

Introduction
TRPV1 is a non-selective nociceptive cationic channel that can be activated by polymodal stimuli such as capsaicin, proton, and noxious heat [10]. TRPV1 is implicated in thermal hyperalgesia under inflammation and tissue injury, and has been identified as a reliable target for anti-hyperalgesic therapy [10; 17]. Besides its well-known function in thermal hyperalgesia in skin, TRPV1 functions as a signal integrator such that multiple nociceptive signals converge onto TRPV1 in nociceptor [24]. Under pathophysiological conditions, multiple inflammatory mediators activate multiple kinases that phosphorylate TRPV1 and enhance its functionality. The phosphorylation-induced upregulation of TRPV1 functions appears to underpin pathological nociception.
Among the protein kinases, PKC is a major player in TRPV1 sensitization [10; 17; 24]. PKC-induced TRPV1 phosphorylation enhances responses to capsaicin, acid, and heat [42]. Previous studies identified three major PKC-phosphorylation residues in rat TRPV1: S502, T704, and S800 [2; 32]. These studies agreed that two serine residues, S502 and S800, are involved in sensitization of capsaicin-evoked responses induced by phorbol myristate acetate (PMA), an agonist of PKC. In contrast, T704 is involved in direct activation of TRPV1 by PMA or basal thermal heat sensitivity rather than hypersensitivity to capsaicin [2; 25]. However, it is uncertain which residues contribute to hypersensitivity to other natural or endogenous-stimuli, such as heat or acid. Understanding the modality-specific basis of TRPV1 sensitization is important since altered ambient temperature and acidity are highly likely to affect TRPV1 under pathological conditions. It has been suggested that capsaicin, heat, and acid activate TRPV1 through distinct structural bases [1; 16; 19; 44], and it is likely that phosphorylation of different combinations of residues are involved in sensitization to different modalities of agonistic stimuli.
Since TRPV1 phosphorylation is a critical mechanism underlying pathological functions of TRPV1, the phosphorylatable residues of TRPV1 may be appropriate targets for antihyperalgesic therapy. However, the contributions of phosphorylation sites to TRPV1 hypersensitivity have been determined only in heterologous systems but not in neuronal context. Given that interactions of TRPV1 with factors specific to sensory neurons or expression of different subtypes of protein kinases in sensory neurons could involve different mechanisms of sensitization of TRPV1 [7; 18; 37], it is important to evaluate the structural contribution of each phosphorylation site in more physiologically relevant contexts. In this study, we first investigated common and distinct structural bases of PKC-induced hypersensitivity to capsaicin, proton, and heat by mutagenic and electrophysiological approaches. Second, we examined the contribution of three PKC-mediated phosphorylation sites to PMA or bradykinin-induced hypersensitivity to capsaicin in sensory neurons. We demonstrated that PKC-induced phosphorylation of TRPV1 functionally enhances sensitivity to different agonists through distinct structural bases and that TRPV1 S800 is a polymodal sensitization residue.
2. Methods
2.1. Cell culture and transfection
Human embryonic kidney (HEK) 293 cells were cultured and transfected using lipofectamine 2000 (Invitrogen) as previously described [20]. Plasmids containing cDNA encoding rat TRPV1 or TRPV1 mutants were co-transfected with cDNA encoding mCherry or green fluorescence protein (GFP). Transiently transfected HEK293 cells were re-plated onto poly-L-ornithine-coated coverslips, kept at 32°C, and used for electrophysiological experiments after 16–26 hours.
2.2. Site-directed mutagenesis
A pcDNA3 vector containing cDNA encoding rat TRPV1 [6] was used for site-directed mutagenesis [20]. Proper mutation of each construct and lack of unintended mutation was confirmed by sequencing.
2.3. Dissociation of mouse sensory neurons and electroporation
All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were performed under a University of Maryland-approved Institutional Animal Care and Use Committee protocol. TRPV1 null mutant mice [5] were purchased from Jackson laboratory. Mice (4–9 weeks old) were anesthetized using a cocktail of ketamine (80 mg/kg) and xylazine (10 mg/kg). Dorsal root ganglia were dissected out and collected in cold Puck’s saline (171 mM NaCl, 6.7 mM KCl, 1.4 mM Na2HPO4, 0.5 mM KH2PO4, 6.0 mM glucose, pH 7.3). The ganglia were incubated in 5 ml DMEM/F12 medium containing collagenase type IV (1 mg/ml) and incubated at 37°C for 30 min. The ganglia were incubated for an additional 15 min following the addition of trypsin (0.25%) and EDTA (0.025%). The tissues were triturated with flame-polished Pasteur pipettes. The Plasmids containing cDNA encoding TRPV1 or TRPV1 mutants were co-transfected with cDNA encoding GFP (GFP in pMax, Lonza) using an electroporator (Nucleofector 4D, X-axis; Lonza) as described [12] with slight modifications. The neurons were plated onto glass coverslips (8-mm) coated with poly-ornithine and laminin. Dissociated neurons were maintained with DMEM/F12 containing 10% FBS, 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator. Electrophysiological recordings or Ca2+ imaging were performed after 2–3 days.
2.4. Whole-cell patch clamp
Whole-cell voltage clamp techniques were performed as described previously [11; 43]. The recording pipettes (2–3 MΩ) were pulled from borosilicate glass using a P-97 (Sutter Instrument). In analyses of HEK293 cells, the pipettes were filled with internal solution (150 mM NaOH, 1 mM MgCl2, 10 mM HEPES, 5 mM EGTA, pH 7.4). Unless otherwise indicated, the recording bath contained an external solution (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4). When we used an acid as an agonist, the external solution contained 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) instead of HEPES. To suppress native acid-evoked currents, all acidic external solutions contained 100 µM amiloride [36]. In analyses of sensory neurons, the pipette was filled with a solution containing 140 mM KCl, 5 mM NaCl, 1 CaCl2, 1 MgCl2, 2.5 Mg-ATP, 10 mM EGTA, and 10 mM HEPES (pH 7.3). Osmolarity of each solution was measured by a vapor pressure osmometer (Wescor Inc), and was adjusted with mannitol to 290 to 310 mOsm as necessary. Unless otherwise indicated, all recordings were performed at room temperature. For evaluating heat activation, we applied a temperature ramp ranging from approximately 18°C to 47°C over approximately 30 sec. The actual temperature was recorded throughout the experiment with a thermocouple (Physitemp Instruments) placed within 4 mm of the patch-clamped cell. Bath temperature was controlled using a combination of an in-line heater/cooler (Warner Instrument Inc) and in-line heater (Warner Instrument Inc).
2.5. Ratiometric Ca2+ imaging
Ratiometric Ca2+ imaging analysis was performed as described previously [23]. Primary TG cultures were loaded with Fura2-AM for 40 minutes at 37°C in a calcium imaging buffer (CIB) containing NaCl 130, KCl 3, 0.5 MgCl2, CaCl2 0.9, HEPES 10, sucrose 10, NaHCO3 1.2 (in mM, pH 7.45, 320 mOsm adjusted with mannitol). Ratiometric Ca2_ imaging was performed using an inverted fluorescence microscope (Nikon Instrument), excitation filter changer (Sutter Instrument), and cooled CCD camera (Nikon Instrument). .After a 15-minute wash period for de-esterification, dual images (510-nm emission) were collected every 2 seconds using NIS Elements (Nikon Instrument). The Fura response was defined as the ratio of emissions measured during excitation at 340 and 380 nm, and the relative Fura response was defined as the ratio normalized to the lowest Fura response upon cooling in each neuron, For assessing heat activation, bath temperature was elevated from approximately 16 to 48 °C slowly over 70 seconds. Temperature was recorded by using thermocouple connected with Thermes USB (Physitemp Instruments). To define TRPV1-dependent heat-evoked responses, we initially assessed heat-induced changes of Fura ratio in TRPV1 null DRG neurons transfected with vector control. For determination of a response level, a relative Fura response histogram was generated in the range of 40 to 45°C. The response level was defined as the level three standard deviation above the mean of the nonresponsive peak, which was 1.61 (61% increase in Fura response relative to the baseline ratio). With this cutoff, vector transfected TRPV1 null DRG neurons showed positive responses only in 3 % of neurons (5/165). Since the TRPV1-independent heat response was negligible, we considered entire heat responses in TRPV1-transfected neurons as TRPV1-dependent responses. Therefore, neurons showing a change greater than the pre-determined response level were further analyzed to obtain threshold temperature of activation and relative response at 37°C.
Data are expressed as the mean±SEM. Statistical comparisons were made as indicated in each figure legend. The level of statistical significance was set at α=0.05.
Results
3.1. TRPV1 T704 and S800, but not S502, contribute to PKC-induced hypersensitivity to heat in HEK293 cells
The functional contribution of phosphorylation sites has been evaluated by ablating the candidate phosphorylation site by mutation to a non-phosphorylatable amino acid such as alanine [2; 32]. Alanine mutation of TRPV1 S502 and S800 individually or in combination attenuates PKC-mediated hypersensitivity of TRPV1 to capsaicin [2; 32]. Although simultaneous alanine mutation of S502 and S800 was demonstrated to attenuate PMA-induced sensitization to heat, the contribution of the individual residue is not known [32]. To examine the contribution of each PKC-phosphorylation residue to the hypersensitivity of TRPV1 to heat, we tested TRPV1 mutants in which S502, T704, or S800 was individually mutated to alanine. The effects of mutation of PKC-phosphorylation residues on induction of hypersensitivity to heat were examined by heterologous transfection in HEK293 cells. Thermal activation was assessed using a temperature ramp. Cells were pretreated with vehicle (0.01% ethanol) or PMA, a PKC activator, for two minutes. To avoid false negative results, we used a high concentration of PMA (1 µM). Following pretreatment with vehicle, current amplitude increased at around 40°C during the temperature ramp (Fig. 1A). In contrast, following pretreatment with PMA, current amplitude increased at approximately 36°C. To compare heat activation, we quantified relative current at 31°C, representing skin surface temperature (Fig. 1F), and relative current at 39°C, representing a subthreshold heat stimulus (Fig. 1G). In vehicle treated TRPV1 wildtype (WT) cells, relative currents at 31°C and 39°C were approximately 2% and 8% of maximal heat currents, respectively. In PMA treated TRPV1 WT cells relative currents at 31°C and 39°C were approximately 10% and 40%, respectively, which were significantly greater than those in vehicle treated cells (Fig. 1F, G). These results are consistent with previous reports regarding PMA-induced hypersensitivity of TRPV1 to heat [32; 42]. Alanine mutation of TRPV1 S502 displayed almost identical heat activation and sensitization as WT following pretreatment with vehicle or PMA (Fig. 1B, E–G), suggesting that TRPV1 S502 is not involved in PMA-induced hypersensitivity to heat. In contrast, alanine mutation of TRPV1 T704 showed no difference in heat activation between vehicle and PMA treated cells (Fig. 1C, E–G). Alanine mutation of TRPV1 S800 significantly decreased heat activation following PMA pretreatment, but the extent of decrease was not as robust as WT (Fig. 1D, E). Relative currents were significantly increased at 39°C but not at 31°C (Fig. 1F, G), suggesting partial impairment of PMA-induced hypersensitivity to heat in TRPV1 S800A. These results indicate that TRPV1 T704 and S800, but not S502, contribute to PKC-induced hypersensitivity to heat.
Fig. 1. The roles of phosphorylation sites in PKC-induced hypersensitivity of TRPV1 to heat in HEK293 cells.
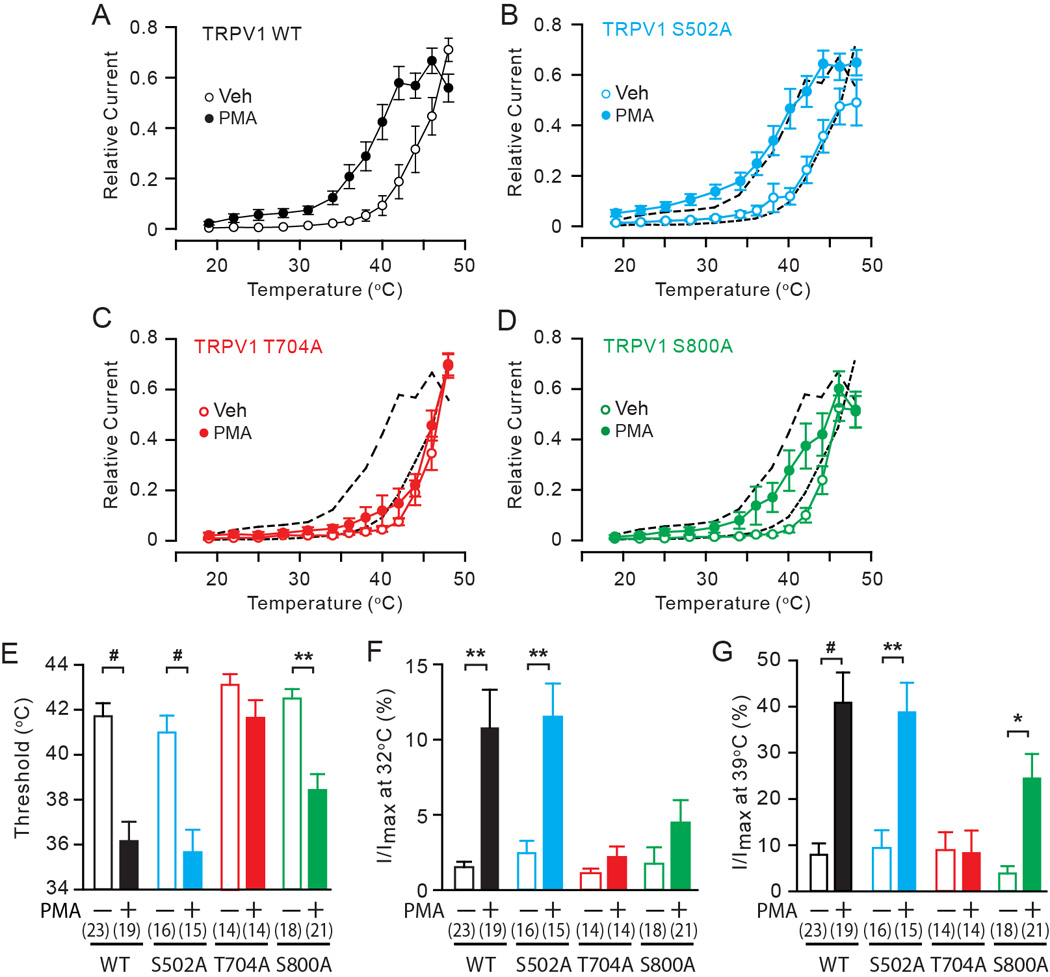
A–D. Averaged relative current density evoked by a temperature ramp from 17 to 48 °C following the exposure to either vehicle (white) or 1 µM PMA (black) in HEK 293 cells transfected by TRPV1 WT (A), S502A (B), T704A (C), or S800A (D). Current amplitudes measured at −50 mV at different temperatures were normalized to the maximal amplitude in each cell. Black dotted lines in B–D represent values of WT.
E–G. Quantification of heat-evoked currents in TRPV1 WT and mutants. Threshold temperature of activation (E), relative currents at 32°C (F) and 39°C (G) were compared between vehicle-treated (open bars) and PMA-treated (filled bars) cells as indicated. Relative currents at 31°C or 39°C were calculated by averaging current amplitudes measured between 29°C to 34°C or 38°C to 40°C in each cell and normalizing to the respective maximal current amplitude in the given cell. Numbers within parenthesis represent the numbers of observations. *p<0.05; **p<10−3; #p<10−4; Student’s t-test.
3.2. PKC-induced hypersensitivity to acid occurs mainly through S800 in HEK293 cells
To quantify PKC-induced hypersensitivity to acid, we applied a submaximal concentration of acid (pH 6.5) followed by a high concentration of acid (pH 5.5) that induces apparent maximal responses with one minute interval. Cells were pretreated with vehicle or PMA (1 µM) for two minutes before the initial application of proton and throughout the interval between acid applications (Fig. 2A). In vehicle-treated cells, TRPV1 WT, S502A, and S800A showed modest acid evoked responses at pH 6.5 and much greater responses at pH 5.5 (Fig. 2B, white bars). In PMA-treated cells, the application of pH 6.5 evoked significantly greater responses than vehicle-treated cells in TRPV1 WT and S502A, but not in S800A (Fig. 2B left). However, the response evoked by pH 5.5 was not significantly different between vehicle and PMA-treated cells (Fig. 2B, right) suggesting that PMA does not functionally sensitize the response to pH 5.5. Thus, the pH 5.5-evoked responses may be considered to be maximal responses. We normalized the pH 6.5-evoked responses to the average pH 5.5 responses in each group to calculate the extent of sensitization (Fig. 2C). Responses to pH 6.5 in TRPV1 WT and S502 were only 5 to 10% of the maximal proton responses of each group in vehicle-treated cells, but were approximately 40% of maximal responses in PMA-treated cells. In contrast, PMA failed to induce significant sensitization of the TRPV1 S800A mutant (Fig. 2C). Unlike TRPV1 WT and other mutants, TRPV1 T704A exhibited little response to pH 6.5 in both vehicle-treated and PMA treated cells (−3.4±2.0 pA/pF in veh, n=7; −4.1±1.8 pA/pF in PMA, n=8). Vehicle-treated TRPV1 T704A cells were robustly activated by pH 5.5 and PMA-treated TRPV1 T704A cells were even more highly activated (−462±138 pA/pF in vehicle, n=7; −1022.3±113 pA/pF in PMA, n=8; p<0.01 in Student’s t-test). These results suggested that sensitivity of TRPV1 T704A to acid is apparently weaker than in WT, and indicated that the response did not reach a maximum at pH 5.5. Therefore, we tested a set of stronger acids, pH 6 followed by pH 5 (Fig. 2D). In vehicle-treated cells, TRPV1 T704A was weakly activated by pH 6, and the response was stronger by pH 5. In PMA-treated cells, pH 6 evoked significantly enhanced responses compared to vehicle-treated cells. However, the response to pH 5 was also greater in PMA-treated cells than in vehicle-treated cells (Fig. 2F). Since TRPV1 T704A showed comparable responses to 30 µM capsaicin in both vehicle and PMA-treated cells (Fig. 2E, F), we normalized the proton responses to the average response evoked by capsaicin in each group. In vehicle-treated TRPV1 T704A cells, responses evoked by two submaximal acid conditions, pH 6 and pH 5, were approximately 1% and 13% of maximal capsaicin-induced responses, respectively. In PMA-treated cells, responses evoked were 6% and 40% of maximal capsaicin induced responses (Fig. 2G). These results suggest that in HEK293 cells PKC-induced hypersensitivity to acid occurs mainly through S800, but not through S502 and T704.
Fig. 2. The roles of phosphorylation sites in PKC-induced hypersensitivity of TRPV1 to acid in HEK293 cells.
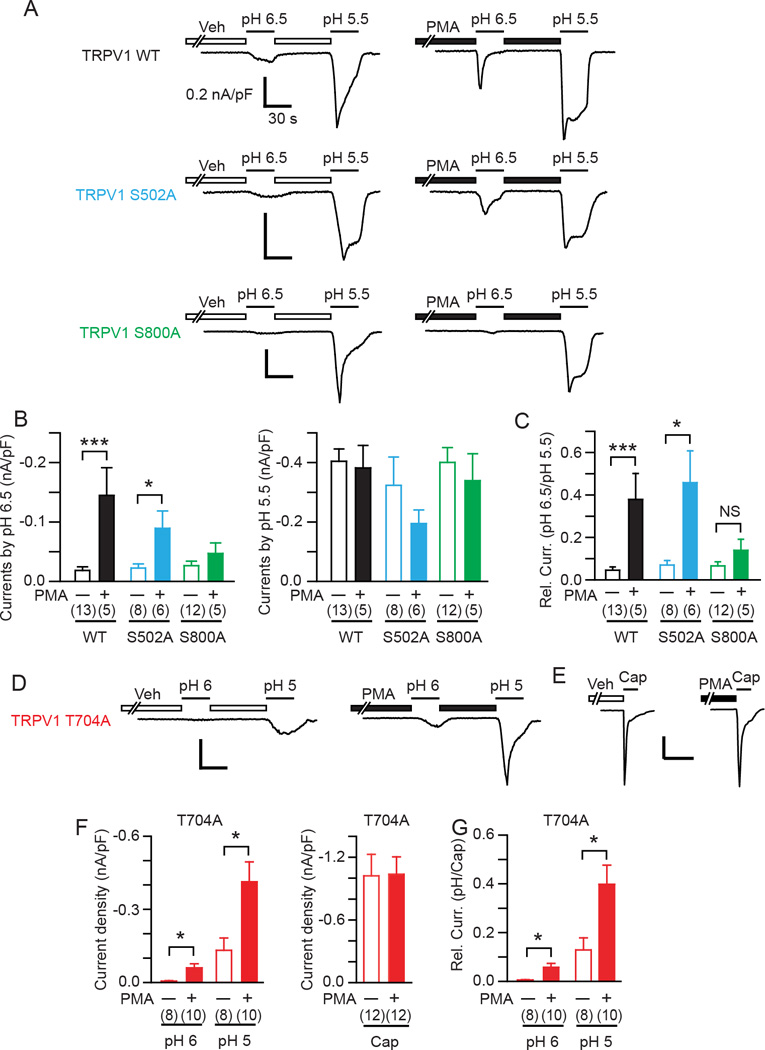
A. Representative acid-evoked current traces evoked by a first application of pH 6.5 and a following application of pH 5.5 in HEK293 cells transfected by TRPV1 WT (top), S502A (middle), or S800A (bottom). Cells were pretreated with PMA (1 µM) or vehicle (ethanol) as indicated. Current amplitudes were evaluated at −60 mV. Scale bar, 0.2 nA/pF-30 sec.
B. Current density evoked by a first application of pH 6.5 (left panel) and a following application of pH 5.5 (right panel). Numbers in parenthesis represent numbers of observations. ***p<0.001; *p<0.05; unpaired Student’s t-test.
C. Relative current quantified by the current density at pH 6.5 normalized to the average current density evoked by pH 5.5 in each group. NS, not significant.
D. Representative acid-evoked current traces evoked by a first application of pH 6 and a following application of pH 5 in HEK293 cells transfected by TRPV1 T704A (bottom). Cells were pretreated with PMA (1 µM) or vehicle (ethanol) as indicated. Current amplitudes were evaluated at −60 mV. Scale bar, 0.2 nA/pF-30 sec.
E. Representative current traces evoked by 30 µM capsaicin following pretreatment with vehicle or PMA in TRPV1 T704A. Scale bar, 2 nA-1 min.
F. Current density evoked by the consecutive application of pH 6 and pH 5 (left panel) or 30 µM capsaicin (right panel) in HEK293 cells transfected by TRPV1 T704A following pretreatment with vehicle or PMA.
G. Relative current quantified by the current density at pH 6 or pH 5 normalized to the average current density evoked by capsaicin in each group.
3.3. TRPV1 S502 and S800, but not T704, contributes to PKC-induced hypersensitivity to capsaicin in sensory neurons
It is known that sensitization of TRPV1 is regulated by multiple environmental factors such as A-kinase anchoring protein 75/150 (AKAP75/150) [18; 37; 45] and specific subtypes of protein kinase such as protein kinase Cε [7; 28]. Although it is known that PKC-induced hypersensitivity of TRPV1 to capsaicin is mediated by the phosphorylation of two residues, S502 and S800, in heterologous TRPV1 expression systems [2; 32], it is not clear whether these residues also contribute to PKC-mediated sensitization in neurons. To address this, we examined phenotypes of TRPV1 WT and alanine mutants lacking individual phosphorylation sites in sensory neurons. To avoid interference from native TRPV1, we expressed TRPV1 WT and alanine mutants in DRG neurons from TRPV1 null mutant mice. The cDNA encoding TRPV1 WT and alanine mutants were transfected by optimized electroporation methods. To quantify the extent of contribution of each phosphorylation residue to hypersensitivity to capsaicin, we followed protocols that have been established previously [2; 11; 32]. Two consecutive applications of a submaximal concentration of capsaicin (100 nM) were applied. The second application was preceded by a two minute treatment with vehicle or PMA. In vehicle-treated cells, TRPV1 WT and all mutants tested showed modest activation by the initial application of 100 nM capsaicin (Fig. 3A, B). Depending on mutational status of TRPV1, the second application either maintained a response similar to the initial response (WT, S502A) or exhibited desensitization (T704A, S800A). In PMA-treated cells, TRPV1 WT and T704A exhibited a robust increase in the ratio of the 2nd/1st response, indicating that exogenously activated TRPV1 was sensitized by the activation of endogenous PKC in sensory neurons. TRPV1 S502A exhibited a marginal response to capsaicin following PMA treatment. Although pretreatment with PMA increased the ratio of 2nd/1st response by approximately 2.5 fold, the increase was not significantly different from vehicle treated cells (p=0.06 in Student’s t-test), suggesting that TRPV1 S502A is not a primary contributor to PMA-induced hypersensitivity of TRPV1 to capsaicin. In TRPV1 S800A, pretreatment with PMA increased the ratio of 2nd/1st response only 1.3 fold, which was not different from vehicle treated cells, suggesting that phosphorylation of S800 is important in PMA-induced sensitization of TRPV1 to capsaicin in sensory neurons.
Fig. 3. The roles of phosphorylation sites in PKC-induced hypersensitivity of TRPV1 to capsaicin in sensory neurons.
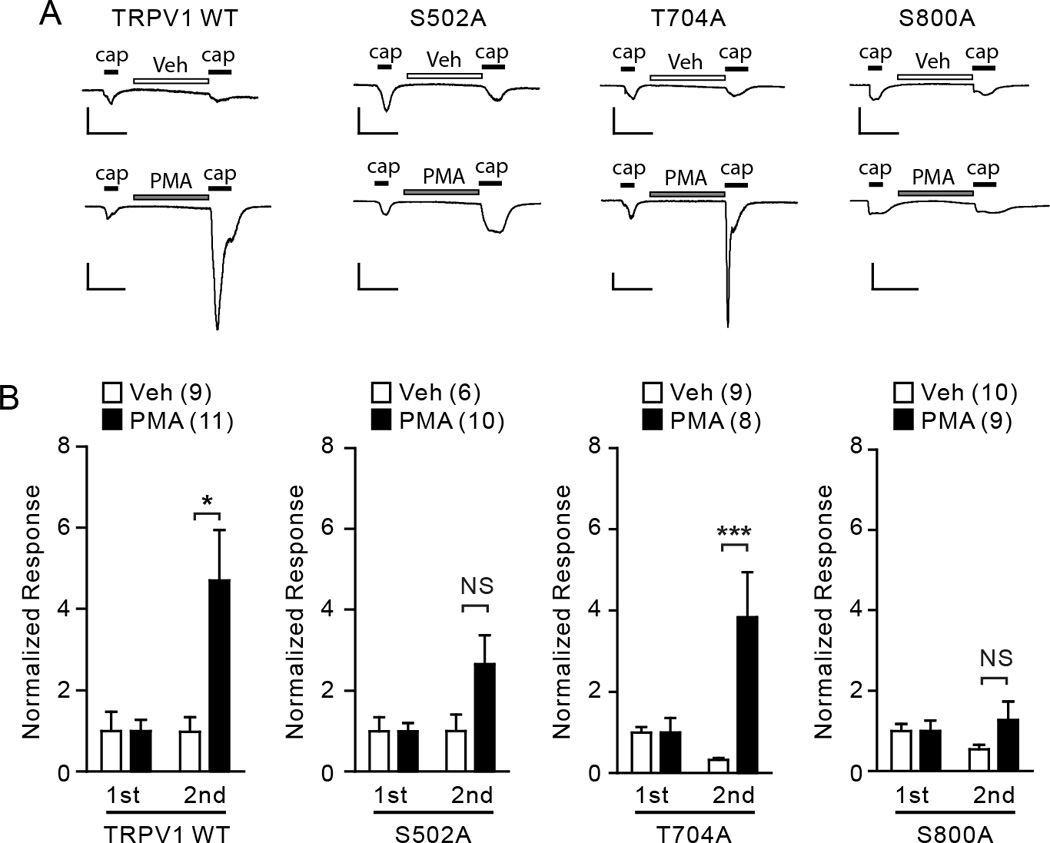
A. Representative current traces evoked by consecutive application of capsaicin (0.1 µM, black bars) following pretreatment with vehicle (ethanol; upper) or PMA (0.3 µM; lower) in DRG neurons dissociated from TRPV1 null mice. Neurons were transfected by electroporation with cDNA encoding TRPV1 WT, TRPV1 S502A, T704A, or S800A as indicated. Current amplitudes were evaluated at −60 mV. Scale bars, 50 pA/pF – 30 sec.
B. Averaged normalized responses. Current density of the 1st and 2nd response from each neuron was normalized to the average of the 1st response in the given group. NS, not significant; *p<0.05; ***p<0.005; Student’s t-test. Values in parentheses represent the number of observations in each group.
3.4. TRPV1 S800 primarily contributes to PKC-induced hypersensitivity to acid in sensory neurons
To further examine modality-dependent contribution of phosphorylation sites to PKC-induced hypersensitivity in sensory neurons, we tested the extent of contribution of each phosphorylation residue to hypersensitivity to acid. Two consecutive applications of a submaximal concentration of acid (pH 6.5 in WT, S502A, S800A; pH 6 in T704A) were applied. The second application was preceded by treatment with vehicle or PMA for two minutes. In vehicle-treated cells, TRPV1 WT and all mutants tested showed modest activation by the initial application of weak acid and substantially reduced response by the second application (Fig. 4A, B). In PMA-treated cells, TRPV1 WT and T704A exhibited a robust increase in the ratio of the 2nd/1st response, indicating that acid potentiates responses of TRPV1 in sensory neurons. TRPV1 S502A also exhibited significant sensitization effects to acid following PMA treatment although the extent of sensitization was slightly lower than WT. In contrast, the ratio of 2nd/1st response was approximately 1 following PMA treatment in TRPV1 S800A, which was not different from vehicle treated cells. These results suggest that phosphorylation of S800 is important in PMA-induced sensitization of TRPV1 to acid as well as capsaicin in sensory neurons.
Fig. 4. The roles of phosphorylation sites in PKC-induced hypersensitivity of TRPV1 to acid in sensory neurons.
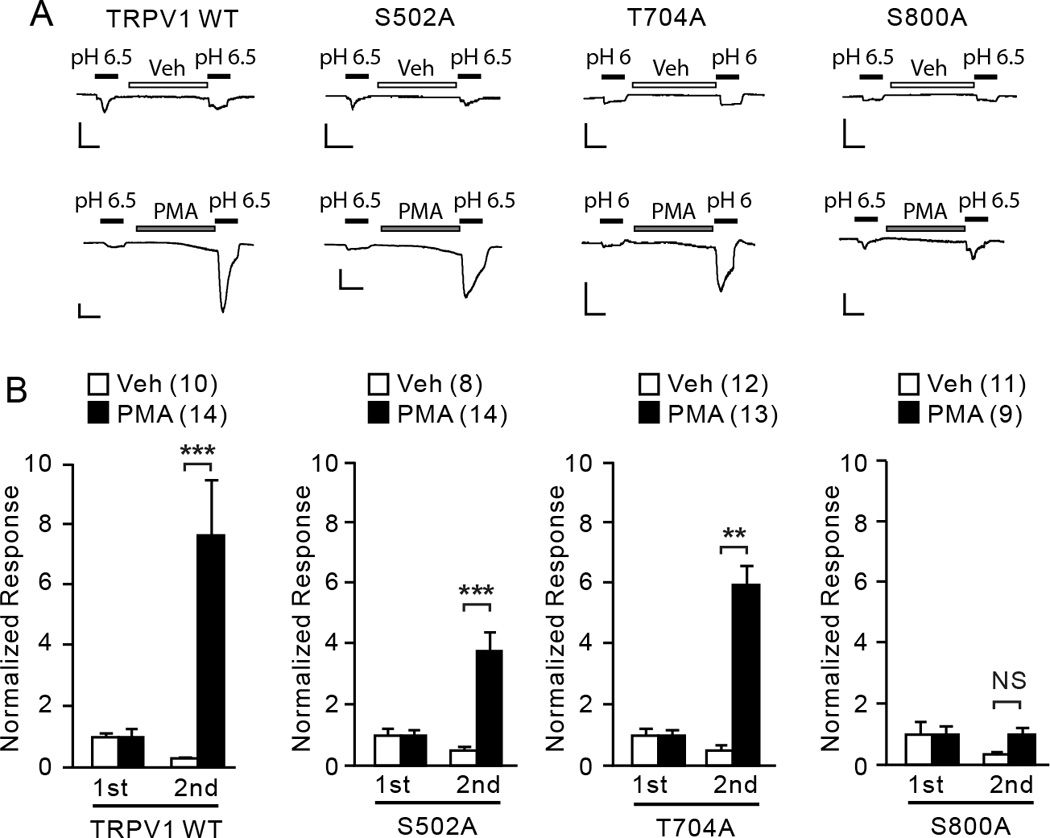
A. Representative current traces evoked by consecutive application of acid (black bars) following pretreatment with vehicle (ethanol; upper) or PMA (0.3 µM; lower) in DRG neurons dissociated from TRPV1 null mice. Neurons were transfected by electroporation with cDNA encoding TRPV1 WT, TRPV1 S502A, T704A, or S800A as indicated. Current amplitudes were evaluated at −60 mV. Scale bars, 10 pA/pF – 30 sec.
B. Averaged normalized responses. Current density of the 1st and 2nd response from each neuron was normalized to the average of the 1st response in the given group. NS, not significant; **p<0.01; ***p<0.001; Student’s t-test. Values in parentheses represent the number of observations in each group.
3.5. TRPV1 T704 and S800, but not S502, contributes to PKC-induced hypersensitivity to heat in sensory neurons
Next we examined differential contribution of phosphorylation sites to PKC-induced hypersensitivity to heat in sensory neurons. Since the electroporated DRG neurons were not optimal for reliable electrophysiological recordings at high temperature employed in our study, we performed Ca2+ imaging analysis using the ratiometric Ca2+ sensitive dye Fura-2AM. TRPV1-mediated heat-evoked Ca2+ responses in dissociated DRG neurons were evaluated using the Ca2+ imaging technique [27]. When dissociated neurons from TRPV1 null mice were transfected with a control plasmid, heat ramps did not induce Fura responses beyond an increase of baseline concomitant with bath temperature increase (Fig. 5A). Based upon the analysis of the temperature-dependent baseline changes, we defined neurons showing increases in Fura ratio in the 40 to 45°C range of more than 61% relative to baseline as responders. When TRPV1 null neurons transfected with TRPV1 WT were exposed to heat ramps, robust Fura responses were induced at approximately 42°C. (Fig. 5B). When TRPV1 WT-transfected neurons were pre-treated with PMA, the mean activation threshold was significantly lower than that of the vehicle-treated group by 2°C (Fig. 5F). In addition, responses at a subthreshold warm temperature (37°C) were also significantly greater in PMA-treated group (Fig. 5G). Neurons transfected with the TRPV1 S502A mutant also displayed a significant PMA-induced decrease in heat activation threshold (Fig. 5C and F). Relative responses at 37°C following PMA treatment were greater than that of vehicle-treated group, but this difference did not reach statistical significance (Fig. 5G; p=0.08). In contrast, alanine mutation of TRPV1 T704 resulted in no difference in heat activation between vehicle and PMA-treated neurons (Fig. 5D, F, G). Alanine mutation of TRPV1 S800 significantly decreased heat activation threshold following PMA pretreatment, to a slightly lesser extent than WT. Relative responses at 37°C in TRPV1 S800A were not significantly different between vehicle and PMA groups (Fig. 5G), suggesting partial impairment of PMA-induced hypersensitivity to heat in TRPV1 S800A. These results are largely consistent with the data obtained in HEK293 cells (Fig. 1) and suggest that TRPV1 T704 and S800, but not S502, contribute to PKC-induced hypersensitivity to heat in sensory neurons.
Fig. 5. The roles of phosphorylation sites in PKC-induced hypersensitivity of TRPV1 to heat in sensory neurons.
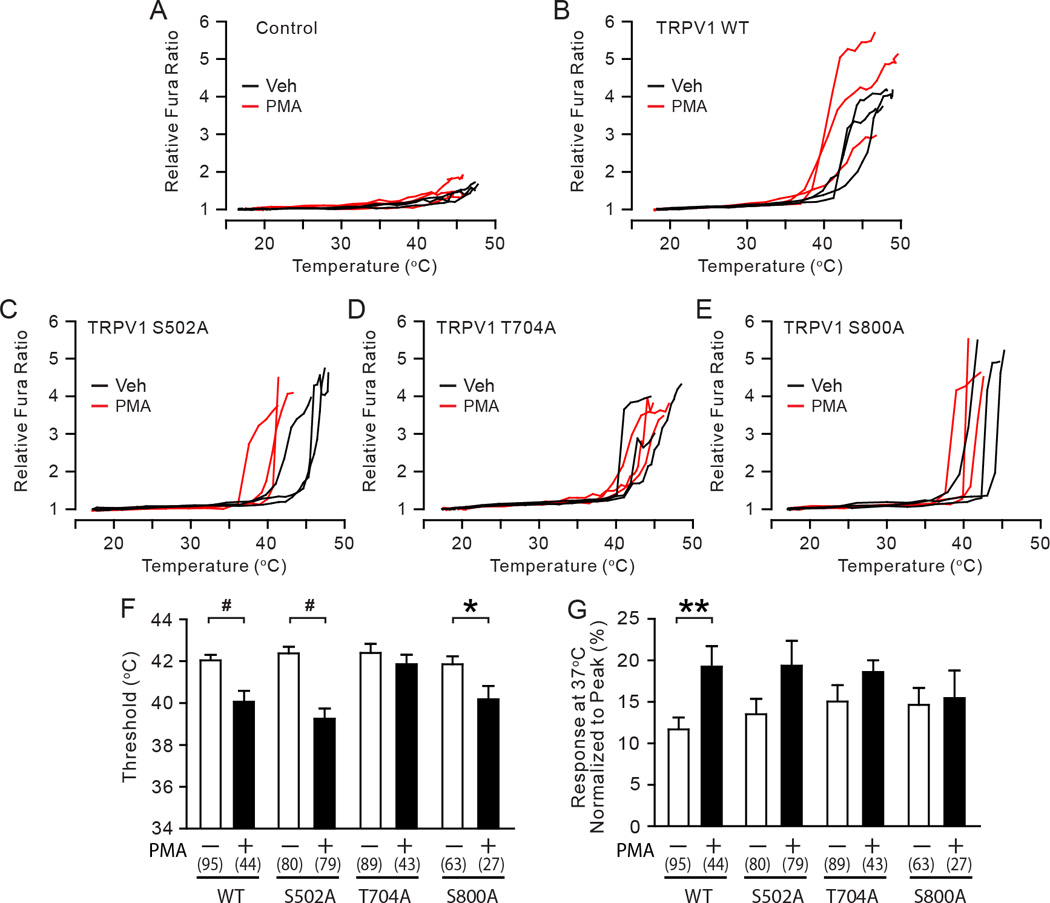
A–E. Representative traces of the relative Fura responses evoked by temperature ramps from 16 to 48 °C following the exposure to either vehicle (black) or 0.3 µM PMA (red) in TRPV1 null DRG neurons transfected by an empty plasmid (A), TRPV1 WT (B), S502A (C), T704A (D), or S800A (E). The Fura ratio was normalized to the minimum value measured at cold temperature range in each cell.
F–G. Quantification of heat-evoked responses in TRPV1 WT and mutants. Threshold temperature of activation (F) and the relative response at 37°C (G) were compared between vehicle-treated (white bars) and PMA-treated (black bars) cells as indicated. The relative responses at 37°C were calculated by averaging the relative Fura responses measured between 36°C to 38°C in each cell and normalizing to the respective peak response in the given cell. Numbers within parenthesis represent the numbers of observations. *p<0.05; **p<0.01; #p<0.0005; **p<0.01; Student’s t-test.
3.6. Preventing phosphorylation of TRPV1 S800 attenuates bradykinin-induced hypersensitivity to capsaicin in sensory neurons
Our data suggest that TRPV1 S800 is an important site for PKC-induced hypersensitivity both in HEK293 cells and sensory neurons. These results were obtained by direct activation of PKC using PMA. Under physiological conditions, however, TRPV1 phosphorylation is primarily regulated by the activation of inflammatory mediators. Therefore, we tested whether prevention of phosphorylation of TRPV1 S800 attenuates hypersensitivity induced by an inflammatory mediator. We tested the effects of bradykinin, which is a chemical mediator that is well known to modulate nociceptor functions and to induce TRPV1 hypersensitivity through the activation of G-protein coupled receptor and PKC [17; 39]. In TRPV1 null DRG neurons expressing TRPV1 WT, pretreatment with bradykinin significantly increased the second capsaicin response by approximately 6 fold, suggesting that exogenously expressed TRPV1 can be sensitized through the activation of endogenous bradykinin receptors (Fig. 6A, B). In TRPV1 null DRG neurons expressing TRPV1 S800A, bradykinin also induced sensitization of the capsaicin response, but the extent of sensitization was only approximately 2.7 fold, which was significantly less than that of WT. These results suggest that TRPV1 S800 is a major contributor to bradykinin-induced hypersensitivity of TRPV1 in sensory neurons.
Fig. 6. Contribution of TRPV1 S800 to bradykinin-induced hypersensitivity of TRPV1 to capsaicin in sensory neurons.
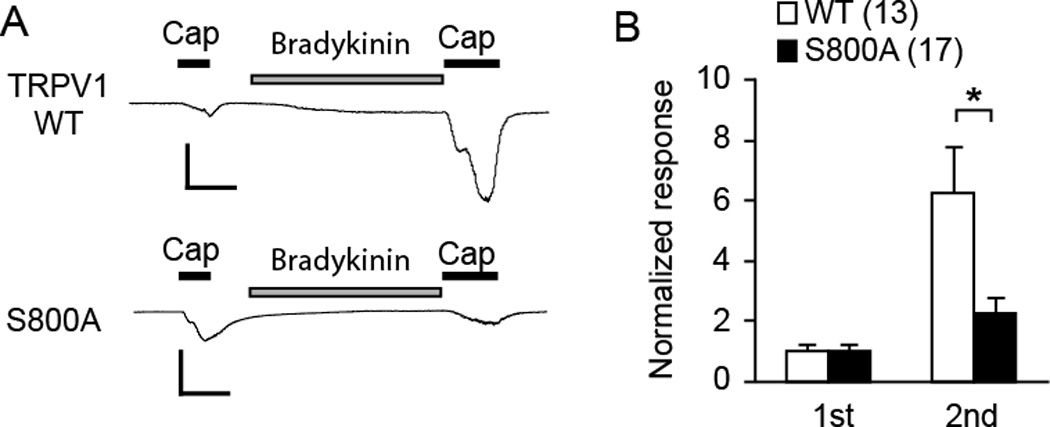
A. Representative current traces evoked by consecutive application of capsaicin (0.1 µM) following pretreatment with bradykinin (1 µM) in DRG neurons dissociated from TRPV1 null mice. The neurons were transfected with cDNA encoding TRPV1 WT (upper) or TRPV1 S800A (lower) by electroporation. Current amplitudes were evaluated at −60 mV. Scale bars, 50 pA/pF – 30 sec.
B. Averaged normalized responses. Current density of the 1st and 2nd response from each neuron was normalized to the average of 1st response in the given group; *p<0.05; Student’s t-test. Values in parentheses represent the number of observations in each group.
4. Discussion
In this study, we demonstrated that PKC-induced hypersensitivity to capsaicin, heat, and acid was mediated by different combinations of phosphorylation sites (Fig. 7) and that TRPV1 S800 is a polymodal sensitization residue.To test the contribution of each phosphorylation residue to functional hypersensitivity following PKC activation, we adopted a conventional combination of pharmacological and mutagenic approaches; testing the effects of PMA in TRPV1 mutants in which the phosphorylation sites were mutated to alanine. The functional effects of the mutants were interpreted as consequences of the lack of phosphorylation at the given phosphorylation site. Alternatively, however, it is possible that the functional effects could be attributed, at least in part, to the mutation of the given residue rather than the lack of phosphorylation. This is particularly important in TRPV1 T704A, which is included in a ‘TRP box’ domain that is critical for channel gating [41]. We found that alanine mutation of T704 substantially reduced the sensitivity to proton, with little change in capsaicin or heat sensitivity, which might be due to dominant effects of the mutation on proton gating. This is consistent with observations that capsaicin sensitivity is abolished by mutation to other amino acid such as aspartate, glutamate (unpublished observation), or isoleucine [30]. Therefore, to examine the contribution of individual residues and to minimize any uncontrollable effects of mutations, we focused on TRPV1 mutants carrying individual mutations, rather than multiple mutations. A similar difficulty arises in the interpretation of data from phosphomimetic mutations, in which altered basal properties rather than PMA-induced changes are evaluated as outcomes. Although we interpreted the effects of PMA as the consequences of phosphorylation, we cannot formally exclude potential contributions of allosteric effects, since PMA is known to directly activate TRPV1 at the concentration used in this study [2; 34]. Under our recording conditions, however, only modest activation was observed upon application of PMA, which occurred only in a minor population of cells. PMA-induced activation occurred transiently and conductance returned to baseline within the two-minute pretreatment. Thus, potential allosteric effects of PMA and agonistic stimuli would have been minimized in our experiments. Furthermore, mutation of S800 largely attenuated hypersensitivity of TRPV1 induced not only by PMA but also by bradykinin. The attenuation of bradykinin induction cannot be explained by the interfered allosteric modulation in TRPV1 S800A. Although we did not try to quantify the extent of direct activation by PMA in different mutants, a previous study indicated that T704 and S800 are involved in PMA-dependent Ca2+ increase [2]. Since our results demonstrated that these two residues are involved in PMA-induced hypersensitivity to heat, it is possible that direct activation of TRPV1 by PMA is partly attributable to the phosphorylation-induced decrease in activation threshold temperature in addition to phosphorylation-independent events. These uncertainties will be addressed in future studies.
Fig. 7. A Schematic of modality-specific contribution of PKC-phosphorylation residues of TRPV1.
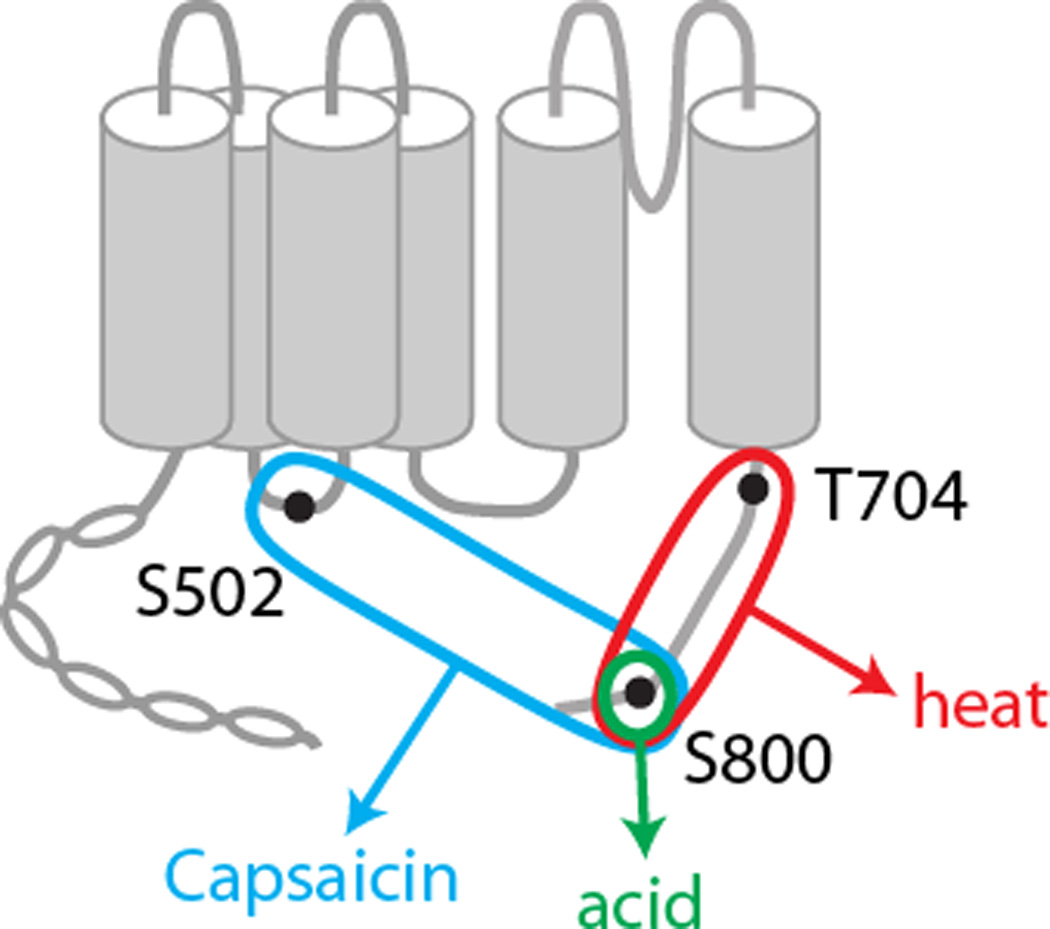
Modality-specific mechanisms of sensitization are consistent with the idea that TRPV1 is activated by multiple agonist modalities through distinct structural bases. Previous studies and recent crystallographic analysis demonstrate that that vanilloids bind to a pocket composed of transmembrane domains [4; 9; 14]. Although a structural change following the phosphorylation of an individual residue is difficult to predict, it is possible that phosphorylation of different residues render differential changes favoring activation by different agonists. TRPV1 S502 is located at the intracellular loop linking transmembrane domain 2 and 3 near the binding pocket [26]. Therefore, the phosphorylation of S502 by the activation of PKC may alter binding of capsaicin in a manner that increases capsaicin sensitivity; if so, mutation of this residue could attenuate hypersensitivity to capsaicin. TRPV1 T704 is located in the ‘TRP box’ domain that is critical for gating and tetramerization [41]. Therefore, it is possible that phosphorylation of T704 may alter gating properties in a manner that sensitizes TRPV1. Although alanine mutation of TRPV1 T704 did not affect hypersensitivity to capsaicin and proton, PKC-induced hypersensitivity to heat was substantially attenuated. It is known that heat-sensitivity of TRPV1 is contributed by a segment within the c-terminal domain that is adjacent to T704 [3], suggesting a potential mechanistic linkage. In contrast to S502 and T704, alanine mutation of S800 attenuated PKC-induced hypersensitivity to capsaicin, heat, and acid. TRPV1 S800 is located in the distal portion of the carboxy terminal domain. This part of the carboxy terminal domain is thought to interact with calmodulin and phosphoinositide, and may play critical roles in the modulation of TRPV1 activation and desensitization [20; 31; 35]. Although it is not known what structural changes are rendered, phosphorylation of S800 may alter the modulatory capacity of the c-terminal domain resulting in hypersensitivity to polymodal stimuli.
Modality-specific sensitization of TRPV1 could also be mediated by the linkage of a specific subtype of PKC with TRPV1. Recently, it was demonstrated that PKCβII contributes to basal thermal sensitivity of TRPV1 through the phosphorylation of T705 of human TRPV1 (the equivalent of rat T704) [25]. Overexpression of PKCβII in HEK293 cells enhances basal thermal sensitivity of TRPV1, which is prevented by the mutation of T705 in human TRPV1 [25]. These results are consistent with our observation that pharmacological activation of PKC enhanced thermal sensitivity of TRPV1, which was attenuated by mutation of T704 or S800. Since PMA is non-specific, our results suggest that activation of PKCβII and PKCε both induce sensitization of TRPV1 to heat. Apparently PKCβII and PKCε determine thermal sensitivity of TRPV1 and, hence, contribute to thermal hyperalgesia in concert [25]. However, it is possible that the two subtypes of PKC and the two phosphorylation residues may play distinct roles. PKCβII may contribute more to basal thermal sensitivity of TRPV1, rather than pathological hypersensitivity, presumably due to its constitutive activation properties [25]. In contrast, PKCε may induce pathological hypersensitivity of TRPV1 due to its association with receptors of inflammatory mediators such as bradykinin [7]. Therefore, PKCε-S800 signaling may be a better target for attenuating pathological hypersensitivity of TRPV1.
Since the extent and mechanisms of functional sensitization of TRPV1 are affected by factors specific to sensory neurons, it is significant to compare the relative contribution of phosphorylation sites in a neuronal context. For example, the protein kinase anchoring by AKAP75/150 facilitates phosphorylation of TRPV1 [18; 37; 45]. Specific subtypes of protein kinases, e.g., PKCε play dominant roles in the sensitization of TRPV1, but the expression of PKC subtypes is heterogeneous among cell types [7; 28; 33]. Moreover, TRPV1 directly interacts with TRPA1 [13; 38], which could also affect overall functions of TRPV1 including sensitization. We tested the contribution of TRPV1 phosphorylation sites in sensory neurons following transfection of TRPV1 and mutants into DRG neurons dissociated from TRPV1 null mice. Our results indicate that differential contribution of PKC-phosphorylation sites to PMA-induced hypersensitivity to heat, capsaicin and proton is largely preserved in the two systems. More importantly, in sensory neurons, the alanine mutation of TRPV1 S800 reduced bradykinin-induced sensitization by more than half compared to WT. These experiments suggest that TRPV1 S800A ablates TRPV1 hypersensitivity induced by the native sensitization machinery, and suggest that TRPV1 S800 is a major site mediating bradykinin-induced sensitization of TRPV1 under physiologically relevant conditions.
TRPV1 was proposed as a functional integrator of nociceptive signals from multiple inflammatory mediators [24]. Our results demonstrate that TRPV1 S800 residue functions as a polymodal sensitization residue commonly mediating hypersensitivity to capsaicin, heat, and acid. Therefore, PKC-mediated signals elicited by multiple chemical mediators may result in phosphorylation of multiple TRPV1 residues, and S800 may play a central role in polymodal hypersensitivity of TRPV1 in nociceptors. Inflammatory mediators elevated at the site of tissue, e.g. substance P and endothelin, injury enhance the function of TRPV1 through PKC [21; 40]. Since these substances also induce PKC-dependent cutaneous thermal hyperalgesia, phosphorylation at TRPV1 S800 was implicated in thermal hyperalgesia in skin. In addition to cutaneous hyperalgesia, phosphorylation of TRPV1 S800 is also implicated in mechanical hyperalgesia in muscle [22; 23]. Activation of peripheral NMDA receptors increases phosphorylation of TRPV1 and evokes mechanical hypersensitivity accompanied by PKC-dependent TRPV1 phosphorylation at S800 [22; 23]. Since inflammatory mediators such as bradykinin, ATP, and glutamate are released upon muscle injury [29], increased phosphorylation of TRPV1 by these substances may sensitize TRPV1 in muscle afferents. Therefore, developing methods of selectively perturbing phosphorylation at TRPV1, especially S800, could be a novel approach for treating both cutaneous thermal hyperalgesia and muscle mechanical hyperalgesia.
In conclusion, PKC-induced hypersensitivity of TRPV1 to capsaicin, heat, and acid involves differential phosphorylation residues, and S800 is a common sensitization site. Given the serious adverse side effects of using systemic antagonism to inhibit entire functions of TRPV1 [15], selective inhibition of phosphorylation at TRPV1 S800 may provide an alternative anti-hyperalgesic approach with fewer side effects. For example, a recent study suggested that effects of a drug attenuating phosphorylation at a specific site of PPARγ are associated with anti-diabetic effects [8]. Further study elucidating phosphorylation-dependent changes in channel structure around S800 or changes in interactions with other molecules may facilitate rationalized designing of compounds selectively attenuating TRPV1 phosphorylation at this site.
ACKNOWLEDGEMENTS
The authors thank Jamila Asgar for technical assistance and Dr. Michael Caterina for helpful discussion. This study was supported by National Institutes of Health grant R01 DE023846 (M.K.C.) and DE16062 (J.Y.R.).
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors disclose no conflict of interest in respect of this work.
REFERENCES
- 1.Aneiros E, Cao L, Papakosta M, Stevens EB, Phillips S, Grimm C. The biophysical and molecular basis of TRPV1 proton gating. EMBO J. 2011;30(6):994–1002. doi: 10.1038/emboj.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RWt. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci U S A. 2003;100(21):12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brauchi S, Orta G, Salazar M, Rosenmann E, Latorre R. A hot-sensing cold receptor: C-terminal domain determines thermosensation in transient receptor potential channels. J Neurosci. 2006;26(18):4835–4840. doi: 10.1523/JNEUROSCI.5080-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao E, Liao M, Cheng Y, Julius D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 2013;504(7478):113–118. doi: 10.1038/nature12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288(5464):306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 6.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389(6653):816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 7.Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23(3):617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- 8.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466(7305):451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou MZ, Mtui T, Gao YD, Kohler M, Middleton RE. Resiniferatoxin binds to the capsaicin receptor (TRPV1) near the extracellular side of the S4 transmembrane domain. Biochemistry. 2004;43(9):2501–2511. doi: 10.1021/bi035981h. [DOI] [PubMed] [Google Scholar]
- 10.Chung MK, Jung SJ, Oh SB. Role of TRP Channels in Pain Sensation. Adv Exp Med Biol. 2011;704:615–636. doi: 10.1007/978-94-007-0265-3_33. [DOI] [PubMed] [Google Scholar]
- 11.Chung MK, Wang S. Cold Suppresses Agonist-induced Activation of TRPV1. J Dent Res. 2011;90(9):1098–1102. doi: 10.1177/0022034511412074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, Waxman SG. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009;4(8):1118–1126. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- 13.Fischer MJ, Balasuriya D, Jeggle P, Goetze TA, McNaughton PA, Reeh PW, Edwardson JM. Direct evidence for functional TRPV1/TRPA1 heteromers. Pflugers Arch. 2014 doi: 10.1007/s00424-014-1497-z. [DOI] [PubMed] [Google Scholar]
- 14.Gavva NR, Klionsky L, Qu Y, Shi L, Tamir R, Edenson S, Zhang TJ, Viswanadhan VN, Toth A, Pearce LV, Vanderah TW, Porreca F, Blumberg PM, Lile J, Sun Y, Wild K, Louis JC, Treanor JJ. Molecular determinants of vanilloid sensitivity in TRPV1. J Biol Chem. 2004;279(19):20283–20295. doi: 10.1074/jbc.M312577200. [DOI] [PubMed] [Google Scholar]
- 15.Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, Alvarez F, Bak A, Darling M, Gore A, Jang GR, Kesslak JP, Ni L, Norman MH, Palluconi G, Rose MJ, Salfi M, Tan E, Romanovsky AA, Banfield C, Davar G. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain. 2008;136(1–2):202–210. doi: 10.1016/j.pain.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 16.Grandl J, Kim SE, Uzzell V, Bursulaya B, Petrus M, Bandell M, Patapoutian A. Temperature-induced opening of TRPV1 ion channel is stabilized by the pore domain. Nat Neurosci. 2010;13(6):708–714. doi: 10.1038/nn.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J, Zhang X, McNaughton PA. Inflammatory pain: the cellular basis of heat hyperalgesia. Curr Neuropharmacol. 2006;4(3):197–206. doi: 10.2174/157015906778019554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeske NA, Diogenes A, Ruparel NB, Fehrenbacher JC, Henry M, Akopian AN, Hargreaves KM. A-kinase anchoring protein mediates TRPV1 thermal hyperalgesia through PKA phosphorylation of TRPV1. Pain. 2008;138(3):604–616. doi: 10.1016/j.pain.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordt SE, Julius D. Molecular basis for species-specific sensitivity to "hot" chili peppers. Cell. 2002;108(3):421–430. doi: 10.1016/s0092-8674(02)00637-2. [DOI] [PubMed] [Google Scholar]
- 20.Joseph J, Wang S, Lee J, Ro JY, Chung MK. Carboxyl-terminal domain of transient receptor potential vanilloid 1 contains distinct segments differentially involved in capsaicin- and heat-induced desensitization. J Biol Chem. 2013;288(50):35690–35702. doi: 10.1074/jbc.M113.513374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawamata T, Ji W, Yamamoto J, Niiyama Y, Furuse S, Namiki A. Contribution of transient receptor potential vanilloid subfamily 1 to endothelin-1-induced thermal hyperalgesia. Neuroscience. 2008;154(3):1067–1076. doi: 10.1016/j.neuroscience.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Lee J, Chung MK, Ro JY. Activation of NMDA receptors leads to phosphorylation of TRPV1 S800 by protein kinase C and A-Kinase anchoring protein 150 in rat trigeminal ganglia. Biochem Biophys Res Commun. 2012;424(2):358–363. doi: 10.1016/j.bbrc.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Saloman JL, Weiland G, Auh QS, Chung MK, Ro JY. Functional interactions between NMDA receptors and TRPV1 in trigeminal sensory neurons mediate mechanical hyperalgesia in the rat masseter muscle. Pain. 2012;153(7):1514–1524. doi: 10.1016/j.pain.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta. 2007;1772(8):989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Hasan R, Zhang X. The Basal Thermal Sensitivity of the TRPV1 Ion Channel Is Determined by PKCbetaII. J Neurosci. 2014;34(24):8246–8258. doi: 10.1523/JNEUROSCI.0278-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504(7478):107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandadi S, Sokabe T, Shibasaki K, Katanosaka K, Mizuno A, Moqrich A, Patapoutian A, Fukumi-Tominaga T, Mizumura K, Tominaga M. TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Arch. 2009;458(6):1093–1102. doi: 10.1007/s00424-009-0703-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandadi S, Tominaga T, Numazaki M, Murayama N, Saito N, Armati PJ, Roufogalis BD, Tominaga M. Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCepsilon-mediated phosphorylation at S800. Pain. 2006;123(1–2):106–116. doi: 10.1016/j.pain.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 29.Mense S. Algesic agents exciting muscle nociceptors. Exp Brain Res. 2009;196(1):89–100. doi: 10.1007/s00221-008-1674-4. [DOI] [PubMed] [Google Scholar]
- 30.Novakova-Tousova K, Vyklicky L, Susankova K, Benedikt J, Samad A, Teisinger J, Vlachova V. Functional changes in the vanilloid receptor subtype 1 channel during and after acute desensitization. Neuroscience. 2007;149(1):144–154. doi: 10.1016/j.neuroscience.2007.07.039. [DOI] [PubMed] [Google Scholar]
- 31.Numazaki M, Tominaga T, Takeuchi K, Murayama N, Toyooka H, Tominaga M. Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc Natl Acad Sci U S A. 2003;100(13):8002–8006. doi: 10.1073/pnas.1337252100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. J Biol Chem. 2002;277(16):13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- 33.Olah Z, Karai L, Iadarola MJ. Protein kinase C(alpha) is required for vanilloid receptor 1 activation. Evidence for multiple signaling pathways. J Biol Chem. 2002;277(38):35752–35759. doi: 10.1074/jbc.M201551200. [DOI] [PubMed] [Google Scholar]
- 34.Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408(6815):985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- 35.Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300(5623):1284–1288. doi: 10.1126/science.1083646. [DOI] [PubMed] [Google Scholar]
- 36.Ryu S, Liu B, Yao J, Fu Q, Qin F. Uncoupling proton activation of vanilloid receptor TRPV1. J Neurosci. 2007;27(47):12797–12807. doi: 10.1523/JNEUROSCI.2324-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schnizler K, Shutov LP, Van Kanegan MJ, Merrill MA, Nichols B, McKnight GS, Strack S, Hell JW, Usachev YM. Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J Neurosci. 2008;28(19):4904–4917. doi: 10.1523/JNEUROSCI.0233-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem. 2010;285(20):15167–15177. doi: 10.1074/jbc.M110.106153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugiura T, Tominaga M, Katsuya H, Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol. 2002;88(1):544–548. doi: 10.1152/jn.2002.88.1.544. [DOI] [PubMed] [Google Scholar]
- 40.Tang HB, Li YS, Miyano K, Nakata Y. Phosphorylation of TRPV1 by neurokinin-1 receptor agonist exaggerates the capsaicin-mediated substance P release from cultured rat dorsal root ganglion neurons. Neuropharmacology. 2008;55(8):1405–1411. doi: 10.1016/j.neuropharm.2008.08.037. [DOI] [PubMed] [Google Scholar]
- 41.Valente P, Garcia-Sanz N, Gomis A, Fernandez-Carvajal A, Fernandez-Ballester G, Viana F, Belmonte C, Ferrer-Montiel A. Identification of molecular determinants of channel gating in the transient receptor potential box of vanilloid receptor I. FASEB J. 2008;22(9):3298–3309. doi: 10.1096/fj.08-107425. [DOI] [PubMed] [Google Scholar]
- 42.Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534(Pt 3):813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Lee J, Ro JY, Chung MK. Warmth suppresses and desensitizes damage-sensing ion channel TRPA1. Mol Pain. 2012;8:22. doi: 10.1186/1744-8069-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang F, Cui Y, Wang K, Zheng J. Thermosensitive TRP channel pore turret is part of the temperature activation pathway. Proc Natl Acad Sci U S A. 2010;107(15):7083–7088. doi: 10.1073/pnas.1000357107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, Li L, McNaughton PA. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron. 2008;59(3):450–461. doi: 10.1016/j.neuron.2008.05.015. [DOI] [PubMed] [Google Scholar]