

Abstract
Pseudoxanthoma elasticum (PXE), an autosomal recessive disorder characterized by ectopic mineralization, is caused by mutations in the ABCC6 gene. We examined clinically 29 Chinese PXE patients from unrelated families, so far the largest cohort of Asian PXE patients. In a subset of 22 patients, we sequenced ABCC6 and another candidate gene, ENPP1, followed by pathogenicity analyses for each variant. We identified a total of 17 distinct mutations in ABCC6, 15 of them being previously unreported, including 5 frame-shift and 10 missense variants. In addition, a missense mutation in combination with a recurrent nonsense mutation in ENPP1 was discovered in a pediatric PXE case. No cases with p.R1141X or del23-29 mutations, common in Caucasian patient populations, were identified. The 10 missense mutations in ABCC6 were expressed in mouse liver via hydrodynamic tail-vein injections. One mutant protein showed cytoplasmic accumulation indicating abnormal subcellular trafficking, while the other nine mutants showed correct plasma membrane location. These nine mutations were further investigated for their pathogenicity using a recently developed zebrafish mRNA rescue assay. Minimal rescue of the morpholino-induced phenotype was achieved with 8 of the 9 mutant human ABCC6 mRNAs tested, implying pathogenicity. This study demonstrates that the Chinese PXE population harbors unique ABCC6 mutations. These genetic data have implications for allele-specific therapy currently being developed for PXE.
INTRODUCTION
Pseudoxanthoma elasticum (PXE), the prototype of heritable ectopic mineralization disorders, is characterized by late-onset, yet progressive, calcium hydroxyapatite deposition on elastic structures in peripheral connective tissues (Neldner, 1988; Uitto et al., 2010). Clinically, PXE manifests with characteristic cutaneous, ocular and cardiovascular findings. The disease is inherited in an autosomal recessive manner with apparently complete penetrance, and ABCC6 has been identified as the gene harboring mutations in most patients with PXE (Bergen et al., 2000; Le Saux et al., 2000; Ringpfeil et al., 2000; Struk et al., 2000). This gene encodes ABCC6, a putative transmembrane efflux transporter protein primarily expressed in the baso-lateral plasma membranes of hepatocytes and to a lesser extent in the proximal tubules of the kidneys.
In addition to ABCC6, recent studies have disclosed mutations in the ENPP1 gene in some patients with PXE-like cutaneous findings, often associated with extensive vascular mineralization (Kalah et al., 2012; Li et al., 2012). ENPP1 mutations also underlie a severe ectopic mineralization disorder, generalized arterial calcification of infancy (GACI), an autosomal recessive disease, which affects primarily the arterial blood vessels (Ruf et al., 2005; Rutsch et al., 2003). This disease is commonly diagnosed by pre- or perinatal ultrasound, and the affected individuals in most cases die within the first year of life from cardiovascular complications. While most cases of GACI are caused by mutations in the ENPP1, ABCC6 mutations have also been demonstrated in some patients (Li et al., 2014; Nitschke et al., 2012). Thus, there is considerable phenotypic and genotypic overlap between PXE and GACI (Li and Uitto, 2013; Nitschke and Rutsch, 2012).
Over 300 distinct mutations in the ABCC6 gene have been identified in patients with PXE, two common recurrent mutations, p.R1141X and genomic deletion of exons 23 through 29 (c.2996-1724_4209-478del; referred to as del23-29), representing 18.5 and 9.9% of all reported mutant alleles, respectively (Pfendner et al., 2007; Terry and Hefferson, 2013; Uitto et al., 2013). However, essentially all published studies have focused on PXE in Caucasian patient populations, and very few mutations have been reported in patients of Asian ancestry. In this study, we have investigated a cohort of 29 Chinese PXE patients from unrelated families, so far the largest cohort of Asian PXE patients. Genetic analysis of 22 patients revealed a mutation profile clearly distinct from that found in Caucasian patients, and the Chinese PXE patients harbor unique mutations.
RESULTS
Identification of ABCC6 Mutations
A cohort of 29 Chinese patients with PXE was examined, and the diagnosis was initially suggested by characteristic cutaneous lesions and histopathology using routine Hematoxylin-Eosin as well as Verhoeff van Gieson and von Kossa stains for elastic structures and mineralization, respectively (Uitto et al., 2014) (Fig. S1). The majority of patients were females (26/29), most of them had the onset at less than 30 years of age, and the majority of patients (~90%) had the disease for over 6 years since diagnosis at the time of the study (Table S1).
In a subset of 22 patients, DNA, isolated from either peripheral blood leukocytes or paraffin-embedded skin biopsies, was available and subjected to mutation analysis first using a strategy and primers that we have previously developed for streamlined mutation detection in the ABCC6 gene (LaRusso et al., 2010; Pfendner et al., 2007). A total of 36 sequence variants in ABCC6 were discovered. These variants included 6 small insertion or deletion mutations resulting in premature termination codon (PTC), and these variants were considered pathogenic (Fig. 1A). Among the 30 single nucleotide substitutions, we identified 7 synonymous mutations while 23 were missense mutations. Among the nonsynonymous substitutions, 9 were present in the single nucleotide polymorphism (SNP) database (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?geneId=368&ctg=NT_010393.16&mrna=NM_001171.5&prot=NP_001162.4&orien=forward) in frequency >1% and were therefore considered to be nonpathogenic polymorphisms. Among the 14 amino acid substitutions not present in the SNP database, one of them, c.3341G>A, has been previously reported as a pathogenic mutation. The remaining 13 amino acid substitutions were examined for potential pathogenicity by PolyPhen-2 and SIFT prediction programs (Table 1), and 10 putative missense variants were examined for subcellular localization in mouse hepatocyte plasma membrane targeting assay and for functional pathogenicity in zebrafish mRNA rescue assay in vivo (see below). Among the putative pathogenic mutations, only two of them, one missense and one single-nucleotide deletion mutation, have been published previously. Most notably, none of the Chinese PXE patients had the recurrent p.R1141X or del23-29 mutation. When examined individually, 13 patients were homozygous or compound heterozygous with mutations in both alleles of ABCC6, while in 7 patients only one mutation was found. In the latter cases, search for ENPP1 mutations was unyielding.
Figure 1. The positions of ABCC6 mutations identified in Chinese patients with PXE.

(a) Intron-exon organization of ABCC6 gene. Vertical boxes represent the 31 exons; Missense mutations are shown above, and insertion or deletion mutations resulting in PTC below the line; Green exons code for the two nucleotide-binding fold domains of the protein; Black, previously reported mutations; Red, to our knowledge previously unreported mutations; *denotes the presence of the mutation in multiple alleles/patients with the number of affected alleles in parenthesis. (b) Positions of the missense variants in the membrane topology model of the ABCC6 protein. The various protein domains are delineated by horizontal arrows above; the positions of amino acid variants investigated in the study are in red; nucleotide binding fold domains and intracellular loops are colored with gray and blue, respectively.
Table 1.
The missense variants of ABCC6 discovered in 22 Chinese patients with PXE, and bioinformatics predictions of the consequence of the mutations
| Mutations | SIFT+ | PolyPhen2+ | |
|---|---|---|---|
| At DNA level* | At protein level | ||
| c.1814T>C | p.Leu605Pro | Damaging (0) | Probably (1) |
| c.373G>A | p.Glu125Lys | Damaging (0.01) | Probably (0.997) |
| c.11C>A | p.Pro4His | Damaging (0) | Probably (0.957) |
| c.1256G>A | p.Arg419Gln | Damaging (0) | Probably (0.994) |
| c.2843T>C | p.Leu948Pro | Damaging (0) | Probably (0.988) |
| c.2126A>G | p. Glu709Gly | Damaging (0) | Probably (0.916) |
|
| |||
| c.2501T>C | p.Met834Thr | Tolerated (0.28) | Benign (0.047) |
| c.61C>T | p.pro21Ser | Tolerated (0.45) | Benign (0.209) |
| c.191G>A | p.Arg64Gin | Tolerated (0.21) | Benign (0.051) |
| c.26C>A | p.Ala9Glu | Damaging (0.02) | Benign (0.109) |
|
| |||
| c.268G>A | p.Ala90Thr | Tolerated (0.5) | Benign (0) |
| c.232G>A (x7) | p.Ala78Thr | Tolerated (0.18) | Benign (0.018) |
| c.4324G>A (x2) | p.Arg1442Thr | Tolerated (0.47) | Benign (0.188) |
The recurrent mutation in multiple alleles is indicated with the number of affected alleles in parentheses.
Indicates the prediction of the consequences of the mutations on the protein function with the score in parentheses.
Assay of membrane targeting of the mutant protein
Among the discovered sequence variants, 13 of them resulted in amino acid substitution (Table 1 and Fig. 1B), and they all were initially considered pathogenic because searches of the SNP database did not report the presence of these variants or they were present in frequency of less than 1%. Analysis of the potential functional consequences of these mutations at the protein level by SIFT and PolyPhen-2 bioinformatics programs predicted that 6 of them were definitely damaging/probably disruptive while the remaining 7 were tolerated or benign (Table 1). In the latter group, three variants, even though not present in the SNP database, were recurrent in the Chinese families with PXE in high frequency, and they were considered nonpathogenic and not studied further.
Theoretically, missense mutations could inactivate the ABCC6 activity by a number of mechanisms. First, it is possible that the mutant protein is mis-localized within the hepatocytes and does not migrate into the appropriate plasma membrane location on the baso-lateral surface of hepatocytes (Aranyi et al., 2013). Alternatively, the protein is appropriately targeted to the correct membrane location but the transporter activity is compromised by inability of the protein to perform its transport function, for example due to deficient binding and hydrolysis of ATP (Ilias et al., 2002). We first determined the subcellular localization of the human mutant protein expressed from an expression vector under the control of a liver-specific mouse albumin promoter delivered to the mouse liver by hydrodynamic injection through the tail vein (Jiang et al., 2010). After 3 days of delivery, the livers were harvested, and the subcellular localization of the human as well as endogenous mouse ABCC6 protein was determined by immuno staining with antibodies that differentially recognize human and mouse protein epitopes (Fig. 2). As demonstrated previously, the endogenous mouse ABCC6 protein resides at the baso-lateral surface of the hepatocyte plasma membrane (Pomozi et al., 2013). Similarly, 9 out of the 10 human mutant proteins tested in this study were co-localized with the mouse protein at the proper membrane location (Fig. 2A). However, one mutation, p.L605P, did not allow the protein to migrate to the plasma membrane, and the mutant protein was localized exclusively in the cytoplasm (Fig. 2A and B). Similarly, another, previously identified mutation, p.R1114P, resulted in partial retention of the protein in the cytoplasm while some cells demonstrated plasma membrane staining (Aranyi et al., 2013) (Fig. 2B).
Figure 2. Subcellular localization of human ABCC6 missense variants expressed in mouse liver, and the effect of 4-phenylbutyrate (4-PBA) on their localization.

(a) The human (red) and mouse (green) ABCC6 proteins were detected on frozen sections of mouse liver by immunofluorescence with species specific primary antibodies three days after hydrodynamic tail vein injection of each ABCC6 missense variant in an expression vector. (b) Mice injected with ABCC6 missense variants were treated with (b, right panels) or without (b, left panels) 4-PBA. Scale bar = 100 mm.
A chaperone compound, 4-phenylbutyrate (4-PBA), has previously been shown to facilitate transfer of some mutant mis-targeted ABCC6 molecules from the cytoplasm to the plasma membrane (Aranyi et al., 2013). Consequently, we tested the effect of 4-PBA on the subcellular localization of the p.L605P and p.R1114P mutants by treating mice with this compound two days prior and four days following the injection of the expression construct for a total of 6 days. 4-PBA clearly facilitated the transfer of the cytoplasmic mutant p.R1114P protein to the plasma membrane, as shown previously (Pomozi et al., 2014) (Fig. 2B). However, this compound had no effect on the subcellular localization of the protein harboring the mutation p.L605P. Thus, 4-PBA may be of help in facilitating the proper targeting of some, but not all, mutant ABCC6 proteins to the plasma membrane.
Demonstration of pathogenicity in zebrafish mRNA rescue assay
The pathogenicity of the missense mutations identified in ABCC6 was further investigated in a zebrafish mRNA rescue assay that we have recently developed (Li et al., 2010a; Zhou et al., 2013). In this assay, zebrafish embryos are injected with an abcc6a morpholino which causes knock-down of the corresponding gene expression. As a consequence, the zebrafish embryos develop a profound phenotype consisting of pericardiac edema, stunted growth and curled tail, and the developing embryos die before the age of 7 days post fertilization (dpf) (Fig. 3). This phenotype can be fully rescued by injection of wild-type human ABCC6 mRNA together with the morpholino (Fig. 3). We consequently injected zebrafish embryos with the morpholino together with human ABCC6 mRNA harboring missense mutations identified in this study. As a negative control, the morpholino was injected with the human mRNA harboring stop codon mutation p.R1141X. As shown in Fig. 3, this mRNA containing the nonsense mutation did not rescue the phenotype. Injection of mutant mRNAs harboring the missense mutations identified in this study together with the morpholino revealed that 8 out of 9 mutations tested did not provide significant rescue as judged by either morphology of the zebrafish embryos (Fig. 3) or by the percent of lethality (Table 2) at 4 dpf, suggesting that they are pathogenic. Only one mutant mRNA, p.R64Q, resulted in rescue comparable to that of the wild-type mRNA (Fig. 3 and Table 2). However, the corresponding mutation, c.191G→A, was not present in the SNP database, and it is unclear whether this is a pathogenic mutation in the 5 patients with PXE.
Figure 3. Morphology of zebrafish 4 days after co-injection of an ABCC6A knock-down morpholino together with different human ABCC6 mRNA variants.
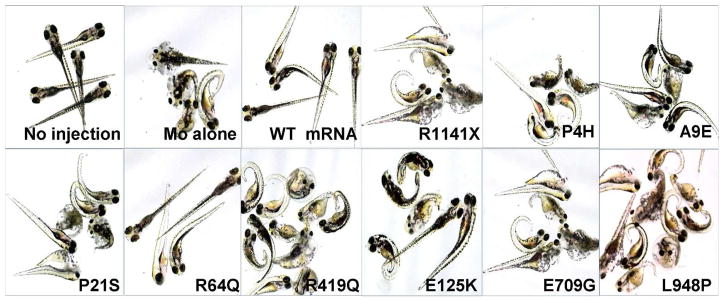
The morpholino-induced phenotype consisting of pericardiac edema, stunted growth and curled tail, similar to zebrafish injected with morpholino (MO) alone, was observed in zebrafish co-injected with human ABCC6 mRNA carrying p.R1141X, p.P4H, p.A9E, p.P21S, p.R419Q, p.E125K, p.E709G or p.L948P mutation, indicating lack of rescue and implying pathogenicity. Zebrafish co-injected with MO and ABCC6 mRNA carrying R64Q mutation showed wild-type phenotype, similar to fish injected with MO together with human wild-type (WT) ABCC6 mRNA.
Table 2.
Zebrafish mRNA rescue assay
| Group* | ABCC6 mRNA variant | No. of embryos injected | Lethality (%)† |
|---|---|---|---|
| No injection | - | 192 | 8.7 |
| MO alone | - | 68 | 79.3 |
| MO+ | p.R1141X | 163 | 73.8 |
| MO+ | WT | 80 | 23.4 |
| MO+ | p.P4H | 71 | 85.9 |
| MO+ | p.A9E | 94 | 83.0 |
| MO+ | p.P21S | 93 | 79.0 |
| MO+ | p.R64Q | 83 | 28.7 |
| MO+ | p.E125K | 109 | 63.2 |
| MO+ | p.R419Q | 48 | 89.1 |
| MO+ | p.E709G | 64 | 65.6 |
| MO+ | p.M834T | 118 | 64.4 |
| MO+ | p.L948P | 109 | 71.1 |
Zebrafish embryos were injected at day 0 with an abcc6a morpholino (MO) alone or with human ABCC6 mRNA, either wild-type (WT) or harboring different mutations.
The cumulative number of dead embryos at 4 days after injection, expressed as % of the total number of embryos injected.
Identification of mutations in the ENPP1 gene
A male patient diagnosed as having PXE by the presence of characteristic cutaneous findings and histopathology of the skin did not reveal the presence of mutations in ABCC6 (Fig. 4). Careful examination of the patient revealed several unusual features. First, the patient’s cutaneous findings had been noted definitely to be present as early as at 8 years of age categorizing him with a diagnosis of pediatric PXE. Furthermore, in addition to characteristic yellowish papules in the axillary fossa, the patient had large areas of hyperpigmented lesions on the trunk, a finding not characteristic of PXE (Fig. 4A). With the notion that mutations have been recently disclosed in the ENPP1 gene in patients with cutaneous PXE-like lesions (Kalah et al., 2012; Li et al., 2012), we next sequenced ENPP1, which identified 2 heterozygous mutations, p.Y261X and p.S479F, and the parents were heterozygous carriers, respectively (Fig. 4B and D). The nonsense mutation, p.Y261X, has been previously reported (Ruf et al., 2005). Sequence alignments indicated that serine at the amino acid position 479 is highly conserved during evolution (Fig. 4C). This sequence variant, p.S479F, was not present in the SNP database, and this mutation was predicted to be probably damaging (0.99) and damaging (0), when analyzed by PolyPhen-2 and SIFT programs, respectively. Collectively, the mutation analysis in the Chinese cohort of PXE demonstrated considerable genetic heterogeneity and identified a number of mutations not previously reported in the literature.
Figure 4. Cutaneous presentation, histopathology and mutation detection in a pediatric patient with PXE.
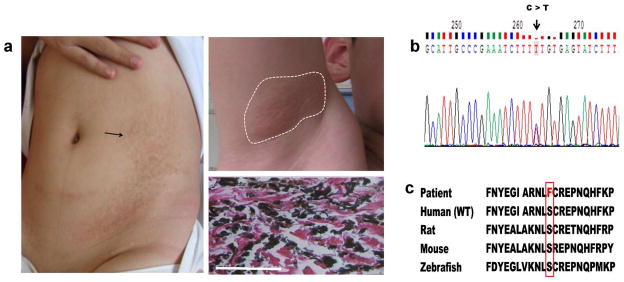
(a) Hyperpigmentation on the trunk (left) and yellowish papules in the axillary fossa (upper right); Aberrant calcification in the dermis detected by von Kossa stain (bottom right); (b) A heterozygous mutation, p.S479F, in the ENPP1 gene revealed by mutation analysis (arrow); (c) Conservation of the serine-479 during evolution from zebrafish to human (outlined). Scale bar = 100 mm.
DISCUSSION
Understanding of the mechanisms leading to aberrant mineralization of connective tissues has been advanced by observations on a group of heritable disorders manifesting with ectopic mineralization. The prototype of such conditions is PXE, an autosomal recessive disorder which affects a number of organs by ectopic mineralization, with primary clinical findings in the skin, the eyes, and the cardiovascular system (Neldner, 1988; Uitto et al., 2010). PXE is a rare disorder, with an estimated prevalence of ~1:50,000 which would imply that there are ~7,000 to 8,000 affected individuals in the United States, and with the same prevalence as many as 50,000 patients in China. The diagnosis of PXE is made by a combination of clinical findings in the skin and the eyes, supported by histopathologic and molecular diagnostic observations (Uitto et al., 2014). While the manifestations of PXE are of late onset and the disease progresses slowly, PXE is associated with major clinical complications, including loss of central vision often leading to blindness, and occurrence of catastrophic cardiovascular events, including early myocardial infarcts and strokes. There is a considerable spectrum of phenotypic presentations and severity of the disease: At one end of the spectrum, young patients in their infancy, with considerable vascular involvement, have been diagnosed with PXE-like cutaneous findings, often classified as pediatric PXE (Li et al., 2013; Li et al., 2014). In addition, patients with GACI, typically caused by mutations in the ENPP1 gene, can demonstrate PXE-like findings, supporting the notion that there is considerable both clinical and genetic overlap between PXE and GACI (Nitschke and Rutsch, 2012).
The classic form of PXE is caused by mutations in the ABCC6 gene, and over 300 distinct mutations have been identified representing well over 1,000 mutant alleles (Terry and Hefferson, 2013). Among the published mutations, two common recurrent mutations, p.R1141X and del23-29, account for up to 30% of all mutant alleles (Pfendner et al., 2007; Terry and Hefferson, 2013). Examination of the ancestry and geographic distribution of patients in the ABCC6 mutation databases reveals that most patients tested are apparently Caucasians from the United States or European countries, and specific reports of mutations in the Italian, French and German cohorts have been published (Chassaing et al., 2007; Gheduzzi et al., 2004; Pfendner et al., 2007; Schulz et al., 2006). Examination of the mutation database indicates that the frequent p.R1141X mutation is distributed widely across Europe, while deletion of exons 23-29 (del23-29) is encountered in Northern Europe and in Northern Mediterranean countries (LaRusso et al., 2010). In addition, limited numbers of patients, with specific mutations, have been reported from Greece, Turkey, South Africa and Brazil (Akoglu et al., 2014; Faria et al., 2013; LaRusso et al., 2010; Le Saux et al., 2002; Ramsay et al., 2009). There is a striking paucity of mutation reports on individuals of Asian ancestry. Specifically, there are only four distinct mutations reported in Japanese patients with PXE, and in addition, six ABCC6 sequence variants have been identified as a cause of angioid streaks in Japanese patients, an eye finding often associated with PXE (Noji et al., 2004; Sato et al., 2009; Tanioka et al., 2014; Yoshida et al., 2005). There is only one ABCC6 mutation reported in a Chinese patient with PXE (Yang et al., 2008).
In the present study, we have clinically examined a cohort of 29 Chinese patients with PXE, and DNA was available to specifically sequence the exons and flanking intronic sequences of ABCC6 in a subset of 22 patients. Among the 36 sequence variants identified in ABCC6, six small insertions or deletions were causing PTCs, five of them being previously unreported. Among the 23 nonsynonymous missense mutations, ten were initially considered potentially pathogenic based on their absence or presence in low frequency (<1%) in SNP database, and as judged by bioinformatics prediction programs PolyPhen-2 and SIFT to be damaging to the protein function. Among the 10 putative pathogenic missense mutations tested in zebrafish mRNA rescue assay, nine of them did not provide rescue, confirming the pathogenic nature of the amino acid substitutions. Only one of the mutant mRNAs, harboring mutation p.R64Q, was able to rescue the zebrafish phenotype, similar to that of wild-type mRNA. However, this mutation was not present in the SNP database and three of the five patients with this sequence variant had another allelic ABCC6 mutation. Therefore, it is unclear whether this mutation, p.R64Q, is pathogenic or not. It should be noted that no ABCC6 or ENPP1 mutations were found in two patients, and the overall rate of detection of mutations in ABCC6 and ENPP1 was 80 percent (35 mutant alleles of a total 44). It should be noted that, the mutation detection strategy utilized PCR amplification of individual exons and flanking intronic sequences. This approach does not detect mutations in the regulatory upstream sequences or in the 3′-UTR, deeper intronic sequences, or large insertions or deletions (Pfendner et al., 2007).
The consequences of missense mutations were also tested in vivo in a mouse system which examines the subcellular targeting of the mutant protein in mouse hepatocytes following hydrodynamic delivery of an expression vectors through the tail vein. The wild-type ABCC6 protein localizes to the basolateral surface of hepatocytes, and 9 out of 10 tested missense mutations allowed the protein to target the physiological plasma membrane location. Only one mutation, p.L605P, resulted in cytoplasmic localization of the mutant protein. Previously, the mutant protein harboring p.R1114P mutation has been shown to remain in cytoplasmic localization which could be corrected by treatment with 4-PBA, a chaperone molecule. However, treatment of mice expressing miss-targeted protein with p.L605P mutation did not result from correction of the subcellular localization. Thus, 4-PBA treatment may be applicable for correction of the subcellular localization only to selected mutant ABCC6 proteins with missense mutations.
One of the patients was diagnosed as PXE manifested with somewhat unusual features, including relatively early age at onset and presence of atypical cutaneous findings, such as extensive hyperpigmentation on the trunk, not a characteristic feature of PXE. Analysis of the ABCC6 gene failed to identify mutations, but subsequent sequencing of the ENPP1, typically associated with GACI, revealed the presence of a nonsense mutation, p.Y261X, and a heterozygous missense mutation, p.S479F. The latter mutation is pathogenic, based on its absence from the SNP database, conservation of the serine-479 through evolution from zebrafish to human, and prediction by PolyPhen-2 and SIFT programs as damaging. This previously unreported missense mutation contributes to the growing database of ENPP1 mutations, and this case also illustrates the phenotypic overlap between PXE and GACI. It should be noted that mutations in the ENPP1 gene have been recently identified in Cole disease, a rare autosomal dominant genodermatosis featuring punctate keratoderma, patchy hypopigmentation, and uncommonly, cutaneous calcifications (Eytan et al., 2013). These observations may have relevance to pigmentary changes noted in our patient diagnosed with pediatric PXE.
Collectively, this study identified 16 mutations, 15 of them in ABCC6 and 1 in ENPP1, in the Chinese PXE population, with implications for accurate diagnosis and subclassification. This information can be used for genetic counseling, and it forms the basis for prenatal testing and preimplantation genetic diagnosis in future pregnancies in families at risk for recurrence. Knowledge of the specific mutations can also be used for presymptomatic testing in families with known history of PXE (Akoglu et al., 2014; Li et al., 2010b). Important for the patients, identification of the precise nature of the mutations underlying the PXE phenotype provides a basis for development of treatment modalities tailored to be allele specific.
MATERIAL AND METHODS
Patient samples
A total of 29 unrelated patients with the putative diagnosis of PXE were investigated. Informed consent was obtained from all subjects, and the present study was approved by the local Medical Research Ethics Committee at Xijing Hospital, Fourth Military Medical University, Xi’an, China. The primary diagnosis of PXE was based on dermatological, ophthalmologic, and/or histopathologic evaluations (Uitto et al., 2014). In each proband, the skin lesions were histologically confirmed to be consistent with the diagnosis of PXE by the observation of calcified elastic fibers in biopsy specimens upon hematoxylin and eosin, Verhoeff van Gieson and/or von Kossa stains with standard protocols.
Mutation analysis
Genomic DNA was isolated from peripheral blood leukocytes or paraffin embedded skin tissues from 22 patients from whom samples were available, according to standard procedures. Mutation detection comprised polymerase chain reaction (PCR) amplification of each of the 31 exons in the ABCC6 gene using primer pairs placed on the flanking intronic sequences (Pfendner et al., 2007). This protocol excludes amplification of the two ABCC6 pseudogenes with sequences corresponding to the 5′end of ABCC6 (Pulkkinen et al., 2001). Purified PCR products were sequenced for variants by comparison with the published cDNA sequence (Gen Bank accession no. NM_001171). The samples in which no mutations were found in the ABCC6 gene were further analyzed for the ENPP1 gene using the same strategy. Evolutionary conservation of the amino acid residue serine-479 in ENPP1 was examined by sequence alignment with Ensemble program.
Mice
Immunodeficient Rag1−/− mice in C57/BL6 background (strain: 002216F; Jackson Labs, Bar Harbor, ME), which are wild-type for Abcc6, were used in this study. The mice were maintained under standard laboratory conditions and were handled in accordance with the guidelines for animal experiments by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Reagents, plasmid and site-directed mutagenesis
Sodium 4-phenylbutyrate (4-PBA) was purchased from Sigma-Aldrich (Deisenhofen, Germany) and dissolved in 0.9% NaCl prior to use. A full-length wild-type human ABCC6 cDNA was cloned into pLIVE™ expression vector purchased from Mirus (MIR5420, Madison, WI). Using this cDNA as a template, ten different ABCC6 missense DNA-constructs were obtained by site-directed mutagenesis following the manufacturer’s instructions (Agilent, Santa Clara, CA).
Liver-specific expression of ABCC6 variants in mice
Liver-specific expression of ABCC6 variants was performed in mice as described in our previous studies (Jiang et al., 2010; Jiang et al., 2006; Pomozi et al., 2014) Briefly, pLIVE expression vector (Mirus Bio, Madison, WI) containing the wild-type or mutant ABCC6 was delivered into the mice by hydrodynamic tail-vein injection of 10% body volume of TransIT-QR hydrodynamic delivery solution (Mirus), as recommended by the manufacturer’s instructions, using a 26-gauge syringe needle. At least three mice were injected with each form of the human ABCC6 cDNA. Mice were sacrificed 3–4 days after hydrodynamic tail vein injections and the livers were harvested for immunofluorescence.
4-PBA treatment of mice
Mice received intraperitoneal injection of 4-PBA (100 mg/kg per day), once a day for 4 days initiated at the time of hydrodynamic tail-vein injection, and they additionally received an approximate dosage of 1000 mg/kg per day in the drinking water, two days prior and four days following the injection, for a total of 6 days.
Immunofluorescence
Immunofluorescence was performed on 8-μm thick frozen liver sections. Slices were fixed in methanol and then washed with PBS. After incubation in blocking buffer for 13hour, the primary antibody recognizing human ABCC6 protein (M6II-7, 1:100; abcam, Cambridge, MA) was added first for 1 hour, followed by incubation with the primary antibody specific for mouse ABCC6 protein (s-20, 1:200; Santa Cruz, Dallas, Texas) for 13hour. After washing with PBS, the sections were incubated with secondary antibodies for 1 hour and the nuclei were stained with 4′, 6-diamidino-2-phenylindole for 53minutes. The stained samples were analyzed using a fluorescent microscope (Zeiss, Göttingen, Germany).
Zebrafish mRNA rescue assay
To test the potential pathogenicity of ABCC6 missense mutations, a zebrafish mRNA rescue assay was performed as described previously (Li et al., 2010a; Zhou et al., 2013). Briefly, human ABCC6 variants were cloned in Bluescript II SK- vector, and mRNA was generated by in vitro transcription using the mMessage mMachine kit (Ambion, Austin, TX). A morpholino specific for zebrafish abcc6a sequence was injected into one- to four-cell-stage embryos either alone or in combination with the human, either mutant or wild-type ABCC6 mRNA (2.4 mmol). The injected zebrafish embryos were followed for their phenotype and survival rate on daily intervals.
Ethics Statement
Informed written consent was obtained from all subjects, and the present study was approved by the local Medical Research Ethics Committee at Xijing Hospital, Fourth Military Medical University, Xi’an, China.
The mice were maintained under standard laboratory conditions and were handled in accordance with the guidelines for animal experiments by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Supplementary Material
Acknowledgments
The authors thank Prof. Donglai Ma (Department of Dermatology, Peking Union Medical College Hospital), Heng Yan (Department of Dermatology, Southwest Hospital), Prof. Songmei Geng (Department of Dermatology, The Second Affiliated Hospital of Xi’an Jiaotong University), Prof. Liuqing Chen (Department of Dermatology, Wuhan No. 1 Hospital), Prof. Deyou Tan (Department of Dermatology, Foshan Shi No.2 People’s Hospital), Yong Li (Department of Dermatology, Yichang Shi No. 2 People Hospital), Prof. Wei Yuan and Tingkai Yan (Department of Dermatology, Affiliated Hospital of Zunyi Medical College) for their assistance in clinical sample collection; Dr. András Várádi for providing plasmids; Dian Wang for technical assistance; and Carol Kelly for manuscript preparation. This study was supported by NIH/NIAMS grants K08 AR057099 (QJ) and R01 AR55225 (JU), and by The National Natural Science Foundation of China (QJ) The Milstein Medical Asian American Partnership Foundation provided generous support.
Abbreviations
- PXE
pseudoxanthoma elasticum
- GACI
generalized arterial calcification of infancy
- Pi
inorganic phosphate
- PPi
inorganic pyrophosphate
- PTC
premature termination codon
- SNP
single nucleotide polymorphism
- 4-PBA
4-phenylbutyrate
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest
References
- Akoglu G, Li Q, Gokoz O, et al. Clinical and histopathological characteristics of a family with R1141X mutation of pseudoxanthoma elasticum - presymptomatic testing and lack of carrier phenotypes. Int J Dermatol. 2014;53:692–8. doi: 10.1111/ijd.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranyi T, Bacquet C, de Boussac H, et al. Transcriptional regulation of the ABCC6 gene and the background of impaired function of missense disease-causing mutations. Front Genet. 2013;4:27. doi: 10.3389/fgene.2013.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen AA, Plomp AS, Schuurman EJ, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25:228–31. doi: 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- Chassaing N, Martin L, Bourthoumieu S, et al. Contribution of ABCC6 genomic rearrangements to the diagnosis of pseudoxanthoma elasticum in French patients. Hum Mutat. 2007;28:1046. doi: 10.1002/humu.9509. [DOI] [PubMed] [Google Scholar]
- Eytan O, Morice-Picard F, Sarig O, et al. Cole disease results from mutations in ENPP1. Am J Hum Genet. 2013;93:752–7. doi: 10.1016/j.ajhg.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria CS, Li Q, Guo H, et al. Clinical phenotypes and ABCC6 gene mutations in Brazilian families with pseudoxanthoma elasticum. Acta Derm Venereol. 2013;93:739–40. doi: 10.2340/00015555-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheduzzi D, Guidetti R, Anzivino C, et al. ABCC6 mutations in Italian families affected by pseudoxanthoma elasticum (PXE) Hum Mutat. 2004;24:438–9. doi: 10.1002/humu.9284. [DOI] [PubMed] [Google Scholar]
- Ilias A, Urban Z, Seidl TL, et al. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6) J Biol Chem. 2002;277:16860–7. doi: 10.1074/jbc.M110918200. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Dibra F, Lee MD, et al. Overexpression of fetuin-A counteracts ectopic mineralization in a mouse model of pseudoxanthoma elasticum (abcc6−/−) J Invest Dermatol. 2010;130:1288–96. doi: 10.1038/jid.2009.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Matsuzaki Y, Li K, et al. Transcriptional regulation and characterization of the promoter region of the human ABCC6 gene. J Invest Dermatol. 2006;126:325–35. doi: 10.1038/sj.jid.5700065. [DOI] [PubMed] [Google Scholar]
- Kalah IG, Seetha D, Panda A, et al. Molecular diagnosis of generalized arterial calcification of infancy (GACI) J Cardiovasc Dis Res. 2012;3:150–4. doi: 10.4103/0975-3583.95373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRusso J, Ringpfeil F, Uitto J. Pseudoxanthoma elasticum: a streamlined, ethnicity-based mutation detection strategy. Clin Transl Sci. 2010;3:295–8. doi: 10.1111/j.1752-8062.2010.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Saux O, Beck K, Sachsinger C, et al. Evidence for a founder effect for pseudoxanthoma elasticum in the Afrikaner population of South Africa. Hum Genet. 2002;111:331–8. doi: 10.1007/s00439-002-0808-1. [DOI] [PubMed] [Google Scholar]
- Le Saux O, Urban Z, Tschuch C, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223–7. doi: 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- Li Q, Baker J, Kowalczyk J, et al. Pediatric pseudoxanthoma elasticum with cardiovascular involvement. Br J Dermatol. 2013;169:1148–51. doi: 10.1111/bjd.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brodsky JL, Conlin L, et al. Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy - genotypic overlap with pseudoxanthoma elasticum. J Invest Dermatol. 2014;134:658–65. doi: 10.1038/jid.2013.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sadowski S, Frank M, et al. The abcc6a gene expression is required for normal zebrafish development. J Invest Dermatol. 2010a;130:2561–8. doi: 10.1038/jid.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Schumacher W, Siegel D, et al. Cutaneous features of pseudoxanthoma elasticum in a patient with generalized arterial calcification of infancy due to a homozygous missense mutation in the ENPP1 gene. Br J Dermatol. 2012;166:1107–11. doi: 10.1111/j.1365-2133.2012.10811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Torok L, Kocsis L, et al. Mutation analysis (ABCC6) in a family with pseudoxanthoma elasticum: presymptomatic testing with prognostic implications. Br J Dermatol. 2010b;163:641–3. doi: 10.1111/j.1365-2133.2010.09856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Uitto J. Mineralization/anti-mineralization networks in the skin and vascular connective tissues. Am J Pathol. 2013;183:10–8. doi: 10.1016/j.ajpath.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neldner KH. Pseudoxanthoma elasticum. Clin Dermatol. 1988;6:1–159. doi: 10.1016/0738-081x(88)90003-x. [DOI] [PubMed] [Google Scholar]
- Nitschke Y, Baujat G, Botschen U, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90:25–39. doi: 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke Y, Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin. Front Genet. 2012;3:302. doi: 10.3389/fgene.2012.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noji Y, Inazu A, Higashikata T, et al. Identification of two novel missense mutations (p.R1221C and p.R1357W) in the ABCC6 (MRP6) gene in a Japanese patient with pseudoxanthoma elasticum (PXE) Intern Med. 2004;43:1171–6. doi: 10.2169/internalmedicine.43.1171. [DOI] [PubMed] [Google Scholar]
- Pfendner EG, Vanakker OM, Terry SF, et al. Mutation detection in the ABCC6 gene and genotype-phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J Med Genet. 2007;44:621–8. doi: 10.1136/jmg.2007.051094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomozi V, Brampton C, Fulop K, et al. Analysis of pseudoxanthoma elasticum-causing missense mutants of ABCC6 in vivo; pharmacological correction of the mislocalized proteins. J Invest Dermatol. 2014;134:946–53. doi: 10.1038/jid.2013.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomozi V, Le Saux O, Brampton C, et al. ABCC6 Is a Basolateral Plasma Membrane Protein. Circ Res. 2013;112:e148–51. doi: 10.1161/CIRCRESAHA.111.300194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkinen L, Nakano A, Ringpfeil F, et al. Identification of ABCC6 pseudogenes on human chromosome 16p: implications for mutation detection in pseudoxanthoma elasticum. Hum Genet. 2001;109:356–65. doi: 10.1007/s004390100582. [DOI] [PubMed] [Google Scholar]
- Ramsay M, Greenberg T, Lombard Z, et al. Spectrum of genetic variation at the ABCC6 locus in South Africans: Pseudoxanthoma elasticum patients and healthy individuals. J Dermatol Sci. 2009;54:198–204. doi: 10.1016/j.jdermsci.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Ringpfeil F, Lebwohl MG, Christiano AM, et al. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A. 2000;97:6001–6. doi: 10.1073/pnas.100041297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf N, Uhlenberg B, Terkeltaub R, et al. The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI) Hum Mutat. 2005;25:98. doi: 10.1002/humu.9297. [DOI] [PubMed] [Google Scholar]
- Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nature Genet. 2003;34:379–81. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- Sato N, Nakayama T, Mizutani Y, et al. Novel mutations of ABCC6 gene in Japanese patients with Angioid streaks. Biochem Biophys Res Commun. 2009;380:548–53. doi: 10.1016/j.bbrc.2009.01.117. [DOI] [PubMed] [Google Scholar]
- Schulz V, Hendig D, Henjakovic M, et al. Mutational analysis of the ABCC6 gene and the proximal ABCC6 gene promoter in German patients with pseudoxanthoma elasticum (PXE) Hum Mutat. 2006;27:831. doi: 10.1002/humu.9444. [DOI] [PubMed] [Google Scholar]
- Struk B, Cai L, Zach S, et al. Mutations of the gene encoding the transmembrane transporter protein ABC-C6 cause pseudoxanthoma elasticum. J Mol Med (Berl) 2000;78:282–6. doi: 10.1007/s001090000114. [DOI] [PubMed] [Google Scholar]
- Tanioka M, Utani A, Tamura H, et al. Calcification of the placenta in a woman with pseudoxanthoma elasticum with a mutation of the ABCC6 gene. J Dermatol. 2014;41:189–91. doi: 10.1111/1346-8138.12360. [DOI] [PubMed] [Google Scholar]
- Terry S, Hefferson T. LOVD Gene Homepage. 2013 http://www.ncbi.nlm.nih.gov/lovd/home.php?select_db=ABCC6.
- Uitto J, Jiang Q, Varadi A, et al. Pseudoxanthoma elasticum: diagnostic features, classification and treatment options. Expert Opin Orphan Drugs. 2014;2:567–577. doi: 10.1517/21678707.2014.908702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J, Li Q, Jiang Q. Pseudoxanthoma elasticum: molecular genetics and putative pathomechanisms. J Invest Dermatol. 2010;130:661–70. doi: 10.1038/jid.2009.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J, Varadi A, Bercovitch L, et al. Pseudoxanthoma elasticum: progress in research toward treatment: summary of the 2012 PXE International Research Meeting. J Invest Dermatol. 2013;133:1444–9. doi: 10.1038/jid.2013.20. [DOI] [PubMed] [Google Scholar]
- Yang MGM, Xiao F, Fang Q, et al. Mutation of ABCC6 gene in a Chinese patient with pseudoxanthoma elasticum. Acta Universitatis Medicinalis Anhui. 2008;43:206–7. [Google Scholar]
- Yoshida S, Honda M, Yoshida A, et al. Novel mutation in ABCC6 gene in a Japanese pedigree with pseudoxanthoma elasticum and retinitis pigmentosa. Eye (Lond) 2005;19:215–7. doi: 10.1038/sj.eye.6701449. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Jiang Q, Takahagi S, et al. Premature termination codon read-through in the ABCC6 gene: Potential treatment for pseudoxanthoma elasticum. J Invest Dermatol. 2013;133:2672–7. doi: 10.1038/jid.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.