

Abstract
Dibenzo[a,l]pyrene (DBP) has been found to be the most potent carcinogen of the polycyclic aromatic hydrocarbons (PAHs). Primary sources for DBP in the environment are combustion of wood and coal burning, gasoline and diesel exhaust, and tires. Given the likelihood of environmental exposure to DBP and strong experimental evidence of its potency, it is likely to contribute to lung cancer development. Intervention with compounds of natural origin (“phytochemicals”) is considered an effective means to prevent cancer development and favorably modulate the underlying mechanisms, including DNA adduct formation. In this study, several agents have been identified that inhibit environmental carcinogen-induced DNA adduct formation using a cell-free microsomal system. Of the ten agents tested, resveratrol (648 ± 26 adducts/109 nucleotides), oltipraz (1007 ± 348 adducts/109 nucleotides), delphinidin (1252 ± 142 adducts/109 nucleotides), tanshinone I (1981 ± 213 adducts/109 nucleotides), tanshinone IIA (2606 ± 478 adducts/109 nucleotides) and diindoylmethane (3643 ± 469 adducts/109 nucleotides) were the most effective compared to vehicle treatment (14,062 ± 1097 adducts/109 nucleotides). DBP is metabolized by phase I metabolizing enzymes CYP1A1, CYP1A2, and CYP1B1. DBP-induced DNA adducts can be inhibited by several mechanisms. We found that all the test agents inhibited DNA adducts by inhibiting one or more of these enzymes. Oltipraz inhibited DNA adducts entirely by inhibiting the CYP450s, while resveratrol and delphinidin inhibited DNA adducts by also interacting directly with the carcinogenic metabolite, anti-dibenzo(a,l)pyrene-11,12-dihydrodiol-13,14-epoxide.
Keywords: Cytochrome P450; Dibenzo[a,l]pyrene; DNA adducts; Polycyclic aromatic hydrocarbons; 32P-postlabeling
1 Introduction
It is clear that the problem of cancer mortality cannot be solved by treatment after diagnosis of cancer alone. Therefore, attention is turning to the detection of early biomarkers and preventive intervention. The focus of this study is to identify effective chemopreventive agents and assess their mechanisms of action.
Polycyclic aromatic hydrocarbons (PAHs) are well known environmental carcinogens and mutagens. They are readily metabolized by oxidation reactions involving phase I metabolizing enzymes including those of the CYP1A, forming DNA adducts. DBP is the most potent PAH tested in rodent mammary tissues, mouse skin and lungs [1, 2] (Fig. 1).
Fig. 1.
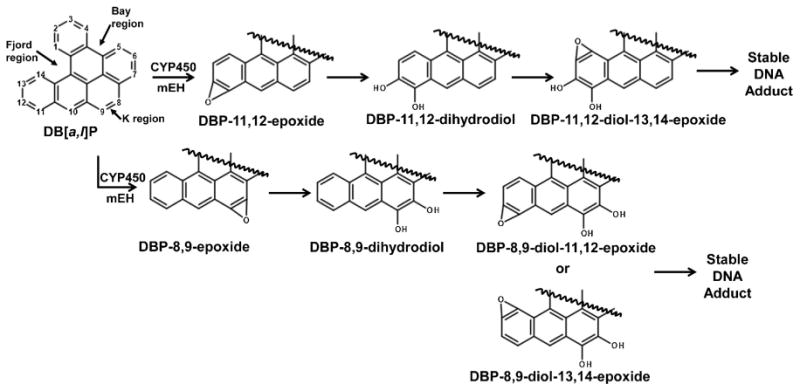
Schematic representation of Phase I metabolism of DBP. Fjord and K region metabolites bind to DNA and lead to adducts, mutagenesis, and eventually carcinogenesis.
It is important to perform trial studies involving the pharmacokinetics and mechanism-based markers of the selected chemopreventive agents before any pre-clinical animal studies [3]. Three major criteria were considered when selecting chemopreventive agents for this study: (1) the agents are effective in diminishing DBP- or other PAH-induced DNA adducts in vitro or in vivo; (2) the agent should favorably modulate DBP metabolizing enzymes CYP1A1, CYP1A2, and 1B1; and (3) the agent should inhibit cancer cell progression. Accordingly, we selected cyanidin, cucurbitacin B, delphinidin, diindoylmethane (DIM), ellagic acid, indole-3-carbinol, resveratrol, tanshinone I, tanshinone IIA, withaferin A and oltipraz based on their chemopreventive effects in various model systems (Fig. 2).
Fig. 2.
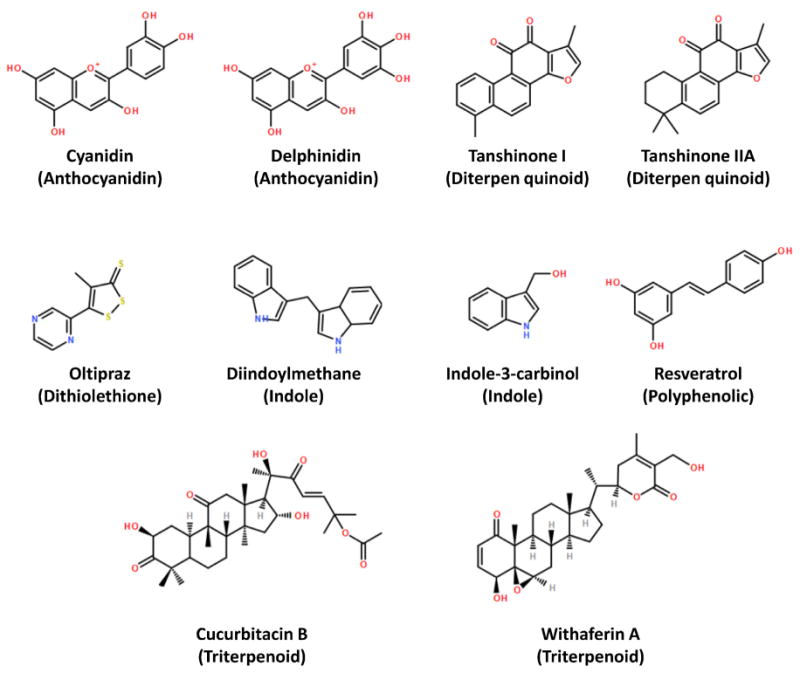
Phytochemicals tested for efficacy in decreasing DBP-induced DNA adducts. Chemical class provided in parentheses. All compounds are naturally occurring, except oltipraz which is a synthetic dithiolethione derivative.
Ellagic acid is a common plant phenolic found in several berries. Ellagic acid inhibits DBP-induced DNA adducts in MCF-7 cells [4]. Oltipraz is a synthetic dithiolthione and has been found to be a highly effective phase II inducer, particularly of glutathione-S-transferase. Indole-3-carbinol (I3C) is a phytochemical found in cruciferous vegetables. I3C has been shown to decrease cell proliferation in vitro and inhibit carcinogen-DNA adducts in vivo [5] and is also an inducer of CYP1A1 [6]. Cucurbitacin B comes from the cucurbita flower of Latin America. Cucurbitacin B inhibited cell proliferation and induced apoptosis in human laryngeal cancer cells [7], and has been shown to be cytotoxic in lung cancer cell lines [8]. Resveratrol is an aphytoalexin compound found in purple-skinned grapes [9]. In some models it has been shown to be a selective human CYP1A1 inhibitor [9] and also inhibits PAH bioactivation through inhibition of CYP1A1 and CYP1B1 gene expression in human bronchial epithelial cells [10]. Tanshinones are the major bioactive compounds of the Salvia miltiorrhiza Bunge (Danshen) roots. Tanshinone I was found to inhibit inflammatory pathways in human non-small-cell lung cancer [11]. Tanshinone IIA is the most abundant phytoconstituent of Danshen. Tanshinone IIA has been found to be anti-carcinogenic through cytotoxic activity [12] and binds to the minor groove of DNA molecules in human carcinoma cell lines [13]. Withaferin A, a triterpinoid, is the root extract of Withania somnifera, and it was shown to exhibit anti-proliferative activity in human cancer cell lines [14]. Delphinidin is a flavonoid from anthocyanins-rich berries such as blueberry and black currant. Following benzo[a]pyrene treatment of MCF-10F cells DNA adduct formation was significantly inhibited by delphinidin [15]. Cyanidin also belongs to this flavonoid family and is present in a wide variety of berries.
All these compounds have shown high potential in vitro and in other models and are suitable candidates for investigating DNA adduct inhibition and cytochrome modulation studies.
2 Materials and Methods
2.1 Chemicals
DBP was purchased from the NCI (National Cancer Institute) Chemical Carcinogen Repository (Bethesda, MD). Magnesium chloride, glucose-6-phophate, glucose-6-phosphate dehydrogenase, Salmon Testes (St) DNA, nicotinamide adenine dinucleotide phosphate-oxidase (NADP+) were purchased from Sigma–Aldrich Corp. (St. Louis, MO). Chemopreventive agents cucurbitacin B, I3C, DIM (3,3′-diindolylmethane), ellagic acid, and resveratrol were purchased from PhytoMyco Research Corporation (Greenville, NC), Sigma–Aldrich Corp. (St. Louis, MO), LKT laboratories (St. Paul, MN), and Biotivia (New York, NY), respectively. Delphinidin and cyanidin (>95%) were isolated in our laboratory from highly enriched black currant extract [16], and withaferin A (>94%) was also isolated in our laboratory from highly enriched root extract of W. somnifera. These compounds were made available for the present study. Oltipraz was provided by Drs. Ronald Lubet and Vernon Steele of the NCI Investigational Studies Branch in Bethesda, MS. Tanshinone I and tanshinone IIA were purchased from PhytoMyco Research Corporation (Greenville, NC). Microsomes were prepared in our laboratory from the liver of 6-week old (150–174 g) male Sprague-Dawley rats (Indianapolis, IN) treated with β-naphthoflavone by intraperitoneal injection (80 mg/kg body weight) daily for 4 days. After treatment, livers were isolated and perfused in situ, and microsomes were isolated as described by Espandiari et al. [17]. CYP1A1, CYP1A2, and CYP1B1 supersomes were purchased from BD Biosciences (San Diego, CA). A racemic mixture of anti-DBPDE (anti-dibenzo(a,l)pyrene-11,12-dihydrodiol-13,14-epoxide) was provided by Dr. Arun Sharma of the Penn State College of Medicine in Hershey, PA. Enzyme substrate 7-ethoxyresorufin was purchased from Anaspec (Fremont, CA). Solvents and chemicals used for the 32P-postlabeling assay were either prepared by our laboratory or purchased commercially as described in Gupta et al. [18].
2.2 Microsomal Assay
St-DNA (300 μg/ml) was incubated with NADPH-regenerating system [MgCl2 (1 mM), glucose-6-phosphate (2.5 mM), glucose-6-phosphate dehydrogenase (1 U/ml), and NADP+ (0.5 mM)] and β-naphthoflavone-induced rat liver microsomes (1 mg/ml) or rat CYP1A1, rat CYP1A2, or human CYP1B1 supersomes (1 μg/ml), and microsomal epoxide hydrolase supersomes (0.25 μg/ml), along with test chemopreventive compounds (150 μM in DMSO). The mixture was incubated for 10 min at 37°C in a shaking water bath. DBP (10 μM in DMSO) was then added to the reaction mixture and the incubation was continued for 1 h at 37°C. The final concentration of DMSO was 1%. The reactions were terminated by the addition of EDTA (20 mM), and DNA was purified as described below.
To generate readily detectable DNA adduct products and obtain reliable quantitative data in the presence of inhibitors, significantly higher levels of DBP and chemopreventives compared to known biological levels were used in these studies. We also maintained the same concentration of test agents (150 μM) as in our published studies for comparison. Human CYP1B1 supersomes were used due to the unavailability of rat-specific supersomes.
2.3 Non-enzymatic assay
St-DNA (300 μg/ml) was added to 50 mM Tris–HC1, pH 8.0 and test chemopreventive compounds (150 μM in DMSO). The mixture was pre-incubated for 10 min at 37°C in a shaking water bath. Anti-DBPDE (1.0 μM) was then added to the reaction mixture and the incubation was continued for 1 h at 37°C. The reactions were terminated by extraction with one volume of ethyl acetate to remove any unbound anti-DBPDE. DNA was then isolated by ethanol precipitation.
2.4 DNA isolation
DNA from microsomal incubations were incubated sequentially with RNases A (150 μg) and T1 (1 U/μl) to digest RNA and proteinase K (150 μg/ml) to digest proteins. This was followed by sequential extractions with phenol, phenol: Sevag (1:1) and Sevag (chloroform: isomly alcohol; 24:1). DNA was then precipitated with ethanol (1 vol) and 5 M NaC1 (0.1 vol). The concentration of DNA was measured by UV spectroscopy at 230, 260, and 280 nm wavelengths. The ratio of 260/280 is ∼1.8 for pure DNA [19].
2.5 32P-Postlabeling analysis
10–20 μg of DNA was digested with a mixture of micrococcal nuclease and spleen phosphodiesterase (enzyme to substrate, 1:5; 37°C; 5 h). An aliquot of the digest (2 μg) was diluted with water (0.5 ng/μl) and the remaining digest was enriched for adducts by treatment with nuclease P1 (enzyme:substrate, 1:2.5; 37°C; 1 h). Dilute DNA digest (2 ng) was labeled by 5′-phosphorylation with T4 polynucleotide kinase with molar excess of [γ-32P]-ATP (80 μCi; 6,000 Ci/mmol). Labeled normal nucleotides were resolved by 1-directional PEI-cellulose thin-layer chromatography (TLC) and quantified by Packard Instant Imager. Enriched adducted nucleotides (∼8 μg) were labeled in parallel with normal nucleotides by T4 polynucleotide kinase and molar excess [γ-32P]-ATP (80 μCi; 6,000 Ci/mmol). Labeled adducts were resolved by multi-directional PEI-cellulose TLC, detected and quantified by Packard InstantImager. Adduct spots were quantified by measuring the radioactivity. The radioactive intensity of each spot was normalized by the radioactivity of total nucleotides. Adducts were finally reported as relative Adduct Labeling (RAL) as follows [18,20]:
2.6 EROD Enzyme Activity Assay
Rat CYP1A1, rat CYP1A2, human CYP1B1 supersomes, or β-naphthoflavone-induced rat liver microsomes (1 μg/μl) were diluted in assay buffer (100 mM potassium phosphate pH 7.4). 7-ethoxyresorufin (12.5 μM) and chemopreventive compounds (150 μM in DMSO) were then added. The plate was warmed to 37°C for 10 min. DBP (10 μM) and NADPH (2.5 mM) were then added to the reaction mixture. The plate was read immediately at 37° C for 30 min spectrofluorometrically at an excitation of 530 nm and an emission of 580 nm for formation of resorufin [21, 22]. The plate and contents were disposed as carcinogen waste.
2.7 Statistics
All data were expressed as mean ± SE and statistically compared using the Student's t-test and one-way ANOVA where appropriate. Data values were considered significantly different if p value <0.05.
3. Results
3.1 Inhibition of DBP-induced DNA adducts in a microsomal cell-free system
Several compounds were tested for their efficacy to inhibit DBP-induced DNA adducts. These phytochemicals were incubated with rat liver microsomes, which contain the phase I metabolizing enzymes. DNA adducts were analyzed by 32P-postlabeling assay (Fig. 3). In comparison to DBP metabolism by microsomes from β-naphthoflavone-treated rat liver (14,062 ± 1097 adducts/109 nucleotides) it was found that at 150 μM the most effective compounds were resveratrol (648 ± 26 adducts/109 nucleotides; p = 0.0001), oltipraz (1007 ± 348 adducts/109 nucleotides; p ≤ 0.0001), and delphinidin (1252 ± 142 adducts/109 nucleotides; p = 0.0001), tanshinone I (1981 ± 213 adducts/109 nucleotides; p < 0.0001), tanshinone IIA (2606 ± 478 adducts/109 nucleotides; p < 0.0001) and DIM (3643 ± 469 adducts/109 nucleotides; p < 0.0001) (Fig. 4).
Fig. 3.
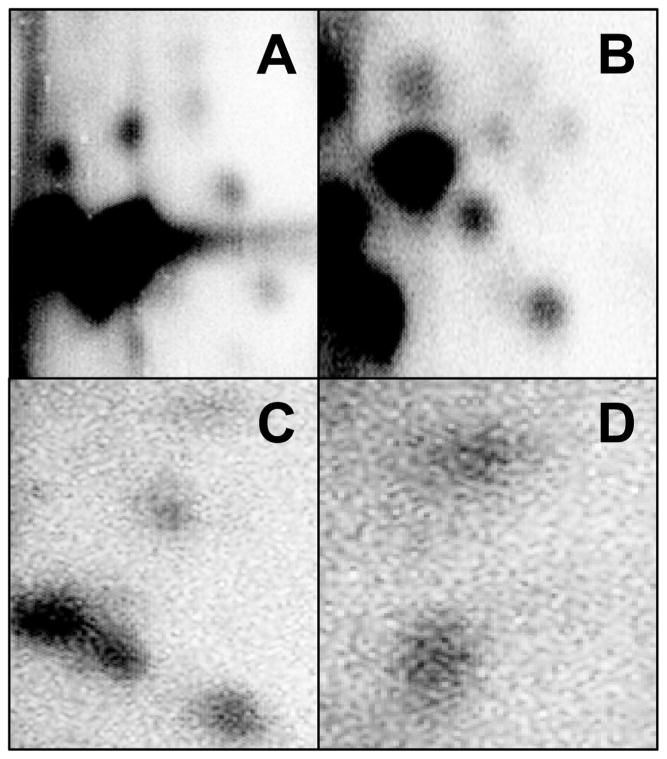
Chromatograms of representative lipophilic DBP-DNA adducts resolved by 32P-postlabeling assay. Adducts were resolved by running in a three step solvent system. (A) DBP + β-napthaflavone-induced microsomes; (B) DBP + CYP1A1 supersomes; (C) DBP + CYP1B1 supersomes; (D) DBP + CYP1A1 supersomes.
Fig. 4.

Effect of test phytochemicals (150 μM) on the modulation of DBP (10 μM)-induced DNA adducts in vitro using salmon testes DNA and β-nathpthaflavone-induced rat liver microsomes by 32P-postlabeling. Total adduct levels in the presence of test agents were compared to vehicle (corn oil + 20% DMSO) control and were significantly different if p < 0.05 (n = 3–8) (**p < 0.001; ***p < 0.0001).
3.2 Inhibition of anti-DBPDE-induced DNA adducts
The potential of test agents to chemically interact with DBP metabolites were tested by analyzing anti-DBPDE-DNA adducts by 32P-postlabeling assay. Compared to anti-DBPDE only-treated DNA (49,806 ± 4647 adducts/109 nucleotides), DNA adducts were decreased moderately by cyanidin (31,204 ± 1951 adducts/109 nucleotides; p = 0.0121), delphinidin (33,409 ± 1512 adducts/109 nucleotides; p = 0.0404), and resveratrol (24,753 ± 2290 adducts/109 nucleotides; p = 0.0079). Ellagic acid, used as positive control [23], inhibited adduct formation substantially (10,185 ± 792 adducts/109 nucleotides; p = 0.0001). Oltipraz showed no significant reduction in anti-DBPDE-induced DNA adducts (46,578 ± 1296 adducts/109 nucleotides; p = 0.6250) (Fig. 5) as compared to control.
Fig. 5.

Modulation of anti-DBPDE-induced DNA adducts in vitro using a non-enzymatic system consisting of salmon testes DNA incubated with anti-DBFDE (1 μM). Anti-DBPDE is the carcinogenic metabolite of DBP. Compounds tested (150 μM) were selected from the most effective agents identified from the micro-somal test system. DNA adducts were analyzed by 32P-postlabeling assay. Total adduct levels in the presence of test agents were compared to vehicle (corn oil + 20% DMSO) control and were significantly different if p < 0.05 (n = 3–5) (*p < 0.05; **p < 0.001; ***p < 0.0001).
3.3 Inhibition of DBP-DNA adducts induced by individual CYP450s
The second criterion was to investigate mechanistically which P450s are involved in inhibiting the formation of DNA adducts by specific chemopreventive agents. DNA adducts induced by CYP1A1 were most significantly reduced by oltipraz (57 ± 4 adducts/109 nucleotides; p = 0.0001) and resveratrol (452 ± 15 adducts/109 nucleotides; p = 0.0015) as compared to control (2648 ± 18 adducts/109 nucleotides) (Fig. 6A). Ellagic acid only moderately decreased CYP1A1-mediated DBP-DNA adducts (1845 ± 154 adducts/109 nucleotides; p = 0.0264). CYP1A2-mediated DBP-DNA adducts were significantly decreased by delphinidin (19 ± 2 adducts/109 nucleotides; p = 0.0013), ellagic acid (33 ± 1 adducts/109 nucleotides; p = 0.0078), and oltipraz (24 ± 1 adducts/109 nucleotides; p = 0.0018) compared to control (51 ± 4 adducts/109 nucleotides) (Fig. 6B). Resveratrol was not effective in decreasing these adducts; in fact, it increased the adduct levels (85 ± 3 adducts/109 nucleotides; p = 0.0015). CYP1B1-mediated DBP-DNA adducts were substantially decreased by ellagic acid (28 ± 2 adducts/109 nucleotides; p = 0.0002), delphinidin (41 ± 7 adducts/109 nucleotides; p = 0.0005), oltipraz (54 ± 9 adducts/109 nucleotides; p = 0.0008), and resveratrol (92 ±11 adducts/109 nucleotides; p = 0.0028) as compared to control (211 ± 14 adducts/109 nucleotides) (Fig. 6C).
Fig. 6.

Effect of test phytochemicals (150 μM) on (A) CYP1A1; (B) CYP1A2 and (C) CYP1B1 activity as assessed by modulation of DBP (10 μM)-induced DNA adducts. DNA adducts formed by in vitro assay using salmon testes DNA incubated with specific rat CYP1A1, rat CYP1A2, and human CYP1B1 supersomes were analyzed by 32P-postlabeling. Total adduct levels in the presence of test agents were compared to vehicle (corn oil + 20% DMSO) control and were significantly different if p < 0.05 (n = 3–4) (*p < 0.05; ** p < 0.001; ***p < 0.0001).
3.4 CYP450 enzyme activity by the EROD assay
To further investigate the role of modulation of enzyme activity on the efficacy of these compounds the ethoxyresorufin O-deethylase (EROD) activity assay was performed. Analyzing total CYP1A enzyme activity in microsomes from β-naphthoflavone-treated rats, ellagic acid and resveratrol seemed not to affect the enzymes (Fig. 7A1 and A2). The effects of the compounds on the individual enzymes were then investigated using supersomes (Fig. 7B1–D2).
Fig. 7.

Effect of phytochemicals (150 μM) on CYP enzyme activity (EROD). (A) Total CYP1A activity from β-naphthoflavone-induced rat liver microsome; (B) CYP1A1 activity from rat supersome; (C) CYP1A2 activity from rat supersome; and (D) CYP1B1 activity from human supersome. Panels A1, B1, C1 and D1 represent native inhibitor activity of the phytochemicals. Significance calculated in comparison to vehicle control (corn oil + 20% DMSO). Panels A2, B2, C2 and D2 represent inhibitor activity of the phytochemicals in the presence of DBP. Inhibitory activity could be either due to scavenging of the intermediate metabolite or competitive inhibition to relevant CYP activity. In these panels, significance was calculated in comparison to DBP (10 μM) only treatment. All data were significantly different if p < 0.05 (n = 3) (*p < 0.05; **p < 0.001; ***p < 0.0001). (Del – delphimdin; EA – ellagic acid; Res – resverstrol; Olt – oltipraz).
CYP1A enzyme activities in β-naphthoflavone-induced rat liver microsomes were 1%, 31%, 17%, and 3% that of vehicle-treated microsomes by delphinidin, ellagic acid, resveratrol, and oltipraz, respectively. In the presence of DBP, the activity was 0%, 38%, 28%, and 17% of the activity of microsomes treated with DBP and delphinidin, ellagic acid, resveratrol, and oltipraz, respectively (Fig. 7A1 and A2)
Compared to vehicle treatment, delphinidin decreased CYP1A1 enzyme activity to 40% of control, ellagic acid increased to 114%, while resveratrol and oltipraz decreased to 4%–5%). CYP1A1 enzyme activity in the presence of the DBP was 35%, 2%, 10%, 3% of DBP and delphinidin, ellagic acid, resveratrol, and oltipraz, respectively (Fig. 7B1 and B2).
Compared to vehicle treatment, delphinidin, ellagic acid, resveratrol, and oltipraz, decreased CYP1A2 enzyme activity to 32%, 8%, 79%, 2%, respectively. In the presence of DBP, ellagic acid modulated activity to 779% compared to DBP alone treatment, while delphinidin, resveratrol, and oltipraz modulated activity to 0%, 77%, and 18%, respectively (Fig. 7C1 and C2).
CYP1B1 activity was nearly completely inhibited by delphinidin, ellagic acid, and oltipraz, but resveratrol showed nearly 80% inhibition. In the presence of DBP also, delphinidin and ellagic acid showed complete inhibition of CYP1B1 activity, and resveratrol and oltipraz decreased the activity by 53% and 90%, respectively (Fig. 7D1 and D2).
The enzyme activity of microsomes treated with vehicle was compared to the enzyme activity of microsomes treated with DBP which showed a significant modulation of all the P450s tested: CYP1A in β-naphthoflavone-induced microsomes (773%), and supersomes CYP1A1 (342%), CYP1A2 (663%) and CYP1B1 (1168%).
4 Discussion
Chemopreventive intervention is a desired approach to reduce cancer burden due to long latency for cancer development. Natural compounds are ideal for such interventions due to their low costs, accessibility via abundant plant source [24] and in most cases, lack of toxicity. However, identification of efficacious natural compounds and their modes of action in in vitro studies is a necessity due to the high cost and time involved in animal studies. Thus, a cell-free microsomal system in conjunction with 32P-postlabeling represents a useful system to test the chemopreventive efficacy of test compounds [25].
A majority of the chemical carcinogens require bioactivation to form an ultimate carcinogen that interacts with the DNA. Biological modifiers that can intervene in this process will be ideal chemopreventive agents. Many chemical carcinogens require phase I cytochrome P450 enzymes, especially CYP1A1, CYP1A2 and CYP1B1, for their activation. Hence compounds that modify, albeit not completely inhibit, the activity of these enzymes can decrease the rate of formation of the electrophilic metabolites and keep the ultimate carcinogen below the threshold [26]. This will allow the phase II enzyme system to detoxify the ultimate carcinogen. We used recombinant rat CYP1A1, CYP1A2, and human CYP1B1 specific microsomes to investigate the individual contribution of each CYP on DBP-induced DNA adducts. Recombinant human CYP1B1 microsomes were the only CYP1B1 recombinant microsomes that were commercially available. We performed preliminary postlabeling studies using recombinant human CYP1B1 microsomes and β-naphthoflavone-induced rat liver microsomes. We found that the DBP-DNA adducts formed by both the activation systems were similar by co-chromatography of DNA adducts (data not shown). From the postlabeling data we found that each of the test compounds modulated the formation of DBP-DNA adducts induced by each individual CYP enzyme tested.
The three approaches used in this study encompass the mechanisms involved in the formation of the ultimate carcinogen. These approaches were: (1) assess the potential of the chemopreventive agent to scavenge the reactive metabolites of DBP; (2) determine which specific cytochrome P450 is targeted by the chemopreventive agent through the formation of various DBP-DNA adducts; and (3) directly deduce the modulation of the activity of the cytochrome P450 enzymes. This way, we were able to test multiple mechanisms by which the test agents could modulate DNA damage. Selected agents that exhibited highest adduct inhibition were further investigated for mechanisms.
Structural analysis showed that delphinidin, an anthocyanidin present in many colored fruits, particularly blueberries, can intercalate with DNA, binding to the major or minor groove and the backbone phosphate group [27]. However, this did not specifically deter the direct binding of anti-DBPDE to DNA. Even though delphinidin demonstrated only moderate effect in modulating the enzyme activity of CYP1A1 and CYP1A2, it was able to inhibit the formation of DNA adducts due to its significant effect on CYP1B1 activity. The moderate effect observed against CYP1A1 is consistent with published reports [15].
Resveratrol, on the other hand, specifically targets CYP1A1 and has a moderate effect on CYP1B1. The moderate effect observed with CYP1B1 inhibition can also be interpreted as competitive inhibition of the substrate since resveratrol is a substrate for CYP1B1 [28]. This is also true with CYP1A2. Both of them convert resveratrol to piceatannol and tetrahydroxystilbene [28,29]. This could reflect in reduced formation of reactive product that was measured based on the fluorescent substrate.
Ellagic acid has no effect on CYP1A1 activity but significantly inhibited both CYP1A2 and CYP1B1. Ellagic acid is also known to scavenge the electrophilic metabolite of PAHs, which was reflected in the direct inhibition of DNA adduct formation when anti-DBPDE was incubated with DNA. These observations also correlated with the modulation of DBP-DNA adduct formation in the presence of ellagic acid with various CYPs. It was intriguing to observe that there was a significant inhibition of enzyme activity in the presence of DBP. A significant reduction in the fluorescent substrate formation was observed in the presence of DBP alone in the EROD assay. This was expected since DBP is a competing substrate for the CYPs, leading to reduced resorufin formation. This can explain the low resorufin formation in the presence of CYP1A1, DBP and ellagic acid. However, in the presence of CYP1A2 and DBP, ellagic acid did not lead to any inhibition of enzyme activity even though it significantly inhibited CYP1A2 activity independently. The rationale behind this property is unclear.
Oltipraz, on the other hand, did not scavenge the electrophilic DBP metabolite but was able to decrease DBP-DNA adduct formation primarily through inhibition of CYP1A1 and CYP1B1, and to a lesser extent, CYP1A2.
The incubation with specific supersomes provided information on the ability of the chemopreventives to inhibit the selective CYP enzymes, either through competition with the substrate or by other means. Thus our data suggest that ellagic acid and delphinidin are potent CYP1B1 inhibitors, while resveratrol is a potent CYP1A1 inhibitor.
In the presence of DBP, it becomes a little complicated to interpret if the inhibition is due to competition to the active site of the enzyme. The inhibition could be explained either due to competition with the substrates, or due to scavenging of the electrophilic DBP metabolite by the chemopreventives. Ellagic acid is a good example for the latter, where the electrophilic DBP metabolite is completely scavenged by it, and hence the CYP1A1 activity is very similar to the vehicle control, while no inhibition was observed by ellagic acid only.
On the other hand, delphinidin demonstrated moderate inhibition of CYP1A1, both by itself and in the presence of DBP, as seen in EROD assay. However, when the data are compared with the DNA adduct study, no significant reduction in DNA adducts was observed. This could be explained by the fact that delphinidin has the ability to inhibit the activity of CYP1A1, but DBP is a more preferred substrate for CYP1A1. Thus in EROD assay, moderate reduction in the formation of resorufin was observed since there was a competitive inhibition with the three substrates. But in DNA adduct study, dephinidin was unable to compete significantly with DBP, hence there was no reduction in the adduct formation. These data clearly demonstrate that there is selective substrate specificity on inhibition based on competition with the active site of the CYP enzyme, but in a complex system, there are other mechanisms also play significant roles, like scavenging of the intermediate electrophilic metabolite.
This study represents a simple and straightforward approach to understand the ability of chemopreventive compounds to inhibit the formation of DNA damaging metabolites, by either inhibiting the enzyme activity, or by scavenging the electrophilic metabolites. The two most efficacious compounds, ellagic acid and oltipraz, need to be taken further for in vivo studies. Additional in vitro work is also needed for the most promising compounds, such as dose–response experiments and kinetic studies to determine the nature of the inhibition. The in vitro studies confirmed the effect on the enzyme activity but do not provide efficacy on induction or reduction of CYP P450 synthesis. Furthermore, the effect on phase II detoxifying enzyme system can also be addressed only in future in vivo studies.
In summary, the proposed approach for measuring the potential of chemopreventive agents to inhibit the formation of DNA adduct can be utilized to screen a large number of potential agents. The approach provides clues to multiple mechanisms by which the test agents can prevent the formation of damaging DNA adducts and identifies potentially efficacious compounds that should be further assessed by additional in vitro and in vivo studies.
Acknowledgments
This work was supported from the USPHS grant CA-118114, Kentucky Lung Cancer Research Program, and Agnes Brown Duggan Endowment. GKR was supported, in part, from the NIEHS trainee grant (ES-011564) and the SREB fellowship program. R.C.G. holds the Agnes Brown Duggan Chair in Oncological Research. The authors would also like to thank Dr. Wendy Spencer for her help in developing this manuscript. The authors also thank Ms. Meghan Hancock, Assistant Director at the Writing Center, for editorial services.
Footnotes
Conflict Of Interest: None
References
- 1.Cavalieri EL, Rogan EG, Higginbotham S, Cremonesi P, Salmasi S. Tumor-initiating activity in mouse skin and carcinogenicity in rat mammary gland of dibenzo[a]pyrenes: the very potent environmental carcinogen dibenzo[a,l]pyrene. J Cancer Res Clin Oncol. 1989;115:67–72. doi: 10.1007/BF00391602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nesnow S, Ross JA, Stoner GD, Mass MJ. Mechanistic linkage between DNA adducts, mutations in oncogenes and tumorigenesis of carcinogenic environmental polycyclic aromatic hydrocarbons in strain A/J mice. Toxicology. 1995;105:403–413. doi: 10.1016/0300-483x(95)03238-b. [DOI] [PubMed] [Google Scholar]
- 3.Gescher AJ, Sharma RA, Steward WP. Cancer chemoprevention by dietary constituents: a tale of failure and promise. Lancet Oncol. 2001;2:371–379. doi: 10.1016/S1470-2045(00)00392-2. [DOI] [PubMed] [Google Scholar]
- 4.Smith WA, Freeman JW, Gupta RC. Effect of chemopreventive agents on DNA adduction induced by the potent mammary carcinogen dibenzo[a,l]pyrene in the human breast cells MCF-7. Mutation research. 2001:480–481. 97–108. doi: 10.1016/s0027-5107(01)00173-7. [DOI] [PubMed] [Google Scholar]
- 5.Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–1215. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- 6.Park JY, Bjeldanes LF. Organ-selective induction of cytochrome P-450-dependent activities by indole-3-carbinol-derived products: influence on covalent binding of benzo[a]pyrene to hepatic and pulmonary DNA in the rat. Chem Biol Interact. 1992;83:235–247. doi: 10.1016/0009-2797(92)90100-y. [DOI] [PubMed] [Google Scholar]
- 7.Liu T, Zhang M, Zhang H, Sun C, Yang X, Deng Y, Ji W. Combined antitumor activity of cucurbitacin B and docetaxel in laryngeal cancer. Eur J Pharmacol. 2008;587:78–84. doi: 10.1016/j.ejphar.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 8.Kongtun S, Jiratchariyakul W, Kummalue T, Tan-ariya P, Kunnachak S, Frahm AW. Cytotoxic properties of root extract and fruit juice of Trichosanthes cucumerina. Planta Med. 2009;75:839–842. doi: 10.1055/s-0029-1185455. [DOI] [PubMed] [Google Scholar]
- 9.Chun YJ, Kim MY, Guengerich FP. Resveratrol is a selective human cytochrome P450 1A1 inhibitor. Biochem Biophys Res Commun. 1999;262:20–24. doi: 10.1006/bbrc.1999.1152. [DOI] [PubMed] [Google Scholar]
- 10.Berge G, Ovrebo S, Eilertsen E, Haugen A, Mollerup S. Analysis of resveratrol as a lung cancer chemopreventive agent in A/J mice exposed to benzo[a]pyrene. Br J Cancer. 2004;91:1380–1383. doi: 10.1038/sj.bjc.6602125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CY, Sher HF, Chen HW, Liu CC, Chen CH, Lin CS, Yang PC, Tsay HS, Chen JJ. Anticancer effects of tanshinone I in human non-small cell lung cancer. Mol Cancer Ther. 2008;7:3527–3538. doi: 10.1158/1535-7163.MCT-07-2288. [DOI] [PubMed] [Google Scholar]
- 12.Yuan SL, Wang XJ, Wei YQ. Anticancer effect of tanshinone and its mechanisms. Ai Zheng. 2003;22:1363–1366. [PubMed] [Google Scholar]
- 13.Zhang Z, Zhang J, Jin L, Song T, Wu G, Gao J. Tanshinone IIA interacts with DNA by minor groove-binding. Biol Pharm Bull. 2008;31:2342–2345. doi: 10.1248/bpb.31.2342. [DOI] [PubMed] [Google Scholar]
- 14.Jayaprakasam B, Zhang Y, Seeram NP, Nair MG. Growth inhibition of human tumor cell lines by withanolides from Withania somnifera leaves. Life Sci. 2003;74:125–132. doi: 10.1016/j.lfs.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Singletary KW, Jung KJ, Giusti M. Anthocyanin-rich grape extract blocks breast cell DNA damage. J Med Food. 2007;10:244–251. doi: 10.1089/jmf.2006.258. [DOI] [PubMed] [Google Scholar]
- 16.Kausar H, Jeyabalan J, Aqil F, Chabba D, Sidana J, Singh IP, Gupta RC. Berry anthocyanidins synergistically suppress growth and invasive potential of human non-small-cell lung cancer cells. Cancer letters. 2012;325:54–62. doi: 10.1016/j.canlet.2012.05.029. [DOI] [PubMed] [Google Scholar]
- 17.Espandiari P, Thomas VA, Glauert HP, O'Brien M, Noonan D, Robertson LW. The herbicide dicamba (2-methoxy-3,6-dichlorobenzoic acid) is a peroxisome proliferator in rats. Fundam Appl Toxicol. 1995;26:85–90. doi: 10.1006/faat.1995.1077. [DOI] [PubMed] [Google Scholar]
- 18.Gupta RC, Spencer-Beach G. Natural and endogenous DNA adducts as detected by 32P-postlabeling. Regul Toxicol Pharmacol. 1996;23:14–21. doi: 10.1006/rtph.1996.0003. [DOI] [PubMed] [Google Scholar]
- 19.Gupta RC. 32P-postlabelling analysis of bulky aromatic adducts. IARC Sci Publ. 1993:11–23. [PubMed] [Google Scholar]
- 20.Arif JM, Gupta RC. Detection of DNA-reactive metabolites in serum and their tissue distribution in mice exposed to multiple doses of carcinogen mixtures: role in human biomonitoring. Carcinogenesis. 1996;17:2213–2219. doi: 10.1093/carcin/17.10.2213. [DOI] [PubMed] [Google Scholar]
- 21.Prough RA, Burke MD, Mayer RT. Direct fluorometric methods for measuring mixed function oxidase activity. Methods Enzymol. 1978;52:372–377. doi: 10.1016/s0076-6879(78)52041-7. [DOI] [PubMed] [Google Scholar]
- 22.Hebbar V, Shen G, Hu R, Kim BR, Chen C, Korytko PJ, Crowell JA, Levine BS, Kong AN. Toxicogenomics of resveratrol in rat liver. Life sciences. 2005;76:2299–2314. doi: 10.1016/j.lfs.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 23.Teel RW, Dixit R, Stoner GD. The effect of ellagic acid on the uptake, persistence, metabolism and DNA-binding of benzo[a]pyrene in cultured explants of strain A/J mouse lung. Carcinogenesis. 1985;6:391–395. doi: 10.1093/carcin/6.3.391. [DOI] [PubMed] [Google Scholar]
- 24.Gullett NP, Ruhul Amin AR, Bayraktar S, Pezzuto JM, Shin DM, Khun FR, Aggarwal BB, Surh YJ, Kucuk O. Cancer prevention with natural compounds. Semin Oncol. 2010;37:258–281. doi: 10.1053/j.seminoncol.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Smith WA, Gupta RC. Use of a microsome-mediated test system to assess efficacy and mechanisms of cancer chemopreventive agents. Carcinogenesis. 1996;17:1285–1290. doi: 10.1093/carcin/17.6.1285. [DOI] [PubMed] [Google Scholar]
- 26.Neumann HG. Ultimate electrophilic carcinogens on cellular nucleophilic reactants. A contribution to the discussion on threshold doses of environmental chemicals. Arch Toxicol. 1974;32:27–38. doi: 10.1007/BF00334609. [DOI] [PubMed] [Google Scholar]
- 27.Kanakis CD, Tarantilis PA, Polissiou MG, Diamantoglou S, Tajmir-Riahi HA. An overview of DNA and RNA bindings to antioxidant flavonoids. Cell Biochem Biophys. 2007;49:29–36. doi: 10.1007/s12013-007-0037-2. [DOI] [PubMed] [Google Scholar]
- 28.Potter GA, Patterson LH, Wanogho E, Perry PJ, Butler PC, Ijaz T, Ruparelia KC, Lamb JH, Farmer PB, Stanley LA, Burke MD. The cancer preventative agent resveratrol is converted to the anticancer agent piceatannol by the cytochrome P450 enzyme CYP1B1. Br J Cancer. 2002;86:774–778. doi: 10.1038/sj.bjc.6600197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piver B, Fer M, Vitrac X, Merillon JM, Dreano Y, Berthou F, Lucas D. Involvement of cytochrome P450 1A2 in the biotransformation of trans-resveratrol in human liver microsomes. Biochem Pharmacol. 2004;68:773–782. doi: 10.1016/j.bcp.2004.05.008. [DOI] [PubMed] [Google Scholar]