

Abstract
Improving our understanding of the role of chromatin regulators in the initiation, development, and suppression of cancer and other devastating diseases is critical, as they are integral players in regulating DNA integrity and gene expression. Developing small molecule inhibitors for this target class with cellular activity is a crucial step toward elucidating their specific functions. We specifically targeted the DNA damage response protein, 53BP1, which uses its tandem tudor domain to recognize histone H4 dimethylated on lysine 20 (H4K20me2), a modification induced by double-strand DNA breaks. Through a cross-screening approach we identified UNC2170 (1) as a micromolar ligand of 53BP1, which demonstrates at least 17-fold selectivity for 53BP1 as compared to other methyl-lysine (Kme) binding proteins tested. Structural studies revealed that the tert-butyl amine of UNC2170 anchors the compound in the methyl-lysine (Kme) binding pocket of 53BP1, making it competitive with endogenous Kme substrates. X-ray crystallography also demonstrated that UNC2170 binds at the interface of two tudor domains of a 53BP1 dimer. Importantly, this compound functions as a 53BP1 antagonist in cellular lysates and shows cellular activity by suppressing class switch recombination, a process which requires a functional 53BP1 tudor domain. These results demonstrate that UNC2170 is a functionally active, fragment-like ligand for 53BP1.
One of the major processes regulating gene expression is the dynamic control of chromatin architecture. A key mechanism involved is the addition, recognition, and removal of post-translational modifications (PTMs) on histone proteins by various enzymes and binding proteins.(1) In particular, the recognition of methylated lysine residues on histone tails by methyl-lysine (Kme) binding proteins is a critical event in chromatin regulation.(2, 3) These binding events recruit effector proteins and their macromolecular complexes to support defined gene activation and repression outcomes. Importantly, histone Kme binding has been shown to be misregulated in diseases including cancer and is essential to critical cellular processes such as DNA damage repair.(4–6)
53BP1 is a Kme binding protein that plays a central role in DNA Damage Repair (DDR) pathways and is recruited to sites of double-strand breaks (DSB). This process is dependent on the activity of ataxia telangiectasia mutated (ATM) kinase that regulates the DNA damage signaling response. Recruitment of 53BP1 can lead to cell cycle arrest and DNA damage repair, or lack thereof if there are defects or deficiencies present in the enzymes or proteins involved in this signaling cascade.(7–19) Cells deficient in 53BP1 prove to be viable but display a phenotype resembling mild ATM kinase-signaling defects due to the cell’s inability to properly respond to and repair DNA damage.(20, 21) 53BP1 knockout mice are viable, but are growth retarded, immune deficient, radiation sensitive, and cancer prone.(20) In addition, these mice display a defect in antibody class switch recombination (CSR) which also requires 53BP1 activity.(22) By testing different fragments of the 53BP1 gene, it has been shown that the region that harbors its Kme binding tandem tudor domain (amino acids 1235–1709) is recruited to ionizing radiation (IR)-induced foci as efficiently as full-length, endogenous 53BP1.(23) Histone H4 lysine 20 dimethyl (H4K20me2) was identified as the primary PTM in histones recognized by the 53BP1 tandem tudor domain (TTD),(24) and this finding was subsequently supported by structural studies.(25) Additionally, the TTD of 53BP1 has been shown to bind to p53K382me2 and thereby increase the concentration of p53 at DNA damage sites.(26) Unlike H2AX phosphorylation, global levels of H4K20me2 do not increase after DNA damage; instead, 53BP1 recognizes histone H2A ubiquitylated at lysine 15 by RNF168 in response to DNA double-strand breaks.(27) 53BP1 recruitment is also facilitated by RNF168 ubiquitylation-dependent removal of L3MBTL1 and JMJD2A from H4K20me2 after DNA damage.(28, 29)
BRCA1 is a checkpoint and DNA damage repair gene that is required for maintenance of genomic integrity, and the inheritance of mutated BRCA1 is a major risk factor for breast and ovarian cancer.(30) BRCA1 knockout in mice is embryonic lethal,(31) and conditional knockout in the mammary gland results in low frequency and long latency of mammary tumor formation.(32) It was recently reported that the BRCA1 knockout developmental phenotype is rescued when placed on a 53BP1-null background, and adult mice that are null for both the 53BP1 and BRCA1 genes age normally and display a very low incidence of tumor formation.(33) The genomic instability in the BRCA1 knockout can be overcome because the homologous recombination (HR) pathway is largely restored in cells lacking both BRCA1 and 53BP1.(34)
We hypothesized that a small molecule ligand that prevents Kme recognition by 53BP1 would antagonize its biological activity in cells possessing BRCA1 mutations and lead to rescue of their genomic stability by restoration of the HR pathway. Notwithstanding the clinical challenges of cancer prevention strategies, the discovery of a small molecule in vivo probe for 53BP1 would enable this hypothesis to be tested preclinically, and the devastating effects of BRCA1 mutations, including prophylactic surgical interventions,(21) could perhaps be diminished. We therefore aimed to synthesize cell penetrant small molecules that would bind selectively to the tandem tudor domain of 53BP1 using structure-based design and iterative medicinal chemistry. Herein we report the initial results of this effort.
RESULTS AND DISCUSSION
A structure-based design approach was initiated by analyzing available crystal structure data of 53BP1 bound to an H4K20me2 peptide (PDB 2IG0).(25) The key interactions noted in this structure were a hydrogen-bond between the Kme basic amine and an aspartic acid (Asp1521), cation-π interactions between Kme2 and phenylalanine, tyrosine, and tryptophan residues within the aromatic binding cage (Tyr1502, Tyr1523, Phe1519, Trp1495), and a cation-π interaction between arginine 19 on the H4 peptide tail and a tryptophan residue (Trp1500).(25) Mutagenesis of the histone peptide had previously demonstrated that H18 also contributes to binding, but this interaction was not clearly defined in the X-ray crystal structure.(25) To develop a more complete understanding of the binding interactions of the H4 peptide (amino acids 14–27) bound to 53BP1, isotope enriched (13C and 15N) NMR spectroscopy(35) was applied to determine the structure of the central residues of the H4K20me2 peptide bound to 53BP1 (PDB 2LVM).(36) It was observed that this central region of the H4 peptide corresponding to residues 15 to 22 adopts a “U-turn” conformation. Notably, in addition to the binding interactions with H4K20me2 and H4R19 that were detected in the crystal structure, further analysis revealed a pocket containing both acidic and hydrophobic residues that accommodates H4R17 and H4V21. It has also been shown that acetylation of H4K16 diminishes 53BP1 binding by disrupting a salt bridge between H4K16 and Glu1551.(36) Therefore, a small molecule that could occupy the methyl-lysine binding cage of 53BP1 and interact favorably with some of the surrounding residues would be expected to block 53BP1 binding to H4K20me2.
Our current efforts utilize a comprehensive cross-screening approach in order to evaluate all synthesized ligands against a panel of Kme reader domains. The Kme reader panel consists of 10 reader proteins from four different families: tudor domains (53BP1, UHRF1, PHF1, PHF19), chromodomains (CBX7), MBT domains (L3MBTL1, L3MBTL3, MBTD1), and PHD fingers (JARID1A, PHF23, UHRF1). These proteins were chosen largely based on available structural information, reader family representation, and biological relevance. Previously our lab reported an AlphaScreen bead-based proximity assay for Kme readers(37) and this screening tool was employed to initially assess 53BP1 binding. UNC2170 (1, Table 1) emerged as a preliminary 53BP1 hit from these cross-screening efforts. Because of the modest affinity of 1 (29 ± 7.4 µM) and its fragment-like nature (MW = 313.24, ligand efficiency = 0.35, lipophilic ligand efficiency = 1.5)(38, 39), we profiled it at concentrations up to 500 µM versus our reader panel to better ascertain its selectivity, as this is of paramount importance for chemical probe development (Supplementary Figure S1).(40) This revealed promising levels of selectivity within this small but diverse sampling of Kme reader proteins, as 1 showed no affinity within the concentrations tested against any other members of the panel, including other tudor domain containing proteins and other readers of methylated H4K20 (Table 1). Additionally, isothermal titration calorimetry (ITC) experiments confirmed ligand binding, revealing a Kd of 22 ± 2.5 µM for UNC2170 against the 53BP1 tandem tudor domain (Figure 1).
Table 1.
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | R | 53BP1 | CBX7 | JARID1A | PHF1 | PHF19 | PHF23 | UHRF1 | L3MBTL1 | L3MBTL3 | MBTD1 |
| 1c (UNC2170) | 29 ± 7.4 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 | |
| 2 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 3 | 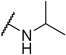 |
> 100 | > 100 | > 100 | 82 ± 13 | 61 ±0.9 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 4 c (UNC2892) | 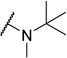 |
> 500 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 | > 500 |
| 5 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 6 | 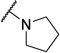 |
> 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | 37 ± 12 | > 100 | 78 ± 16 |
IC50 values are the average of at least 3 values ± the standard deviation as determined by AlphaScreen.
The maximum concentration in the assay was 100 µM and compounds showing less than 50% inhibition at this concentration are labeled >100 µM.
Compounds 1 and 4 were tested at a higher concentration (500 µM) against the panel to further evaluate selectivity.
Figure 1.

ITC analysis of 53BP1 binding to UNC2170 (1), revealing a Kd of 22 ± 2.5 µM.
Based on this initial data, we initiated synthetic efforts to prepare analogues of UNC2170 in order to evaluate structure-activity relationships (SAR) within this series and improve the affinity for 53BP1. For these SAR studies, all synthesized ligands were tested at concentrations up to 100 µM against our Kme panel to evaluate both their selectivity and potency. Analysis of UNC2170 suggested three primary areas for synthetic modification: the basic amine, the linker region, and the aromatic ring. SAR for each of these regions are discussed below.
It was hypothesized that the basic amine of UNC2170 would bind within the Kme binding pocket of 53BP1, similar to dimethyl-lysine. Exploration of the steric tolerance of the basic amine in 1 began by replacement of the secondary N-tert-butyl amine group with dimethyl amine (2) to more closely mimic the endogenous H4K20me2 substrate (Table 1). Interestingly, this compound showed no binding activity by AlphaScreen and this finding was confirmed by ITC (Supplementary Figure S2).
This result led us to hypothesize that the 53BP1 Kme binding pocket may have a preference for secondary amines over tertiary amines in the context of the UNC2170 framework, in contrast to its preference for dimethylated versus monomethylated lysine peptides.(25) Preparation of the isopropyl amino compound, 3, showed no activity against 53BP1 but interestingly, it was weakly active against some of the other tudor domain containing proteins in the panel (Table 1). Incorporation of other tertiary amines as in compound 4, where the secondary amine of UNC2170 is methylated, also showed no appreciable binding activity up to 500 µM as measured by AlphaScreen (Table 1 and Supplementary Figure 1a). ITC studies further confirmed that 4 has no measurable affinity for 53BP1, and therefore compound 4 (UNC2892) was selected as a negative control compound due to its inactivity and structural similarity to UNC2170 (1).
Further exploration of the ligand tolerance within the Kme binding pocket by replacement of the basic amine with an oxygen (5) resulted in no activity, as anticipated. We also investigated whether 53BP1 has similar Kme mimetic preferences as the MBT containing protein, L3MBTL1, as both bind the H4K20me2 mark. Compounds incorporating pyrrolidine have been shown to serve as effective ligands for such MBT domain containing proteins.(41–45) To test this we introduced a pyrrolidine amine to give compound 6 and found that this modification did not improve affinity towards 53BP1, confirming that Kme readers of the same histone mark do not necessarily have similar small molecule ligand preferences. Overall, our SAR exploration of the basic amine of UNC2170 (1) shows that a sterically bulky, lipophilic secondary amine is preferred for binding in the Kme pocket of 53BP1.
Prior to expanding our SAR studies to other portions of the molecule, we first sought to confirm that the N-tert-butyl amine of UNC2170 (1) was mimicking the endogenous Kme2 ligand and binding within the Kme binding pocket. We prepared an active site mutant of 53BP1 (D1521A) that no longer binds H4K20me2 due to loss of a salt bridge and hydrogen bond with Asp1521.(25) ITC demonstrated that binding of UNC2170 (1) was abrogated by the 53BP1 D1521A mutant (Supplementary Figure S2) which is consistent with the basic amine of UNC2170 (1) occupying the Kme reader pocket.
Various linkers to modify the distance and flexibility between the basic amine of UNC2170 and the aromatic functional group were explored next (Table 2). Initial SAR efforts began with the substitution of a sulfonamide linker for the amide bond to give compound 7, resulting in loss of 53BP1 binding affinity. Conversion of the amide to a benzylamine as in compound 8 similarly abrogated binding. When the linker region between the amide bond and basic nitrogen was rigidified using two different cyclic variants (compounds 9 and 10), 53BP1 activity was again lost. Additionally, lengthening the aliphatic chain between the amide bond and the amine as in compounds 11 and 12 resulted in inactivity, while shortening the linker (13) had a similar effect. Within this series, the propyl alkyl chain in compound 1 was found to be optimal.
Table 2.
| ID | R | 53BP1 | CBX7 | JARID1A | PHF1 | PHF19 | PHF23 | UHRF1 | L3MBTL1 | L3MBTL3 | MBTD1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 8 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 9 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 10 | 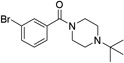 |
> 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 11 | 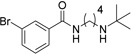 |
> 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 12 | 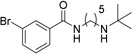 |
> 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 13 | 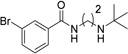 |
> 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
IC50 values are the average of at least 3 values ± the standard deviation as determined by AlphaScreen.
The maximum concentration in the assay was 100 µM and compounds showing less than 50% inhibition at this concentration are labeled >100 µM.
We extended our SAR studies to the aromatic ring of UNC2170 by first assessing the contribution of the bromine to binding via preparation of compound 14, which showed no measurable binding to 53BP1 (Table 3). Furthermore, the bromine regioisomers, compounds 15 and 16, were similarly inactive, suggesting that the bromine of UNC2170 is making a specific interaction with 53BP1. A halogen scan demonstrated that chlorine (17) and fluorine (18) provided no improvement in affinity, but replacement of the bromine with iodine was well tolerated (19, IC50 = 13 ± 4.8 µM). Consistent with this, we found that binding affinity is maintained when an isopropyl group (20) or a trifluoromethyl substituent (21) occupies the 3-position, suggesting that larger, more lipophilic substituents are preferred. The binding of 21 to 53BP1 was further confirmed by ITC (Supplementary Figure S2), which resulted in a Kd that correlated well with the AlphaScreen results (Kd = 10 ± 1.0 µM). While the 3-trifluoromethyl analog was roughly equipotent to UNC2170 (1), activity was also specific to this regioisomer, as compound 22 with CF3 para to the amide is inactive. Replacement of the Br of 1 with a nitro group at the 3-position (23) also results in a loss in potency.
Table 3.
 | |||||||||||
| ID | R | 53BP1 | CBX7 | JARID1A | PHF1 | PHF19 | PHF23 | UHRF1 | L3MBTL1 | L3MBTL3 | MBTD1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 14 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 15 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 16 | 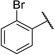 |
> 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 17 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 18 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 19 | 13 ± 4.8 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 20 | 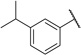 |
14 ± 7.6 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 21 | 22 ± 14 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 22 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 23 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 24 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
| 25 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | |
IC50 values are the average of at least 3 values ± the standard deviation as determined by AlphaScreen.
The maximum concentration in the assay was 100 µM and compounds showing less than 50% inhibition at this concentration are labeled >100 µM.
We hypothesized the halogenated 4-substituted pyridine derivatives, compounds 24 and 25, would increase the positive charge of the σ-hole of the bromine in UNC2170 and the CF3 in compound 21, in turn both increasing the likelihood of a halogen-hydrogen bond with the protein and improving binding.(46) These pyridine analogs resulted in a decrease in potency relative to their phenyl counterparts, suggesting that a potential halogen bond with 53BP1 is unlikely and instead favorable hydrophobic interactions are contributing to binding.
A number of other modifications to the aromatic ring were tested, including the addition of amino and hydroxyl groups at the 2-, 4-, and 5-positions while maintaining a 3-bromo substituent. UNC2170 derivatives incorporating functionalized lactones, furans, benzofurans, and pyridines, as well as variable methylene linkers between the amide bond and aromatic ring were also prepared. All of these analogs resulted in compounds that were inactive against 53BP1. Overall, these findings demonstrate that limited manipulations can be performed to the aromatic core of UNC2170 while maintaining potency, and that 53BP1 generally prefers analogs of UNC2170 that possess a lipophilic substituent at the 3-position.
To better understand the structural basis for UNC2170 binding, we co-crystallized the 53BP1 tandem tudor domain with UNC2170 and solved its structure by X-ray crystallography at 1.5 Å resolution (PDB 4RG2). Interestingly, we found that under these conditions UNC2170 is bound to a 53BP1 dimer, making distinct interactions with both tudor domains (Figure 2a). This correlates well with the molar ratio observed by ITC (Figure 1), which consistently produced an N value less than 1, suggesting a 2:1 ratio of protein to ligand. The structure revealed that the N-tert-butyl amine of UNC2170 is buried in the Kme binding pocket of one tudor domain (Figure 2b; magenta), driven largely by van der Waals interactions, hydrogen bonds, and electrostatic interactions. Due to the fact that the aromatic residues of both Kme pockets are greater than 5 Å from the amine, we anticipate that cation-π interactions are a minor contribution to binding relative to the hydrogen bonds formed between the ligand and protein.
Figure 2.

Crystallography and NMR evidence for UNC2170 (1) binding to 53BP1. (a) Co-crystal structure of 1 (cyan) bound to the 53BP1 tudor domain dimer (PDB 4RG2). One 53BP1 protein unit is shown in light gray and the other in dark gray, with the residues that interact with UNC2170 shown in fuchsia and green, respectively. (b) View of the protein surface of each 53BP1 tudor domain that interacts with UNC2170 (the two domains shown in (a) are separated and rotated); color coding is the same as in (a). (c) Overlay of the 1H-15N HSQC correlation spectra of 53BP1 in the free state (black) and in the presence of 10-fold molar excess UNC2170 (red).
The basic amine of UNC2170 is about equidistant from Asp1521 in each protein binding pocket (2.8 Å and 2.6 Å), and is well positioned to interact with and make a critical hydrogen bond to Asp1521 in both proteins. The importance of this interaction is consistent with ITC data that shows that the Kme pocket mutant, D1521A, does not bind to UNC2170. Additionally, the amide nitrogen is also within hydrogen bonding distance of the Asp1521 residue of one tudor domain, which is likely to further facilitate binding. The distance between the amine and the amide of UNC2170 therefore appears to be quite important in enabling a number of key interactions, and this sheds light on why changing the length of the aliphatic linker (compounds 11 – 13) impairs binding. The aliphatic linker and aromatic ring of UNC2170 is also nicely sandwiched between the dimer interface, packing against the surface of each protein unit.
Various series of analogs were prepared around UNC2170 beyond those presented above, many of which contained less subtle modifications; however, they all revealed a minimal tolerance to modification of the original ligand structure. The co-crystal structure assists in explaining this lack of SAR, as the interaction of UNC2170 with the 53BP1 dimer reveals that the ligand is encircled by both proteins and that there is limited space for modification. This is in contrast to our preliminary structural predictions based on UN2170 bound to a single 53BP1 tudor domain. We plan to utilize this novel binding mode in future efforts aimed at designing both UNC2170 related and structurally distinct inhibitors of 53PB1.
In addition to the X-ray structure, a 1H-15N HSQC NMR correlation spectrum of the tudor domain of 53BP1 was also obtained in the absence and presence of a 10-fold excess of UNC2170. Extensive changes in chemical shifts were observed (Figure 2c), further confirming specific binding of the ligand to 53BP1. Exchange between the bound and unbound state was observed to be slow on the NMR chemical shift time scale which is surprising given the measured affinity of UNC2170.
To further evaluate whether UNC2170 is able to compete with Kme2 peptide substrates for binding, H4K20me2 and p53K382me2 peptide pull down assays were conducted. UNC2170 was able to successfully displace the tandem tudor domain of a His-53BP1 fusion protein from both H4K20me2 and p53K382me2 peptides (Figure 3a). Additionally, UNC2170 displaces His-53BP1 from immobilized p53K382me2 in a dose dependent fashion (Figure 3b), resulting in an apparent IC50 of about 30 µM which is consistent with the results from other in vitro assays described above.
Figure 3.

Western blot analysis following competitive in-solution peptide pulldown assays with His-53BP1 TTD and (a) 500 µM UNC2170 or (b) the indicated concentration of UNC2170 shows inhibition of the interaction between the 53BP1 TTD and H4K20me2 and p53K382me2 peptides. UNC2170 displaces His-53BP1 from immobilized p53K382me2 in a dose dependent fashion.
Additional chromatin release assays were conducted in HEK293 cell lysates to determine if UNC2170 could influence the amount of endogenous 53BP1 bound to chromatin. Purification of chromatin from HEK293 cells was followed by compound treatment for two hours at room temperature and subsequent separation of the solubilized proteins from those remaining on chromatin. Western blot analysis revealed that treatment with UNC2170 (500 µM) resulted in a significant increase in soluble 53BP1 as compared to untreated lysates or lysates treated with UNC2892, the negative control compound (Figure 4a). This effect was found to be concentration dependent, as an increase in soluble 53BP1 was observed with increasing concentrations of UNC2170 (Figure 4b), and partial release of 53BP1 was detected at concentrations as low as 10 µM. This result is consistent with UNC2170 antagonizing the interaction between the tudor domain of full-length 53BP1 and chromatin.
Figure 4.

Western blot analysis of HEK293 cell lysates biochemically separated into chromatin and soluble fractions after treatment with (a) 500 µM UNC2170 or UNC2892 or (b) the indicated concentration of UNC2170 for 2 hours at room temperature. Anti-Histone 3 is shown as an input control.
We next sought to evaluate the potential of UNC2170 to engage 53BP1 in a cellular context. The low molecular weight (313.24 g/mol) and clogP (~3.27) of UNC2170 suggested a priori that UNC2170 would be cell permeant. To confirm this and assess the suitability of the compound for cell studies, the cell permeability of UNC2170 was determined. We found that UNC2170 was indeed highly cell permeant with no significant measureable cellular efflux (efflux ratio = 1.2) as determined by a bi-directional Caco-2 cell permeability assay.(47) Furthermore, UNC2170 has no measurable toxicity within the desired concentrations for cellular experiments, with cell toxicity only beginning to take effect at >10 mM as measured by a CellTiter-Glo luminescent cell-viability assay (Supplementary Figure S4).
Although the tandem tudor domains of 53BP1 are required for foci formation after ionizing radiation,(23) pretreatment of U2OS cells with UNC2170 (1 hour, 100 – 300 µM) did not significantly decrease the extent of endogenous 53BP1 foci formed following 5 Gy irradiation, and similar results were observed with UNC2982 (Supplementary Figure S5). To further support this, γH2AX foci were also quantified after compound treatment and no changes were observed. In addition, UNC2170 did not affect the apparent residence time of a GFP fusion protein containing the 53BP1 tudor domains as measured by fluorescence recovery after photobleaching (FRAP) experiments before or after ionizing radiation (10 Gy). Recent evidence suggesting that 53BP1 is a bivalent histone modification reader due to its recognition of H2AK15ub by its ubiquitination-dependent recruitment (UDR) modif(27) is one possible explanation for the maintenance of 53BP1 recruitment to DSB sites upon treatment with UNC2170 in these assays. Overall, UNC2170 treatment does not appear to be sufficient to block 53BP1 recruitment after ionizing radiation and a more potent ligand may be required for such activity.
As efficient B cell antibody class switch recombination (CSR) requires 53BP1 to have an active tudor domain and 53BP1 knockout mice are severely impaired in their ability to undergo CSR,(48, 49) we sought to determine whether UNC2170 would induce this same defect. Naive IgM+ B cells were stimulated to undergo isotype switching to IgG1 with LPS and Il-4, treated with UNC2170, UNC2892, or mock treated, and then the relative switching to IgG1 was determined by FACS analysis 3.5 days after compound treatment. B cells treated with UNC2170 (75 µM) underwent CSR 64% as effectively as untreated cells (Figure 5, Supplementary Figure S6), while treatment with UNC2892 had a much less significant inhibitory effect on CSR (89%). Although slightly higher and lower doses were tested (100 µM and 30 µM, respectively), treatment at 75 µM appears to be optimal in that the effects observed are specific to the active compound. Our attempts to probe the effect on CSR upon treatment with 300 µM of UNC2170 were unsuccessful, as this resulted in a reduction in cell viability. However, this effect could be mechanism related as UNC2892 does not have the same anti-proliferative effects, and B cells undergoing CSR endure a high rate of DNA damage while proliferating rapidly and therefore represent a particularly sensitive cell subset. It has been reported previously that B cells stimulated from mice bearing a single 53BP1 D1518R amino acid substitution (this mutant is equivalent to D1521R in humans) switch at about 10% of wild type levels.(48) While the effect of UNC2170 is not as pronounced, treatment with UNC2170 clearly phenocopies the reduction in CSR seen in 53BP1 mutant B cells.
Figure 5.

Summary of relative CSR to IgG1 switching in naive splenocytes cultured with LPS and IL-4 for 3.5 days in the presence of UNC2170 or UNC2892 at the indicated concentrations as compared to untreated cells. Averages represent triplicate cultures from four independent experiments. P value (* p<0.0001) was determined by a two-tailed t test assuming unequal variance.
CONCLUSIONS
We have shown that the 53BP1 ligand, UNC2170, while only modestly potent, demonstrates at least 17-fold selectivity for 53BP1 as compared to nine other Kme reader proteins. The binding of UNC2170 is also dependent upon a functional tudor domain as demonstrated by site directed mutagenesis and ITC studies. A co-crystal structure revealed that the small molecule ligand binds within the same Kme binding pocket as endogenous Kme peptides. However, distinct from Kme peptide ligands, UNC2170 engages the binding pockets of two 53BP1 tudor domains. This simple, relatively non-toxic ligand also exhibits modest activity as a 53BP1 antagonist in cellular lysates and has functional consequences in CSR assays consistent with its weak in vitro activity. A closely related negative control compound, UNC2892, lacks both in vitro and cellular effects, suggesting that the results observed with UNC2170 are indeed due to the inhibition of 53BP1. Thus, UNC2170 is a novel small molecule ligand of 53BP1, a Kme reader protein that has gained much attention recently due to its integral role in DNA damage repair and its link to BRCA1. Our preliminary SAR studies revealed only modest improvements in potency, and further exploitation of the structural data should facilitate the development of a high-quality chemical probe for 53BP1. This work also aids in demonstrating the potential for Kme readers to be modulated via small molecule intervention, despite the limited research in this field to date.
METHODS
AlphaScreen
The AlphaScreen assay was generally performed as previously described.(37) In brief, compound plates (1 µL at 10 mM highest concentration; 3-fold, 10-point dilutions in DMSO) were diluted in 1× assay buffer (20 mM TRIS pH 8.0, 25 mM NaCl, 2 mM DTT and 0.05% Tween-20) to 1 mM using a Multimek robotic pipettor (Nanoscreen) and 1 µL was spotted into the wells of 384-well low-volume Proxiplates (PerkinElmer). To these plates 9 µL of protein-peptide mix in 1× assay buffer was added by Multidrop (Thermo) to bring the final compound concentration to 100 µM and incubated for 30 min at room temperature. Next, 2 µL of a 1:1 mixture of streptavidin-conjugate donor and nickel-chelate acceptor beads (45 µg/mL in 1× assay buffer) were added and the plates were allowed to incubate for an additional 30 min in the dark at room temperature. After incubation, the plates were read on an EnVision multi-label reader equipped with an HTS AlphaScreen laser (Perkin Elmer). The IC50 values reported are the average of at least 3 values ± the standard deviation. When IC50 values for a single compound were not all active (< 100 µM) or inactive (> 100 µM), the IC50 values were calculated using 4-paramter curve fitting (GraphPad Prism 5) from replicate runs using averaged response values for each compound concentration.
Chromatin Fractionation Experiments
Chromatin was isolated from HEK293 cells. Cells were lysed in EBC-1 buffer (Tris pH 7.5, 100mM NaCl, 0.5% NP-40, 1mM EDTA), nuclear pellets were collected, washed in EBC-1 and resuspended in EBC-2 (Tris pH7.5, 300mM NaCl, 5mM CaCl2) for 30 minutes. The insoluble chromatin was pelleted and resuspended in EBC-2 for use in the release assay. This chromatin was incubated with the indicated concentration of compounds for 2 hrs at RT. The supernatant was collected and analyzed as the soluble fraction while the remaining pellet was resuspended in loading buffer, sonicated, and analyzed as the chromatin bound fraction. Anti-53BP1 (Sigma B4436) and anti-histone 3 (loading control) were used in western blots.
Crystallization
Purified 53BP1 (42 mg/mL) in 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM TCEP was pre-incubated with 10 mM UNC2170 (dissolved in water) and the best crystals were obtained by vapor diffusion technique at 20 °C in sitting drops by mixing 1 µL of protein solution with 1 µL of reservoir solution containing 19% PEG3350, 150 mM DL-malic acid pH 7.2. For cryoprotection, the crystals were soaked in the reservoir solution supplemented with 15% ethylene glycol (v/v) for 60 s before flash freezing in liquid N2.
CSR Experiments
B cell isolation and culture have been previously described.(50) In brief, primary naive B-lymphocytes from WT C57/BL6 mouse spleens were purified by negative selection with anti-CD43 beads (Miltenyi Biotec) and cultured in RPMI 1640, 1 mM sodium pyruvate, 10% fetal bovine serum (Atlanta Biologicals,), 50 uM 2-mercaptoethanol, 25 µg/ml Lipopolysaccharide(LPS) and 5 ng/ml IL-4 (Sigma-Aldrich). Inhibitors were added at 12 hours culture and cells were analyzed 72 hours later. Cultured splenocytes were stained with anti–mouse IgG1 antibodies (BD). Dead cells were excluded on the basis of forward-side scatter and propidium iodide staining. Cells were analyzed on a LSRFortessa (BD) and data was analyzed with FloJo software. Data is the summary of triplicate culture analysis of 100uM (n=4), 75uM (n=3) and 30uM (n=3) independent experiments. Statistical significance was determined by a two-tailed Student's t test assuming unequal variance, p values indicated.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Hull-Ryde and N. Cheng for support with the CellTiter-Glo cell viability assay and G. Wang for providing PHF1, PHF19, PHF23, and JARID1A protein constructs. We thank M. Bedford for helpful discussions. The research described here was supported by the National Institute of General Medical Sciences, US National Institutes of Health (NIH, grant R01GM100919), the Carolina Partnership and the University Cancer Research Fund, University of North Carolina at Chapel Hill, and the Welch Foundation (G-1847). The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Boehringer Ingelheim, the Canada Foundation for Innovation (CFI), the Canadian Institutes of Health Research (CIHR), Genome Canada, Ontario Genomics Institute Grant OGI-055, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome Trust Grant 092809/Z/10/Z. G.M. acknowledges support from NIH grant R01 CA132878 and Mayo Clinic NCI SPORE programs P50 CA116201 and P50 CA108961. G.C. is supported by a Mayo Clinic Cancer Center Fraternal Order of Eagles Fellowship. B.D.S. acknowledges support from the W.M. Keck Foundation. S.B.R. acknowledges support from NIH grant K99CA181343.
ABBREVIATIONS
- Kme
methyl-lysine
- Kme2
dimethyl-lysine
- H4K20me2
histone H4, lysine 20 dimethyl
- H4K20me1-2
mono- or dimethylation of lysine 20 on histone 4
- SAR
structure activity relationships
- PTM
post-translational modification
- 53BP1
Tumor suppressor p53 binding protein 1
- PHD
Plant homeodomain
- MBT
malignant brain tumor
- L3MBTL3
Lethal(3)malignant brain tumor-like protein 3
- L3MBTL1
Lethal(3)malignant brain tumor-like protein 1
- MBTD1
MBT domain containing 1
- PHF1
PHD finger protein 1
- PHF19
PHD finger protein 19
- PHD23
PHD finger 23
- JARID1A
Lysine (K)-specific demethylase 5A
- CBX7
Chromobox homolog 7
- UHRF1-TTD
Ubiquitin-like, containing PHD and RING finger domains, 1- tandem tudor domain
- UHRF1-TTD
Ubiquitin-like, containing PHD and RING finger domains, 1- tandem tudor domain + PHD finger domain
- ITC
isothermal titration calorimetry
- DDR
DNA Damage Response
- CSR
Class Switch Recombination.
Footnotes
Supporting Information. Supporting figures and tables, a detailed description of the chemical synthesis and characterization, and the materials and experimental methods are described in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes. The structure of 53BP1 bound to UNC2170 (1) has been deposited with the Protein Data Bank as entry 4RG2.
The authors declare no competing financial interest.
Contributor Information
Lindsey I. James, Email: ingerman@email.unc.edu.
Stephen V. Frye, Email: svfrye@email.unc.edu.
REFERENCES
- 1.Huang J, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins. Current opinion in genetics & development. 2008;18:152–158. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nature biotechnology. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Portela A, Esteller M. Epigenetic modifications and human disease. Nature biotechnology. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 7.Shibata A, Barton O, Noon AT, Dahm K, Deckbar D, Goodarzi AA, Lobrich M, Jeggo PA. Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest. Mol Cell Biol. 2010;30:3371–3383. doi: 10.1128/MCB.01644-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 9.Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, Nussenzweig A, Casellas R, Robbiani DF, Nussenzweig MC. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, Xu D, Durocher D. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mochan TA, Venere M, DiTullio RA, Jr, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer research. 2003;63:8586–8591. [PubMed] [Google Scholar]
- 13.Kousholt AN, Fugger K, Hoffmann S, Larsen BD, Menzel T, Sartori AA, Sorensen CS. CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. The Journal of cell biology. 2012;197:869–876. doi: 10.1083/jcb.201111065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. The Journal of cell biology. 2005;170:201–211. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, Lukas J, Lukas C. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 18.Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, Zaheen A, Durocher D, Scharff MD, Martin A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:809–814. doi: 10.1073/pnas.0913790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Molecular cell. 2005;20:801–809. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narod SA. BRCA mutations in the management of breast cancer: the state of the art. Nature reviews. Clinical oncology. 2010;7:702–707. doi: 10.1038/nrclinonc.2010.166. [DOI] [PubMed] [Google Scholar]
- 22.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 23.Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, Date T, Doherty AJ. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem. 2003;278:36487–36495. doi: 10.1074/jbc.M304066200. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Botuyan MV, Lee J, Ward IM, Kim J-E, Thompson JR, Chen J, Mer G. Structural Basis for the Methylation State-Specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kachirskaia I, Shi X, Yamaguchi H, Tanoue K, Wen H, Wang EW, Appella E, Gozani O. Role for 53BP1 Tudor domain recognition of p53 dimethylated at lysine 382 in DNA damage signaling. J Biol Chem. 2008;283:34660–34666. doi: 10.1074/jbc.M806020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol. 2011;18:1345–1350. doi: 10.1038/nsmb.2188. [DOI] [PubMed] [Google Scholar]
- 29.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012 doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, Firpo E, Hui CC, Roberts J, Rossant J, Mak TW. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- 33.Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, Deng CX, Finkel T. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Molecular cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui G, Botuyan MV, Mer G. Preparation of recombinant peptides with site- and degree-specific lysine (13)C-methylation. Biochemistry. 2009;48:3798–3800. doi: 10.1021/bi900348z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20:317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wigle TJ, Herold JM, Senisterra GA, Vedadi M, Kireev DB, Arrowsmith CH, Frye SV, Janzen WP. Screening for inhibitors of low-affinity epigenetic peptide-protein interactions: an AlphaScreen-based assay for antagonists of methyl-lysine binding proteins. J Biomol Screen. 2010;15:62–71. doi: 10.1177/1087057109352902. [DOI] [PubMed] [Google Scholar]
- 38.Shultz MD. Setting expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorganic & medicinal chemistry letters. 2013;23:5980–5991. doi: 10.1016/j.bmcl.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 39.Reynolds CH, Tounge BA, Bembenek SD. Ligand binding efficiency: trends, physical basis, and implications. Journal of medicinal chemistry. 2008;51:2432–2438. doi: 10.1021/jm701255b. [DOI] [PubMed] [Google Scholar]
- 40.Frye SV. The art of the chemical probe. Nature chemical biology. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 41.James LI, Korboukh VK, Krichevsky L, Baughman BM, Herold JM, Norris JL, Jin J, Kireev DB, Janzen WP, Arrowsmith CH, Frye SV. Small-Molecule Ligands of Methyl-Lysine Binding Proteins: Optimization of Selectivity for L3MBTL3. J Med Chem. 2013;56:7358–7371. doi: 10.1021/jm400919p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, MacNevin CJ, Norris JL, Sagum CA, Tempel W, Marcon E, Guo H, Gao C, Huang XP, Duan S, Emili A, Greenblatt JF, Kireev DB, Jin J, Janzen WP, Brown PJ, Bedford MT, Arrowsmith CH, Frye SV. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nature chemical biology. 2013;9:184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herold JM, Wigle TJ, Norris JL, Lam R, Korboukh VK, Gao C, Ingerman LA, Kireev DB, Senisterra G, Vedadi M, Tripathy A, Brown PJ, Arrowsmith CH, Jin J, Janzen WP, Frye SV. Small-molecule ligands of methyl-lysine binding proteins. Journal of medicinal chemistry. 2011;54:2504–2511. doi: 10.1021/jm200045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kireev D, Wigle TJ, Norris-Drouin J, Herold JM, Janzen WP, Frye SV. Identification of Non-Peptide Malignant Brain Tumor (MBT) Repeat Antagonists by Virtual Screening of Commercially Available Compounds. J Med Chem. 2010;53:7625–7631. doi: 10.1021/jm1007374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herold JM, James LI, Korboukh VK, Gao C, Coil KE, Bua DJ, Norris JL, Kireev DB, Brown PJ, Jin J, Janzen WP, Gozani O, Frye SV. Structure-activity relationships of methyl-lysine reader antagonists. Med chem comm. 2012;3:45–51. [Google Scholar]
- 46.Wilcken R, Zimmermann MO, Lange A, Zahn S, Boeckler FM. Using halogen bonds to address the protein backbone: a systematic evaluation. Journal of computer-aided molecular design. 2012;26:935–945. doi: 10.1007/s10822-012-9592-8. [DOI] [PubMed] [Google Scholar]
- 47.Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C, Bricogne G. Global Phasing Ltd. 2011 [Google Scholar]
- 48.Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, Nussenzweig A, Nussenzweig MC. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Molecular cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, Nussenzweig MC, Chen J. 53BP1 is required for class switch recombination. The Journal of cell biology. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gazumyan A, Timachova K, Yuen G, Siden E, Di Virgilio M, Woo EM, Chait BT, Reina San-Martin B, Nussenzweig MC, McBride KM. Amino-terminal phosphorylation of activation-induced cytidine deaminase suppresses c-myc/IgH translocation. Mol Cell Biol. 2011;31:442–449. doi: 10.1128/MCB.00349-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.