

Abstract
IL-25 is a member of the IL-17 family of cytokines that promotes Th2 cell-mediated inflammatory responses. IL-25 signals through a heterodimeric receptor (IL-25R) composed of IL-17RA and IL-17RB, which recruits the adaptor molecule Act1 for downstream signaling. Though the role of IL-25 in potentiating type 2-inflammation is well characterized by its ability to activate the epithelium as well as T cells, the components of its signaling cascade remain largely unknown. Here we found that IL-25 can directly activate STAT5 independently of Act1. Furthermore, conditional STAT5 deletion in T cells or epithelial cells led to a defective IL-25-initiated Th2 polarization as well as defective IL-25-enhancement of Th2 responses. Finally, we found that STAT5 is recruited to the IL-25R in a ligand dependent manner through unique tyrosine residues on IL-17RB. Together, these findings reveal a novel Act1-independent IL-25 signaling pathway through STAT5 activation.
Introduction
Chronic pulmonary inflammation associated with allergic asthma is characterized by the recruitment of CD4-positive Th2 cells, eosinophils, and mast cells. These events are orchestrated systemically via the production of Th2-derived cytokines (IL-4, IL-5, IL-9, IL-13) and locally with elevated IgE in the serum. IL-17E (also known as IL-25) is a member of the IL-17 cytokine family. Unlike the other prototypic IL-17-family cytokines, IL-25 distinctly promotes Th2 cell-mediated inflammatory responses (1–4). IL-25 is produced by the epithelium in response to allergen exposure and can promote the recruitment of Th2 cells, eosinophils, innate lymphoid cells, and mast cells to the site of inflammation. Over-expression of IL-25 can result in enhanced Th2 responses that promote allergic inflammation that manifests into airway eosinophilia and IgE production (1). Intriguingly, epithelial-derived IL-25 acting via an autocrine feed-forward mechanism, can induce the epithelium to produce more IL-25 as well as other potent Th2-driving cytokines like IL-33 and TSLP, thus amplifying the allergic response (4–6). In murine models, IL-25-deficiency results in a defective Th2 response, rendering mice susceptible to helminth infections, yet protected against allergic diseases (4, 6, 7).
IL-25 signals through a heterodimeric receptor complex (IL-25R) composed of two subunits, IL-17RA and IL-17RB, both of which contain a conserved SEFIR domain at the cytoplasmic region (8, 9). Upon ligand binding, IL-25R recruits the adaptor molecule Act1 through the homotypic interactions of the SEFIR domains, which also exists on Act1 (10, 11). Act1-deficiency results in a loss of IL-25-dependent gene expression and renders mice resistant to IL-25-mediated allergic airway inflammation (10, 12, 13). Using cell-specific deletion of Act1, we have shown that Act1 in the epithelium and in T cells play a critical role in IL-25-dependent type 2 responses for allergic lung inflammation (10, 13). Aside from signaling through Act1, IL-25 has been shown to activate MAP kinases like P38 and JNK as well as NF-κB (14). Though the function of IL-25 in inducing type 2 allergic inflammatory responses is well recognized, the components of its signaling cascade remain largely unknown.
In this report, we identified a novel IL-25-signaling pathway through the activation of STAT5. STAT5 activation is a critical for T-cell development as well as during Th2 cell differentiation via its cooperation with GATA-3 to prime T cells to the Th2 phenotype (15, 16). We found that IL-25-dependent STAT5 activation was independent of Act1. IL-25-mediated STAT5 activation in T cells and epithelial cells are required for the induction of Th2 cytokines. Moreover, STAT5 is recruited to the IL-25R in a ligand dependent manner that relies on the phosphorylation of tyrosine 444 and 454 of IL-17RB. These findings demonstrate that IL-25 signaling through STAT5 can potentiate type 2 responses and initiate T cell polarization towards a Th2-like phenotype.
Materials and Methods
Mice
ERT2Cre (purchased from The Jackson Laboratory; B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J) was crossed with STAT5f/f (generated by Lothar Hennighausen, NIDDK, NIH)(17). The Il-17rb −/− mice were generated by Dr. Wenjun Ouyang (Genentech). All animal procedures were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic Foundation.
T cell polarization
Naïve T cells were isolated from the spleen and lymph nodes of mice with a Miltenyi CD4+CD62L+ isolation kit and were cultured for 4 days on plate-bound anti-CD3 (3μg/mL) and anti-CD28 (3μg/mL) ; BD Pharmingen Cats. 553057 and 553294. Th0: anti-IL-4 (10μg/mL); BD Pharmingen Cat. 554432, anti-IFN-γ (10μg/mL) ; BD Pharmingen Cat. 554408; Th1: anti-IL-4 (10μg/mL), IL-12 (10ng/mL); Th17: anti-IL4 (10μg/mL), anti-IFN-γ (10μg/mL), IL-6 (20ng/mL), TGF-β (5ng/mL); Th2: anti-IFN-γ (10μg/mL), IL-4 (10ng/mL), IL-2 (10ng/mL); Th25: anti-IFN-γ (10μg/mL), IL-25 (100ng/mL). For intracellular staining, cells were stimulated with PMA and ionomycin for 5 hours, GolgiStop was added during the last 2 hours of stimulation.
Cell culture and Immunoprecipitation
HEK293 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. HEK293 cells were transfected with 1μg of DNA using Lipofectamine 2000. Primary kidney epithelial cells isolated from mice were infected with Adenovirus encoding Cre-recombinase or control to mediate deletion for 2 days followed by stimulation with IL-25. Adenoviruses expressing Cre-recombinase or empty (GFP) control were purchased from VectorBioLabs, and used at 1 × 106 PFU/ml. IL-17RB wild-type and tyrosine-mutants were subcloned into pAd/CMV/–V5 adenovirus vectors and viral generation and tittering done according to the manufacturers’ instructions (Invitrogen). Cells were lysed in lysis buffer (0.5% Triton X-100, 20mM HEPES (pH 7.4), 150mM NaCl, 2mM EGTA, 1.5 mM MgCl2, 10mM NaF, 12.5 mM β-glycerophosphate, 1mM sodium orthovanadate, 20mM aprotinin and 1mM phenylmethyl-sulfonyl fluoride).
Results
IL-25 activates STAT5 independently of Act1
IL-17RB is basally expressed on naïve T cells but is highly induced upon Th2 polarization (4). We polarized T cells to the Th2 phenotype followed by IL-25 stimulation in order to elucidate components activated upon IL-25 stimulation. IL-25 stimulation in polarized Th2 cells led to an Act1-dependent activation of NF-κB (indicated by the phosphorylation of IκBα) as well as the activation of MAPKs such as P38 and JNK (Fig. 1A). It is well established that STAT proteins play critical roles in T cell polarization. Therefore, we screened whether IL-25 stimulation had any effect on the phosphorylation of STAT proteins. While IL-25 had no effect on the activation of STAT6 and STAT3 (data not shown), IL-25 stimulation led to the activation of STAT5, as indicated by the phosphorylation of STAT5 (Fig. 1A). Furthermore, IL-25 treatment led to the phosphorylation of JAK2. In contrast to the other pathways, where Act1 was required for their activation, the phosphorylation of STAT5 was Act1-independent. Act1-deficient Th2 cells exhibited no defect in the ability to phosphorylate STAT5 in response to IL-25 stimulation. In fact, in the absence of Act1, IL-25-induced STAT5 activation was stronger than in its presence. These findings suggest for the first time, that STAT5 plays a role in the IL-25 signaling cascade.
Figure 1. IL-25 activates STAT5 independently of Act1.

A) T cells isolated from WT and Act1−/− mice were polarized ex vivo to Th2 cells for 3 days. Serum starved T cells were then stimulated with IL-25 for the indicated times followed by immunoblotting for p-STAT5, Act1, p-JAK2, p-IκBα, p-P38, p-JNK, and Actin. B) WT and Act1−/− mouse embryonic fibroblasts (MEFs) that stably express Myc-tagged IL-25R were treated with IL-25 for the indicated times. Cell lysates were subjected to immunoprecipitation of IL-25R-Myc with anti-Myc antibodies followed by western blotting for p-STAT5, p-JAK2, Act1, and Myc. Immunoprecipitated (IP) lysates, top; Whole cell lysates (WCL), bottom. Immunoblots shown are representative data of 3 independently performed experiments.
STAT5 is recruited to the IL-25R in a ligand-dependent manner
To further study IL-25-dependent STAT5 activation, we utilized WT and Act1−/− mouse embryonic fibroblasts (MEFs) that stably express a Myc-tagged IL-25R (11). IL-25 stimulation led to the recruitment of p-STAT5 to the IL-25R (Fig. 1B). Pull-down of the Myc-tagged IL-25R resulted in the co-immunoprecipitation of Act1, p-STAT5, as well as p-JAK2 in a ligand dependent manner, suggesting that the association of JAK2 and STAT5 with the receptor complex can be induced upon IL-25 stimulation. Moreover, in Act1−/− MEFs, immunoprecipitation of IL-25R also resulted in the co-immunoprecipitation of p-STAT5 and p-JAK2, indicating that the interaction of JAK2 and STAT5 with the IL-25R is independent of Act1.
IL-25 induces Th2 cytokines through STAT5 activation
Previous reports have shown that T cells forced to express a constitutively active STAT5 under non-polarizing conditions (anti-CD3 and anti-CD28 only) results in a Th2-skewed cytokine profile, implying that STAT5 activation is sufficient to generate a Th2 profile (15, 18, 19). In line with this, committed Th1 cells expressing a constitutively active STAT5 will acquire the capacity to produce IL-4 and IL-9, suggesting that STAT5 activation results in a bias towards a Th2 phenotype. Previous studies from our group and others have shown that the presence of IL-25 is sufficient to polarize naive T cells to a Th2-like phenotype (Th25 polarization) (4, 13).
Since STAT5 is an important mediator of Th2 responses, we sought to determine the role of STAT5 in Th25 polarization. As an initial test we examined P-STAT5 in the Th25-culture conditions at two doses of IL-25 (Fig. 2A). The IL-25 cultured cells exhibited an approximate 2-fold increase in P-STAT5 compared to unstimulated conditions (Fig. 2A). The basal activation is likely due to paracrine IL-2 signaling. While these data clearly show that IL-25 promotes P-STAT5 activation during in vitro polarization, we next we sought to determine the functional role of IL-25-mediated STAT5 activation in these cells. To test this we crossed tamoxifen-inducible Cre (ERT2Cre+) mice with Stat5f/f mice to generate ERT2Cre+ Stat5f/f and ERT2Cre+ Stat5f/+ mice. Naïve CD4+ T cells isolated from ERT2Cre+ Stat5f/f mice or littermate controls (ERT2Cre+ Stat5f/+) were treated with 10nM of the metabolically active 4-hydroxy-tamoxifen (4-OHT) in vitro for two days to mediate STAT5-deletion, followed by T cell activation with plate bound anti-CD3 and anti-CD28 under Th0, Th1, Th2, Th17 and Th25 polarizing conditions (Fig. 2B–C). STAT5-deletion had no effect on Th1 and Th17 polarization (Fig. 2C). Consistent with what has been previously reported, STAT5-deficient Th2 cells produced less IL-4 and IL-5 than STAT5-sufficient Th2 cells (7–8) (Fig. 2C). Similarly, STAT5-deficiency resulted in a defect in Th25 polarization with less expression of type 2 cytokines such as Il4, Il5, Il9, and Il13 than STAT5-sufficient Th25 cells (Fig. 2C–F). These findings suggest that IL-25-dependent Th2 polarization requires STAT5. We previously demonstrated that Act1 is required for Th25 polarization (13). Since IL-25 stimulation can lead to STAT5 activation independently of Act1, our data here suggests that in addition to Act1, STAT5 is required to fully mediate IL-25-dependent Th2 responses.
Figure 2. T cell-derived STAT5 is necessary for Th25 polarization.

A) Naïve CD4-positive T-cells were isolated from WT mice. Cells were incubated on CD3/CD28-coated plates in the indicated conditions. On Day 5, cells were fixed and P-STAT5 analyzed by flow cytometry. B) Naïve T cells isolated from Stat5f/f ERT2Cre or Stat5f/+ ERT2 Cre mice were treated with 10nM of 4-hydroxy-tamoxifen (4-OHT) for 2 days with 10ng/ml of IL-7 to mediate STAT5 deletion. Cell lysates were then separated on SDS-PAGE for immunoblot analysis. C) Naïve CD4+ T cells isolated from Stat5f/f ERT2Cre or Stat5f/+ ERT2 Cre mice were treated with 4-OHT for 2 days followed by T cell activation under the indicated polarizing conditions for 4 days. Cells were then collected for flow cytometry analysis for IL-4, IL-5, and IFN-γ producing cells. Flow graphs are gated on CD4+ T cells. D) Percent cytokine producing cells from (B) in Th25 conditions. Results shown are from 3 experiments. E) STAT5 WT or STAT5-deleted T cells from (B) were cultured on plate-bound anti-CD3 and anti-CD28 for 48hrs with or without IL-25 (100ng/ml). Cells were then collected for RNA isolation and RT-qPCR analysis for gene expression. Gene expression is graphed as fold induction over naïve CD4 T cells relative to Actin. F) ELISA was performed from supernatants from Th25 conditions shown in D. Data shown are representative results from 2 (A) or 3 independently performed experiments. Error bars represent mean ± S.E.M., ** or *** indicate p-vlalue ≤ 0.01 or 0.001, respectively.
Epithelial-derived STAT5 is required for response to IL-25
One important function of IL-25 in allergic inflammation is its ability to promote type 2 inflammation in various cells. We and others have reported that epithelial cells, T cells and innate lymphoid cells are highly responsive to IL-25 stimulation that results in the production of type 2 cytokines such as IL-4, IL-5, IL-13, and IL-9 (5–7, 10, 12, 13, 20). Given that the epithelium plays an essential role in promoting and potentially initiating allergic inflammation, we sought to determine whether STAT5 plays a role in IL-25-dependent effects in epithelial cells. We treated human airway epithelial cells (Bet1A cells) and mouse kidney epithelial cells (KECs) with IL-25 and found that while IL-25 induced modest phosphorylation of IkBα, the IL-25-STAT5 pathway is induced within 15 minutes of treatment in the epithelial cell compartment (Figure 3A–B). Adenovirus-Cre mediated deletion of STAT5 in Stat5f/f KECs indicated that STAT5 in epithelial cells is required for IL-25-dependent induction of type 2 cytokines (Figures 3C–E). While the expression of Il5, Il9, Il13, and Il25 were induced as early as 2hrs in Ad-Ctrl-infected cells, STAT5-deficient KECs exhibited an attenuated or delayed response to IL-25 (Figure 3E). Together, these data suggest a critical role for STAT5 in potentiating the IL-25 response in epithelial cells.
Figure 3. STAT5 is required for IL-25-dependent type 2 cytokines in epithelial cells.

A) Human airway Bet1A epithelial cell line were stimulated with IL-25 for the indicated times followed by western blot analysis for p-STAT5, p-ERK1/2, and actin. B) Kidney epithelial cells (KECs) isolated from WT mice were stimulated with IL-25 (100ng/ml) for the indicated times and immunoblotted for p-STAT5, STAT5, and actin. C) Stat5f/f KECs were infected with adenovirus expressing Cre (AdCre) or GFP (AdCtrl) to mediate STAT5 deletion in the epithelial cells. Lysates were immunobloted for STAT5 and Actin. D) Stat5f/f KECs infected with AdCre or AdCtrl as in C were then stimulated with IL-25 for 24 hours after which ELISA for IL-5 was performed. E) Stat5f/f KECs infected as in C were stimulated with IL-25 for the indicated times followed by RT-qPCR analysis for gene expression. Gene expression is graphed as fold induction over unstimulated cells relative to Actin. Data shown are representative results from 3 independently performed experiments. Error bars represent mean ± S.E.M.
IL-17RB recruits STAT5 through novel Tyrosine residues
STAT recruitment to a receptor requires the phosphorylation of specific tyrosine residues on the receptor to serve as docking sites for STAT proteins (18, 21). Within the intracellular domain of the IL-25-specific receptor subunit, IL-17RB, there are six tyrosine residues (Fig. 4A). Indeed, IL-25 stimulation led to a ligand-dependent association of IL-17RB with P-Tyr (Fig. 4B). Next, we sought to test whether the tyrosine residues within the IL-17RB were required for the STAT5 interaction. As an initial approach we mutated all of the tyrosines within the intracellular portion of IL-17RB to a structurally similar amino acid lacking the hydroxyl group, phenylalanine. While immunoprecipitation of wild-type IL-17RB resulted in the co-immunoprecipitation of STAT5, mutation of all six tyrosine residues to phenylalanine (IL-17RB all Tyr) resulted in a loss of interaction between IL-17RB with STAT5, suggesting that the tyrosine residues on IL-17RB are required for STAT5 binding (Figure 4C).
Figure 4. IL-17RB recruits STAT5 through tyrosine residues.
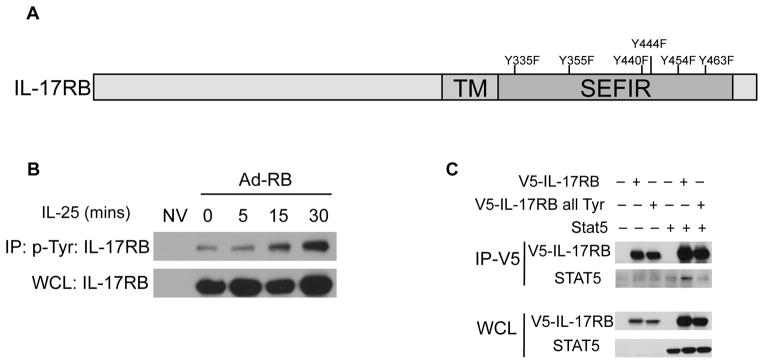
A) Schematic representation of IL-17RB with six tyrosine residues within the intracellular domain. B) KECs were left uninfected or infected with Adenovirus encoding IL-17RB followed by IL-25 stimulation. Cell lysates were incubated with anti-p-Tyr antibodies followed by immunoblot analysis. C) HEK293 cells were transfected with 1μg each of constructs expressing V5-tagged IL-17RB or V5-tagged IL-17RB all Tyr, and STAT5. Cell lysates were immunoprecipitated with anti-V5 antibody followed by western blotting analysis for STAT5 and V5. Co-immunoprecipitated products, top; Whole cell lysate (WCL), bottom. Results shown are representative data of 3 independently performed experiments.
Tyrosine residues Y444 and Y454 are critical for STAT5 recruitment
Next, we sought to determine which of the six tyrosine residues on IL-17RB is/are required for STAT5 interaction. To test this, individual tyrosine residues were mutated to phenylalanine. Immunoprecipitation of STAT5 resulted in the pull-down of WT IL-17RB, Y335F, Y355F, Y440F, and Y463F (Fig. 5A). However, mutating tyrosine residues 444 and 454 to phenylalanine resulted in a decreased interaction of IL-17RB with STAT5, suggesting that these tyrosine residues may be utilized for STAT5 binding.
Figure 5. IL-17RB tyrosines Y444 and Y454 are critical for STAT5 interaction and the IL-25 response.

A) HEK293 cells were transfected with constructs expressing STAT5, V5-IL-17RB, or V5-tagged IL-17RB individual tyrosine mutants. Cell lysates were then immunoprecipitated with anti-STAT5 antibodies and subjected to western blot analysis for V5 or IL-17RB expression. Co-immunoprecipitates, top; Whole cell lysate (WCL), bottom. B) Il17rb −/− KECs were left uninfected or infected with adenovirus encoding IL-17RB or IL-17RB Y444F or Y454F followed by stimulation with IL-25. Cell lysates were incubated with anti-P-Tyr antibodies followed by western blotting analysis for IL-17RB. C) Il17rb −/− KECs were infected with adenovirus encoding IL-17RB or IL-17RB Y444F or Y454F then stimulated with IL-25, followed by western blot analysis. D) Il17rb −/− KECs were infected with adenovirus encoding IL-17RB or IL-17RB Y444F and Y454F then stimulated with IL-25 for the indicated times, followed by RT-qPCR analysis for gene expression. Data shown are representative results from 3 independently performed experiments. Error bars represent mean ± S.E.M.
Database searches revealed that IL-17RB phosphorylation has been observed in mass spectrometry analysis (Phosphosite Plus). Thus we generated adenoviruses expressing either WT IL-17RB or IL-17RB Y444F or Y454F and infected Il-17rb −/− KECs. Cells were then stimulated with IL-25 followed by immunoprecipitation for p-Tyr. While mutation of the Y454F attenuated the IL-25-induced IL-17RB association with tyrosine phosphorylation, the difference with Y444F was more modest (Fig. 5B). However, mutation of either tyrosine resulted in diminished IL-25-dependent STAT5 activation (Fig. 5C). Furthermore, IL-25-dependent expression of Il5 was reduced in IL-17RB Y444F or Y454F-reconstituted KECs compared to IL-17RB WT-reconstituted cells (Fig. 5D). Together these results indicate that STAT5 recruitment through tyrosine residue 444 and 454 of the IL-17RB subunit is essential to fully mediate IL-25-induced cytokine expression (Fig. 6).
Figure 6. Model of IL-25-induced STAT5 pathway activation.
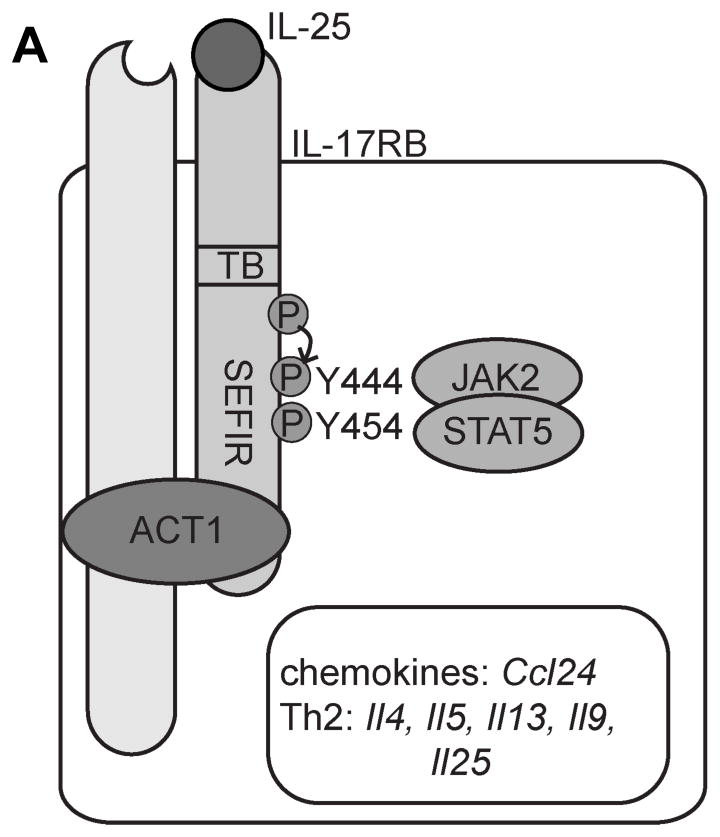
IL-25 signals through IL-17RA and IL-17RB. Upon ligand stimulation STAT5 is recruited to two tyrosine residues (indicated) and then mediates downstream Th2-cytokine expression.
Discussion
Here we demonstrated an Act1-independent pathway through STAT5 by which IL-25 can promote Th2 responses. Our observations revealed that in addition to Act1, IL-25 stimulation can lead to the recruitment of STAT5 through tyrosine 444 and 454 of IL-17RB, which are required to mediate IL-25-dependent type 2 cytokine expression. Ablation of either of these two independent pathways (by Act1-deletion or by STAT5-deletion) can result in the loss of IL-25 responses suggesting that both pathways are required to fully mediate IL-25-dependent expression of type 2 cytokines (10, 12). Given that STAT5 activation can promote Th2 responses by increasing Th2 gene accessibility during T cell differentiation, IL-25-dependent STAT5 phosphorylation likely functions similarly to increase Th2 gene accessibility (15, 22). Thus, Act1 and STAT5 may act synergistically to mediate IL-25-dependent type-2 cytokine expression.
IL-25 is the most structurally and biologically divergent member of the IL-17 cytokine family. Comparing to the other IL-17 cytokine family members, the mechanisms of the IL-25–unique capacity to promote Th2 responses are likely attributable to two critical factors; cell-type specific expression of IL-17RB and unique molecular signaling events. Numerous studies have demonstrated that epithelial, T-cells, monocytes, MPP-type 2 cells and innate lymphoid cells (ILC2) are critical responders to IL-25 (7, 10, 12, 13, 20, 23). As demonstrated in murine models of allergic inflammatory diseases, IL-25-mediated Type-2 responses are heavily reliant on the temporal and spatial orientation of the IL-17RB+ cellular milieu. In spite of these findings one remaining question has been what molecular components are involved in an IL-17RB response, especially in cellular targets with diverse IL-17R-expression, namely epithelial and T-cells.
Here we found that IL-25 stimulation can lead to activation of STAT5 in T-cells as well as in epithelial cells. It is important to note that IL-25 did not activate STAT3 or STAT6 (data not shown). Furthermore, P-STAT5-positive T-cells appear to be the main IL-5-producing cells following IL-25 treatment (unpublished observations). Importantly, this activation event was independent of Act1 since Act1-deficient cells exhibited robust P-STAT5 expression in response to IL-25. STAT5 has two isoforms A and B, whether one or both are sensitive to IL-25 stimulation will be an interesting line for future studies. Since Act1 has been shown to be a key component in the IL-25 signaling cascade, together these data suggest that Act1 and STAT5 are both required for an effective IL-25 response. STAT5 has been shown to be required for the expression of type-2 associated cytokines including Il-4, Il-9 and Il-13. Mechanistically, STAT5 has been shown to facilitate GATA3 promoter access to these genes (15, 22). One possibility is that Act1 and STAT5 may also cooperate to regulate Th2-associated cytokine expression. Further experimentation would be needed to elucidate the exact mechanism(s).
Since IL-25 activates STAT5 independently of Act1, these data suggested to us that IL-25-dependent STAT5 activation may be instigated at the level of the receptor. Indeed co-immunoprecipitation of STAT5 with IL-17RB was observed in a ligand-dependent manner. Furthermore, six tyrosines reside within the intracellular portion of IL-17RB. With IL-25 stimulation we observed an increased association of IL-17RB co-immunoprecipitated with P-Tyr specific antibody. Specific deletion of tyrosine residues Y444 and Y454 to phenylalanine abrogated the STAT5 interaction as well as IL-25-induced type-2 cytokine expression. Interestingly the IL-17RB/P-Tyr association was different between the Y444F or Y454F mutant, suggesting that the activation on each tyrosine may be temporally regulated. Whether each site has any overlapping consequences in addition to STAT5 activation is an intriguing line for future studies. It is also important to note that these two tyrosine residues are not present in the IL-17A specific receptor subunit, IL-17RC. Moreover, we also identified JAK2 as a possible kinase linking IL-17RB to STAT5. While these data imply that Y444 and Y454 are critical residues for IL-17RB-dependent STAT5 recruitment, mass spectrometry would be needed to confirm whether these amino acids are phosphorylated following IL-25 stimulation.
While much progress has been made in identifying signaling components activated by IL-17A, very little has been known for IL-25. In a separate report, our group has also identified a negative regulatory network specific for the IL-17RB subunit by a novel P-Tyr binding protein, Dazap2. Notably this negative regulation impairs Act1 and STAT5 recruitment and is immediately removed following ligand stimulation. Thus unlike for IL-17A, the IL-25 receptor is under much stricter control. Taking into consideration our present observations that IL-25 utilizes the STAT5 pathway; it is obvious that the molecular diversity of IL-25 activated signaling pathways imparts its Th2-promoting activity but it also offers possible therapeutic targets in Th2-elicited maladies.
Acknowledgments
This work was supported by NIH grants R01-NS071996 (NINDS) and P01-HL103453 (NHLBI) to X.L.
References
- 1.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, Muchamuel T, Hurst SD, Zurawski G, Leach MW, Gorman DM, Rennick DM. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 2.Pan G, French D, Mao W, Maruoka M, Risser P, Lee J, Foster J, Aggarwal S, Nicholes K, Guillet S, Schow P, Gurney AL. Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. Journal of immunology. 2001;167:6559–6567. doi: 10.4049/jimmunol.167.11.6559. [DOI] [PubMed] [Google Scholar]
- 3.Tamachi T, Maezawa Y, Ikeda K, Kagami S, Hatano M, Seto Y, Suto A, Suzuki K, Watanabe N, Saito Y, Tokuhisa T, Iwamoto I, Nakajima H. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. The Journal of allergy and clinical immunology. 2006;118:606–614. doi: 10.1016/j.jaci.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 4.Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, Liu YJ, Zhu Z, Dong C. Interleukin 25 promotes the initiation of proallergic type 2 responses. The Journal of experimental medicine. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, Yao Z, Ying S, Huston DP, Liu YJ. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. The Journal of experimental medicine. 2007;204:1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang Z, Swaidani S, Yin W, Wang C, Barlow JL, Gulen MF, Bulek K, Do JS, Aronica M, McKenzie AN, Min B, Li X. Epithelial cell-specific Act1 adaptor mediates interleukin-25-dependent helminth expulsion through expansion of Lin(−)c-Kit(+) innate cell population. Immunity. 2012;36:821–833. doi: 10.1016/j.immuni.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF, Jr, Tocker JE, Budelsky AL, Kleinschek MA, Kastelein RA, Kambayashi T, Bhandoola A, Artis D. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464:1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends in biochemical sciences. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- 9.Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, Anders PM, Tocker JE, Comeau MR, Budelsky AL. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. Journal of immunology. 2008;181:4299–4310. doi: 10.4049/jimmunol.181.6.4299. [DOI] [PubMed] [Google Scholar]
- 10.Swaidani S, Bulek K, Kang Z, Liu C, Lu Y, Yin W, Aronica M, Li X. The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. Journal of immunology. 2009;182:1631–1640. doi: 10.4049/jimmunol.182.3.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu C, Swaidani S, Qian W, Kang Z, Sun P, Han Y, Wang C, Gulen MF, Yin W, Zhang C, Fox PL, Aronica M, Hamilton TA, Misra S, Deng J, Li X. A CC’ loop decoy peptide blocks the interaction between Act1 and IL-17RA to attenuate IL-17- and IL-25-induced inflammation. Science signaling. 2011;4:ra72. doi: 10.1126/scisignal.2001843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, Chen A, Ren NY, Wang H, Siebenlist U. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. Journal of immunology. 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swaidani S, Bulek K, Kang Z, Gulen MF, Liu C, Yin W, Abbadi A, Aronica M, Li X. T cell-derived Act1 is necessary for IL-25-mediated Th2 responses and allergic airway inflammation. Journal of immunology. 2011;187:3155–3164. doi: 10.4049/jimmunol.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong CK, Cheung PF, Ip WK, Lam CW. Interleukin-25-induced chemokines and interleukin-6 release from eosinophils is mediated by p38 mitogen-activated protein kinase, c-Jun N-terminal kinase, and nuclear factor-kappaB. American journal of respiratory cell and molecular biology. 2005;33:186–194. doi: 10.1165/rcmb.2005-0034OC. [DOI] [PubMed] [Google Scholar]
- 15.Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–748. doi: 10.1016/s1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
- 16.Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, Hennighausen L, O’Shea JJ. Stat5a/b are essential for normal lymphoid development and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Molecular and cellular biology. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flores-Morales A, Pircher TJ, Silvennoinen O, Gustafsson JA, Sanchez-Gomez M, Norstedt G, Haldosen LA, Wood TJ. In vitro interaction between STAT 5 and JAK 2; dependence upon phosphorylation status of STAT 5 and JAK 2. Molecular and cellular endocrinology. 1998;138:1–10. doi: 10.1016/s0303-7207(98)00054-9. [DOI] [PubMed] [Google Scholar]
- 19.Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, Zhu J, Paul WE. Interleukin 2 plays a central role in Th2 differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie AN. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell research. 2006;16:3–10. doi: 10.1038/sj.cr.7310002. [DOI] [PubMed] [Google Scholar]
- 23.Dolgachev V, Petersen BC, Budelsky AL, Berlin AA, Lukacs NW. Pulmonary IL-17E (IL-25) production and IL-17RB+ myeloid cell-derived Th2 cytokine production are dependent upon stem cell factor-induced responses during chronic allergic pulmonary disease. Journal of immunology. 2009;183:5705–5715. doi: 10.4049/jimmunol.0901666. [DOI] [PubMed] [Google Scholar]