrates (retinol, cholesterol, diacylglycerols, serine) in both developing and adult tissues. These results further our understanding of the physiological actions that the β-carotene symmetric cleavage enzyme may perform in mediating a cross-talk between micro- and macro-nutrients metabolism at different stages of the life cycle. This action may be very relevant to human health given the high genetic variability that exists in the human BCO1 gene [49], including a loss of function mutation [50].
Highlights.
BCO1 mRNA, but not BCO2, is well-detectable in embryonic liver at mid-gestation.
BCO1 and BCO2 mRNA expression is negligible in embryonic brain at mid-gestation.
BCO1 affects embryonic retinoid and lipid homeostasis in a tissue-specific manner.
In adult mice, BCO1 influence on retinoid homeostasis is tissue- and sex-specific.
Acknowledgments
We are indebted to Dr. Anita Brinker and the Core LC-MS Facility of the Institute of Food, Nutrition and Health (IFNH) at Rutgers University for the invaluable assistance and support in performing quantitative lipid analysis in various mouse tissues. We would also like to thank Dr. Elizabeth Spiegler for her critical reading of the manuscript. This work was supported by grants R01HD057493, R01HD057493-02S1 and R01HD057493-05S1 from the U.S. National Institute of Health.
Abbreviations
- ACAT1 and ACAT2
Acyl CoA:cholesterol acyltransferase 1 and 2
- ARAT
acyl CoA:retinol acyltransferase
- DGAT1 and DGAT2
acyl CoA:diacylglycerol acyltransferase 1 and 2
- BCO1
β-carotene-15,15′-oxygenase
- BCO2
β-carotene-9′,10′-oxygenase
- dpc
days post coitum
- LCAT
lecitin:cholesterol acyltransferase
- LRAT
lecithin:retinol acyltransferase
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PS
phosphatidylserine
- SPT1 and SPT2
serine palmitoyl transferase 1 and 2
- TAGs
triacylglycerols
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spiegler E, Kim YK, Wassef L, Shete V, Quadro L. Biochim Biophys Acta. 2012;1821:88–98. doi: 10.1016/j.bbalip.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shete V, Quadro L. Nutrients. 2013;5:4849–4868. doi: 10.3390/nu5124849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Lintig J. Am J Clin Nutr. 2012;96:1234S–1244S. doi: 10.3945/ajcn.112.034629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kedishvili NY. J Lipid Res. 2013;54:1744–1760. doi: 10.1194/jlr.R037028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Byrne SM, Blaner WS. J Lipid Res. 2013;54:1731–1743. doi: 10.1194/jlr.R037648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross AC. J Lipid Res. 1982;23:133–144. [PubMed] [Google Scholar]
- 7.Yen CL, Monetti M, Burri BJ, Farese RV., Jr J Lipid Res. 2005;46:1502–1511. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP, Li E, Blaner WS. J Biol Chem. 2008;283:13510–13519. doi: 10.1074/jbc.M800777200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Wongsiriroj N, Blaner WS. Hepatobiliary Surg Nutr. 2014;3:126–139. doi: 10.3978/j.issn.2304-3881.2014.05.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quadro L, Hamberger L, Colantuoni V, Gottesman M, Blaner WS. Mol Aspect Med. 2003;24:421–430. doi: 10.1016/s0098-2997(03)00038-4. [DOI] [PubMed] [Google Scholar]
- 11.Amengual J, Widjaja-Adhi MA, Rodriguez-Santiago S, Hessel S, Golczak M, Palczewski K, von Lintig J. J Biol Chem. 2013;288:34081–34096. doi: 10.1074/jbc.M113.501049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palczewski G, Amengual J, Hoppel CL, von Lintig J. FASEB J. 2014;28:4457–4469. doi: 10.1096/fj.14-252411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lobo GP, Isken A, Hoff S, Babino D, von Lintig J. Development. 2012;139:2966–2977. doi: 10.1242/dev.079632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YK, Wassef L, Chung S, Jiang H, Wyss A, Blaner WS, Quadro L. FASEB J. 2011;25:1641–1652. doi: 10.1096/fj.10-175448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SA, Jiang H, Trent CM, Yuen JJ, Narayanasamy S, Curley RW, Jr, Harrison EH, Goldberg IJ, Maurer MS, Blaner WS. Am J Physiol Heart Circ Physiol. 2014 Sep 26; doi: 10.1152/ajpheart.00548.2014. ajpheart.00548.2014. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon JL, Kim YK, Brinker A, Quadro L. Biochim Biophys Acta. 2014;1841:34–43. doi: 10.1016/j.bbalip.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindqvist A, Andersson S. J Histochem Cytochem. 2004;52:491–499. doi: 10.1177/002215540405200407. [DOI] [PubMed] [Google Scholar]
- 18.Lindqvist A, He YG, Andersson S. J Histochem Cytochem. 2005;53:1403–1412. doi: 10.1369/jhc.5A6705.2005. [DOI] [PubMed] [Google Scholar]
- 19.Lobo GP, Amengual J, Palczewski G, Babino D, von Lintig J. Biochim Biophys Acta. 2012;1821:78–87. doi: 10.1016/j.bbalip.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Ambrosio DN, Clugston RD, Blaner WS. Nutrients. 2011;3:63–103. doi: 10.3390/nu3010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Innis SM. Matern Child Nutr. 2011;7(Suppl 2):112–123. doi: 10.1111/j.1740-8709.2011.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gil-Sanchez A, Demmelmair H, Parrilla JJ, Koletzko B, Larque E. Front Genet. 2011;2:57. doi: 10.3389/fgene.2011.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quadro L, Hamberger L, Gottesman ME, Wang F, Colantuoni V, Blaner WS, Mendelsohn CL. Endocrinology. 2005;146:4479–4490. doi: 10.1210/en.2005-0158. [DOI] [PubMed] [Google Scholar]
- 24.Hessel S, Eichinger A, Isken A, Amengual J, Hunzelmann S, Hoeller U, Elste V, Hunziker W, Goralczyk R, Oberhauser V, von Lintig J, Wyss A. J Biol Chem. 2007;282:33553–33561. doi: 10.1074/jbc.M706763200. [DOI] [PubMed] [Google Scholar]
- 25.O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K, Blaner WS. J Biol Chem. 2005;280:35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim YK, Quadro L. Methods Mol Biol. 2010;652:263–275. doi: 10.1007/978-1-60327-325-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wassef L, Quadro L. J Biol Chem. 2011;286:32198–32207. doi: 10.1074/jbc.M111.253070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Summers SA. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Liu L, Tang XH, Gudas LJ. Biochem Pharmacol. 2008;75:2316–2324. doi: 10.1016/j.bcp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Gudas LJ. J Biol Chem. 2005;280:40226–40234. doi: 10.1074/jbc.M509643200. [DOI] [PubMed] [Google Scholar]
- 31.Amengual J, Gouranton E, van Helden YG, Hessel S, Ribot J, Kramer E, Kiec-Wilk B, Razny U, Lietz G, Wyss A, Dembinska-Kiec A, Palou A, Keijer J, Landrier JF, Bonet ML, von Lintig J. PLoS One. 2011;6:e20644. doi: 10.1371/journal.pone.0020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shmarakov I, Fleshman MK, D’Ambrosio DN, Piantedosi R, Riedl KM, Schwartz SJ, Curley RW, Jr, von Lintig J, Rubin LP, Harrison EH, Blaner WS. Arch Biochem Biophys. 2010;504:3–10. doi: 10.1016/j.abb.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eroglu A, Hruszkewycz DP, Dela Sena C, Narayanasamy S, Riedl KM, Kopec RE, Schwartz SJ, Curley RW, Jr, Harrison EH. J Biol Chem. 2012;287:15886–15895. doi: 10.1074/jbc.M111.325142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun J, Narayanasamy S, Curley RW, Jr, Harrison EH. J Biol Chem. 2014;289:33118–24. doi: 10.1074/jbc.M114.610501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lobo GP, Amengual J, Li HN, Golczak M, Bonet ML, Palczewski K, von Lintig J. J Biol Chem. 2010;285:27891–27899. doi: 10.1074/jbc.M110.132571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granados N, Amengual J, Ribot J, Musinovic H, Ceresi E, von Lintig J, Palou A, Bonet ML. Int J Obes. 2013;37:1169–1176. doi: 10.1038/ijo.2012.190. [DOI] [PubMed] [Google Scholar]
- 37.Kiefer C, Hessel S, Lampert JM, Vogt K, Lederer MO, Breithaupt DE, von Lintig J. J Biol Chem. 2001;276:14110–14116. doi: 10.1074/jbc.M011510200. [DOI] [PubMed] [Google Scholar]
- 38.Zolfaghari R, Ross AC. J Lipid Res. 2000;41:2024–2034. [PubMed] [Google Scholar]
- 39.Chen F, Marquez H, Kim YK, Qian J, Shao F, Fine A, Cruikshank WW, Quadro L, Cardoso WV. J Clin Invest. 2014;124:801–811. doi: 10.1172/JCI70291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang TY, Li BL, Chang CC, Urano Y. Am J Physiol Endocrinol Metab. 2009;297:E1–9. doi: 10.1152/ajpendo.90926.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wassef L, Varsha S, Hong A, Spiegler E, Quadro L. J Nutr. 2012;142:1456–1462. doi: 10.3945/jn.112.162677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, Shulman GI, Blaner WS, Goldberg IJ. J Biol Chem. 2010;285:37976–37986. doi: 10.1074/jbc.M110.174458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Helden YG, Godschalk RW, von Lintig J, Lietz G, Landrier JF, Luisa Bonet M, van Schooten FJ, Keijer J. Mol Nutr Food Res. 2011;55:1466–1474. doi: 10.1002/mnfr.201100194. [DOI] [PubMed] [Google Scholar]
- 44.Petrosino JM, Disilvestro D, Ziouzenkova O. Nutrients. 2014;6:950–973. doi: 10.3390/nu6030950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trasino SE, Benoit YD, Gudas LJ. J Biol Chem. 2014 Dec 1; doi: 10.1074/jbc.M114.616763. pii: jbc.M114.616763. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yasmeen R, Reichert B, Deiuliis J, Yang F, Lynch A, Meyers J, Sharlach M, Shin S, Volz KS, Green KB, Lee K, Alder H, Duester G, Zechner R, Rajagopalan S, Ziouzenkova O. Diabetes. 2013;62:124–136. doi: 10.2337/db11-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reichert B, Yasmeen R, Jeyakumar SM, Yang F, Thomou T, Alder H, Duester G, Maiseyeu A, Mihai G, Harrison EH, Rajagopalan S, Kirkland JL, Ziouzenkova O. Mol Endocrinol. 2011;25:799–809. doi: 10.1210/me.2010-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tourniaire F, Gouranton E, von Lintig J, Keijer J, Bonet ML, Amengual J, Lietz G, Landrier JF. Genes Nutr. 2009;4:179–187. doi: 10.1007/s12263-009-0128-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lietz G, Lange J, Rimbach G. Arch Biochem Biophys. 2010;502:8–16. doi: 10.1016/j.abb.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 50.Lindqvist A, Sharvill J, Sharvill DE, Andersson S. J Nutr. 2007;137:2346–2350. doi: 10.1093/jn/137.11.2346. [DOI] [PubMed] [Google Scholar]