

Abstract
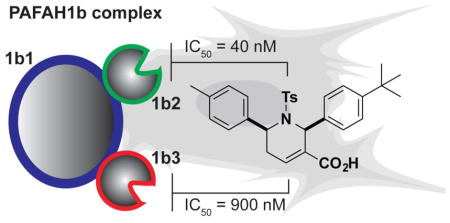
Platelet-activating factor acetylhydrolases (PAFAHs) 1b2 and 1b3 are poorly characterized serine hydrolases that form a complex with a non-catalytic protein (1b1) to regulate brain development, spermatogenesis, and cancer pathogenesis. Determining physiological substrates and biochemical functions for the PAFAH1b complex would benefit from selective chemical probes that can perturb its activity in living systems. Here we report a class of tetrahydropyridine reversible inhibitors of PAFAH1b2/3 discovered using a fluorescence polarization-activity-based protein profiling (fluopol-ABPP) screen of the NIH 300,000+ compound library. The most potent of these agents, P11, exhibited IC50 values of ~40 and 900 nM for PAFAH1b2 and 1b3, respectively. We confirm selective inhibition of PAFAH1b2/3 in cancer cells by P11 using an ABPP protocol adapted for in situ analysis of reversible inhibitors and show that this compound impairs tumor cell survival, supporting a role for PAFAH1b2/3 in cancer.
The platelet-activating factor acetylhydrolase 1b (PAFAH1b) complex is a ~100 kDa heterotrimeric enzyme composed of catalytic 1b2 (30 kDa) and 1b3 (29 kDa) subunits (PAFAH1b2 and PAFAH1b3, or PAFAH1b2/3) that associate with a non-catalytic protein, 1b1 (45 kDa).1–4 Genetic disruption studies in mice support a role for PAFAH1b in diverse physiological processes, including brain formation and spermatogenesis.5, 6 RNA interference-mediated knockdown of PAFAH1b2 in Drosophila and human cells have further implicated this enzyme in the regulation of β-amyloid generation.7 PAFAH1b2 and 1b3 share high sequence similarity (~66% identity) and are members of the serine hydrolase class that adopt an unusual GTPase-like fold that contrasts with the α/β-hydrolase fold more commonly observed for enzymes from this family.4 Despite their unusual three-dimensional structures, PAFAH1b2/3 possess a serine-histidine-aspartic acid triad similar to most other serine hydrolases, and the serine nucleophile of this triad can be covalently modified by fluorophosph(on)ate (FP) inhibitors.3, 8–10
Recombinant PAFAH1b2 and 1b3 both hydrolyze the bioactive lipid platelet-activating factor (PAF) in vitro;1 however, the physiological substrates and functions of these enzymes remain poorly understood. Recently, PAFAH1b3 has been implicated as a metabolic driver of breast cancer progression,10 but again, the precise biochemical functions played by PAFAH1b3 in tumorigenesis are unclear, as cells with perturbed PAFAH1b2/3 expression in this study did not show alterations in PAF content. PAFAH1b2/3 may also have non-catalytic functions as part of the larger PAFAH1b complex. Indeed, deleterious mutations in the PAFAH1b1 subunit (also called LIS1) cause lissencephaly, a severe neuronal migration defect that leads to seizures and mental retardation in humans.2 Biochemical studies suggest that lissencephaly-linked mutations in PAFAH1b1 impair its association with PAFAH1b2/3.6
Selective small-molecule inhibitors of PAFAH1b2/3 are lacking, and the development of such chemical probes would constitute an important step towards elucidating the catalytic and non-catalytic functions of the PAFAH1b complex in mammalian physiology and disease. As serine hydrolases, PAFAH1b2/3 are assayable by activity-based protein profiling (ABPP) methods that use reporter-tagged FP probes to label, detect, enrich, and identify serine hydrolases from native biological systems.11 When performed in a competitive mode, where proteomes are pre-treated with inhibitors, ABPP provides a versatile assay for the discovery and optimization of serine hydrolase inhibitors.12 During the course of performing competitive ABPP screens with diverse libraries of serine hydrolase-directed inhibitors, including carbamates,9 heterocyclic ureas,13 and lactones,14 we were surprised to find that none of these compound collections furnished hits for PAFAH1b2/3. Confronted with this outcome, we turned to high-throughput screening (HTS) as an alternative strategy to discover inhibitors of PAFAH1b2/3.
We have previously shown that ABPP, when coupled with fluorescence polarization (fluopol) as a readout, can provide a robust HTS assay.15 In this fluopol-ABPP method, compounds are evaluated for their ability to block increases in fluopol signal generated by reaction of a fluorescent FP probe with the active site of a purified serine hydrolase target. We recombinantly expressed mouse PAFAH1b2 in E. coli as a His6-tagged protein and confirmed that purified enzyme reacted with a FP-rhodamine (FP-Rh16) probe to generate time-dependent increases in fluopol signal (Figure 1A). At a 90 min time point, where the PAFAH1b2-FP-rhodamine reaction still showed time-dependent increases in signal, suitable Z′ (0.61) and signal-to-background ratio (S:B 3.3) values were obtained in comparison to reactions without enzyme or with a catalytic serine mutant S48A-PAFAH1b2 protein. In collaboration with TSRI’s Screening Center (part of the Molecular Libraries Probe Production Centers Network (MLCPN)), we used our optimized fluopol-ABPP assay conditions to screen the NIH public 300,000+ compound library for PAFAH1b2 inhibitors. 1,118 compounds were identified as active in the screen (defined as showing > 50 percent inhibition of the PAFAH1b2 fluopol signal; 0.37% hit rate; see Figure 1B for a representative subset of primary screening data), and, from these hits, we selected compounds for follow-up studies that had: 1) a < 4% hit rate in all other bioassays reported; and 2) were not active in previous screens performed against other serine hydrolases, including an additional PAF hydrolase (pPAFAH; http://pubchem.ncbi.nlm.nih.gov/). These initial filters yielded 172 candidate PAFAH1b2 inhibitors.
Figure 1.

A Fluopol-ABPP assay identifies PAFAH1b2 inhibitors from the NIH compound library. (A) Time course for fluorescence polarization (fluopol) signal generated by reaction of recombinant mouse PAFAH1b2 (1 μM) with the serine hydrolase-directed activity-based probe FP-Rh (375 nM). No time-dependent increase in fluopol signal was observed in the absence of enzyme or with the catalytically inactive S48A-PAFAH1b2 mutant. The indicated time point (90 min, Z′ = 0.61) was selected for HTS. Data are presented as mean values ± SD for 60–80 wells per group performed in a 384-well plate assay. (B) Representative screening data for 3,200 compounds from the NIH validation set. Compounds that decreased fluopol signal by >50% were designated as hits for PAFAH1b2 (red squares). (C) Structures of tetrahydropyridine hits identified from a full-deck screen of the NIH compound library. (D) Gel-based competitive ABPP counter-screen of hit compounds from HTS. Mouse brain soluble proteome was doped with 1 μM recombinant mouse PAFAH1b2 and treated with compounds (10 μM, 30 min) followed by labeling with FP-alkyne (1 μM, 10 min). Click chemistry with a Rh-azide reporter tag allowed visualization of probe-labeled enzymes by in-gel fluorescence scanning. Fluorescent gels are shown in grayscale.
Filtered hit compounds were next counter-screened by gel-based competitive ABPP at 20 μM using mouse brain soluble proteomes doped with recombinant PAFAH1b2. This secondary assay allowed us to rapidly eliminate compounds that were either false-positives (i.e., showed no evidence of PAFAH1b2 inhibition in the gel-based ABPP assay) or non-selective (i.e., inhibited many serine hydrolase activities in the mouse brain proteome) and focused our attention on a structurally related set of 2,6-cis-diaryl-substituted, 1-tosyl 1,2,5,6-tetrahydropyridine-3-carboxylic acids (referred to hereafter as tetrahydropyridines)17, 18 that showed strong and selective PAFAH1b2 inhibition (Figure 1C and SI Table S1). A subset of compounds bearing diverse aryl groups at the 2- and 6-position was rescreened at a lower concentration of 10 μM against both PAFAH1b2 and 1b3 (Figure 1D and SI Figure S1). Analogues containing aryl groups with bulky para-substituents showed improved PAFAH1b2/3 inhibition (e.g., compare activity of C11 and P11; Figure 1D and SI Figure S1). We next measured IC50 values for a subset of compounds by gel-based ABPP, which revealed that P11 showed particularly good potency against PAFAH1b2 (IC50 value of 0.8 μM; Figure 2A and Table 1). P11 did not show evidence of cross-reactivity with other serine hydrolases, as measured by gel-based ABPP (Figure 2A) or reflected in previous high-throughput screens performed on enzymes from this class (including two other PAF hydrolases, PLA2G7 and PAFAH2, assayed by fluopol-ABPP; SI Table S2). We should note that the IC50 values for P11 and a few other analogues (e.g., B6, H4, K9) approximated the concentration of recombinant PAFAH1b2 (1 μM) in the gel-based ABPP assay, indicating that the potency of these compounds could have been underestimated in this experiment.
Figure 2.

Potency and selectivity of the PAFAH1b2/3 inhibitor P11. (A) Evaluation of potency and selectivity of P11 (A; IC50 = 0.8 μM; 95% confidence interval (C.I.) of 0.4 – 1.6 μM) by gel-based competitive ABPP in the mouse brain soluble proteome doped with recombinant mouse PAFAH1b2 (1 μM). (B, C) PAF hydrolysis substrate assays using purified mouse PAFAH1b2 (B) and human PAFAH1b3 (C) treated with P11, where conversion of PAF to lyso-phosphatidylcholine was measured by LC-MS. See Supporting Information for more details on the PAF substrate assay. For A-D, IC50 values represent the average values and 95% C.I.s determined from three independent experiments. (D) A gel-filtration assay indicates that P11 acts as a reversible inhibitor of PAFAH1b2. Recombinant mouse PAFAH1b2 protein was incubated with DMSO, P11 (10 μM), and the irreversible inhibitor FP-alkyne (5 μM) for 30 min, and each reaction was then split into two fractions. One fraction was reacted directly with FP-Rh (1 μM, 15 min; left panels), and the other was filtered and then reacted with FP-Rh (1 μM, 15 min; right panels) to assess the reversibility of inhibition. (E) ABPP-MudPIT analysis of mouse brain proteomes treated with P11 (5 μM, 30 min, 37 °C, red) or DMSO (black) revealed that P11 significantly inhibited PAFAH1b2 and PAFAH1b3, but not other brain serine hydrolases. **p < 0.01 for P11- versus DMSO-treated samples. Data are presented as mean values ± SEM for three independent experiments performed on brain membrane and soluble proteomes (spectral count signals shown in the graph are summed from both proteomic fractions).
Table 1.
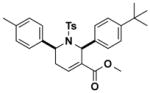
Structures and IC50 values of functionalized tetrahydropyridine PAFAH1b2 inhibitors. Inhibitors were screened by gel-based ABPP with FP-alkyne in mouse brain soluble proteomes doped with recombinant mouse PAFAH1b2 (1 μM). IC50 values are reported as means and 95% confidence intervals from three independent experiments. Ts, tosyl.
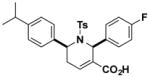
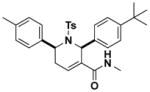
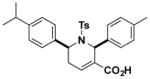
| Compound | Structure | IC50 μM (95% C.I.) | Compound | Structure | IC50 μM (95% C.I.) |
|---|---|---|---|---|---|
| B6 |
|
1.1 (0.6–2.4) | JW1036 |
|
> 10 |
| F5 |
|
4.5 (2.3–7.1) | JW1038 |
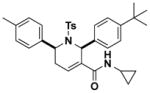
|
9.3 (6.2–13.3) |
| H4 |
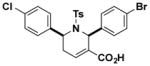
|
1.1 (0.2–3.2) | JW1039 |
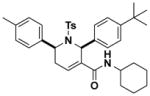
|
> 50 |
| J8 |
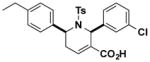
|
9.4 (7.1–14.8) | JW1041 |
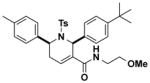
|
1.7 (1.2–2.3) |
| J13 |
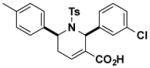
|
8.2 (5.8–12.3) | JW1043 |
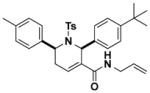
|
7.5 (5.2–10.1) |
| K9 |
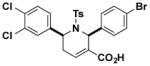
|
1.4 (1.1–2.0) | JW1058 |
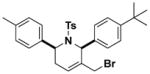
|
4.6 (2.3–7.2) |
| O8 |
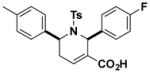
|
2.0 (1.3–2.8) | JW1061 |
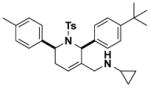
|
7.6 (1.7–15.2) |
| P11 |
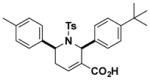
|
0.8 (0.4–1.6) | JW1068 |
|
> 10 |
Most of the tetrahydropyridines deposited in the NIH library differed in their aromatic substituents (Figure 1C and Table 1). We therefore next synthesized a series of analogues of P11, where the carboxylic acid in the 3 position of the 1,2,5,6-tetrahydropyridine ring was modified (Table 1 and SI Table S3). In general, conversion of the carboxylic acid to an amide or ester diminished or abolished activity towards PAFAH1b2 (Table 1, SI Figure S2 and Table S3). Some of the analogues containing smaller amide substituents on the tetrahydropyridine ring, such as JW1041, retained inhibitor activity against PAFAH1b2 (Table 1), but replacement of the carboxylate with a larger amide groups yielded inactive compounds (e.g., JW1039; IC50 > 10 μM, Figure 2B, SI Figure S2B, and Table 1). The poor solubility of analogues with bulky amide substituents, however, limited their use as inactive control probes.
We next confirmed that P11 blocks PAFAH1b2- and PAFAH1b3-mediated hydrolysis of the substrate PAF to lyso-PAF with IC50 values of 36 (Figure 2C) and 880 nM (Figure 2D), respectively. The predicted CLogP value of P11 is high (7.8), and we therefore wanted to provide further evidence that P11 inhibits PAFAH1b2 by a specific mechanism (as opposed to non-specific aggregation19). Compounds that produce activity through aggregation often show steep dose-response curves reflected in high Hill coefficients20 and reduced activity in the presence of detergents.21 The calculated Hill slope for P11 inhibition of PAFAH1b2, however, was 0.99 (0.81–1.2 95% confidence limits), which is a value consistent with single-site binding of the inhibitor. Additionally, the inhibitory activity of P11 for PAFAH1b2, as measured by competitive gel-based ABPP, was unaffected by the presence of detergent (0.01% Triton X-100; compare data in Figure 2A versus SI Figure S3A). These data, taken together, indicate that P11 inhibits PAFAH1b2 through a specific mechanism, rather than by a non-specific aggregation effect.
Because P11 possesses an α,β-unsaturated carboxylic acid moiety, which could, in principle, serve as a site for covalent reactivity, we next tested whether this compound acted as a reversible or irreversible inhibitor of PAFAH1b2. Reversibility of inhibition was evaluated using a gel filtration assay, where recombinant PAFAH1b2 was incubated with P11 (10 μM) or the irreversible probe FP-alkyne22 (5 μM) for 30 min, after which time, the samples were split and one portion was treated directly with the FP-Rh probe and the second portion was subject to gel filtration to remove inhibitors prior to FP-Rh treatment. This experiment showed that the FP-Rh reactivity of PAFAH1b2 was blocked by P11 prior to, but not after gel filtration (Figure 2E), a profile that is consistent with reversible inhibition of the enzyme. In contrast, FP-alkyne retained inhibitory activity of PAFAH1b2 after gel filtration, as expected for an irreversible inhibitor (Figure 2E).
Our initial gel-based ABPP experiments suggested that P11 selectively inhibited PAFAH1b2 compared to other serine hydrolase activities in the mouse brain (Figure 1D), and we next wanted to confirm and extend these findings using the more in-depth mass spectrometry (MS) platform ABPP-MudPIT.23 Mouse brain proteome was treated with DMSO or P11 (5 μM) followed by FP-biotin (1 μM, 30 min), and FP-biotin-labeled proteins were then enriched by streptavidin chromatography, digested on-bead with trypsin, and the resulting peptides analyzed by multidimensional LC-MS. Comparison of spectral count values for enriched serine hydrolases from DMSO- versus P11-treated brain proteomes enabled detection of targets of P11. Across the 35 mouse brain serine hydrolases measured in this study, only PAFAH1b2 and 1b3 were inhibited by P11 (Figure 2F and SI Table S4). P11 also selectively inhibited the human PAFAH1b2 enzyme compared to other human serine hydrolases as measured by competitive ABPP of PC3 prostate cancer cell proteomes (SI Figure S3B and SI Table S5). Human PAFAH1b3 was not detected in PC3 cells, consistent with previous ABPP and gene expression studies indicating that PAFAH1b3 is expressed at very low levels in this cell line.24 These results indicate that P11 acts as a selective inhibitor of human and mouse PAFAH1b2/3 enzymes.
We next set out to determine whether P11 selectively inactivated PAFAH1b2/3 in cells. Confirming in situ target engagement for reversible inhibitors can be challenging, especially for enzymes like PAFAH1b2/3, for which endogenous substrates and products have not yet been discovered.25 While conventional ABPP methods have been used for ex vivo assessment of interactions between reversible inhibitors and enzymes in living systems by exposing proteomic lysates from inhibitor-treated cells26 or animals27 to ABPP probes, we worried that the dilution effect caused by cell lysis might distort the detection of P11-binding to PAFAH1b2/3. To address this issue, we performed competitive ABPP studies in situ by treating cells sequentially with P11 (1 or 10 μM) and the cell-permeable FP-alkyne probe22 (1 μM, 30 min) (Figure 3A). We anticipated that limiting the incubation time of cells with FP-alkyne would preserve a window for measuring reversible inhibition of PAFAH1b2/3 and other serine hydrolases.
Figure 3.

Assessment of PAFAH1b2/3 inhibition in cancer cells. (A) Schematic representation of an in situ competitive ABPP assay for measuring target engagement and selectivity for reversible inhibitors of serine hydrolases. (B) Concentration-dependent inhibition of PAFAH1b2/3 by P11 in Neuro2A cells as measured by competitive ABPP. FP-alkyne-labeled proteins were visualized by click chemistry conjugation of Rh-azide and detection by in-gel fluorescence scanning. Note that we could not confidently assign the PAFAH1b2 and PAFAH1b3 signals by gel-based ABPP, so the gel band signals have been designated as PAFAH1b2/3. (C) Degree of PAFAH1b2/3 inhibition calculated from integrated band intensities measured in part (B). Data are presented as mean values ± SEM for three independent experiments. (D) ABPP-SILAC analysis of inhibitor-treated Neuro2A cells. Isotopically labeled light and heavy amino acid-labeled cells were treated with DMSO or P11 (10 μM), respectively, for 4 h prior to treatment with FP-alkyne (1 μM, 30 min). Data are reported as mean values ± SEM of heavy/light ratios for multiple peptides detected for each enzyme (minimum of 2 unique peptides per enzyme) in combined soluble and membrane proteomes from two independent experiments. (E, F) P11 (10 μM) impaired serum-free survival of Neuro2A (E) and PC3 cells (F) compared to cells treated with DMSO as a control. **p < 0.01 for P11 versus DMSO treatment groups.
We first confirmed that FP-alkyne could detect endogenous PAFAH1b2/3 activity in mouse neuroblastoma Neuro2A proteomes. We treated Neuro2A soluble proteome in vitro with P11 followed by FP-alkyne, and FP-labeled proteins were visualized by copper-catalyzed azide-alkyne cycloaddition (or click) chemistry28, 29 conjugation with an Rh-azide reporter tag and SDS-PAGE analysis.30 P11 selectively inhibited in a concentration-dependent manner the FP-labeling of two ~28–30 kDa proteins, which match the mass of PAFAH1b2/3 (SI Figure S3C). We next performed an in situ competitive ABPP study by treating Neuro2A cells with DMSO or varying concentrations of P11 for 4 h followed by FP-alkyne (1 μM, 30 min). Cells were then lysed and FP-labeled proteins detected as described above. Again, we observed that P11 selectively inhibited two 28–30 kDa serine hydrolase activities predicted to represent PAFAH1b2/3 in Neuro2A cells (Figure 3B, C). To confirm these protein assignments, we analyzed inhibitor-treated cells using the quantitative MS proteomic method ABPP-SILAC13 (Figure 3A), which revealed that P11 (10 μM, 4 h) near-completely blocked FP-alkyne labeling of PAFAH1b2 and 1b3 in Neuro2A cells, while showing negligible effects on the 30+ other detected serine hydrolases (Figure 3D and SI Table S5). Treatment of Neuro2A cells with a lower concentration of P11 (1 μM, 4 h) produced partial (~50%) PAFAH1b2/3 inhibition as measured by ABPP-SILAC (SI Figure S3D and Table S5), matching the concentration-dependent inhibition profiles observed for the 30 kDa PAFAH1b2/3 band observed by gel-based ABPP (Figure 3B, C). These data indicate that P11 is a selective, in situ-active inhibitor of PAFAH1b2/3 that can be used to investigate the cellular functions of PAFAH1b2/3.
PAFAH1b2 and PAFAH1b3 have recently been shown to support cancer pathogenicity.10 Specifically, RNA interference-mediated knockdown of either serine hydrolase impaired cancer cell survival and tumor growth in vivo. Consistent with these findings, P11 significantly decreased the survival of multiple human and mouse cancer cell lines grown under serum-starvation conditions (Figure 3E, F). These studies suggest that the pro-tumorigenic functions of PAFAH1b2/3 depend on the activity, rather than just expression, of these enzymes.
In summary, we used a fluopol-ABPP assay to screen the NIH 300,000+ compound library and discovered a set of functionalized 1,2,5,6-tetrahydropyridines that reversibly inhibit the poorly characterized serine hydrolases PAFAH1b2 and 1b3. One of the most potent inhibitors P11 near-completely and selectively blocked both PAFAH1b2 and 1b3 in cancer cells. SAR studies identified features of P11 required for potent PAFAH1b2/3 inhibition, the presence of bulky para-substituents on the aromatic rings in the 2 and 6 positions of the tetrahydropyridine scaffold and a carboxylic acid in the 3 position. Using P11, we found that pharmacological inhibition of PAFAH1b2/3 activity impairs cancer cell survival.
The tetrahydropyridine inhibitors of PAFAH1b2/3 discovered herein do not, to our knowledge, share structural similarity to other classes of serine hydrolase inhibitors and are also minimally related to the rest of the NIH library (SI Figure S4). Notably, the tetrahydropyridines reflect a contribution to the public library by an academic synthetic chemistry lab – Dr. Ohyun Kwon (UCLA)17, 18 – providing another compelling example of how the structural and functional diversity of the NIH compound collection is enriched by deposition of compounds from academic chemistry groups.31
Projecting forward, we believe that P11 will serve as a valuable chemical probe for the investigation of PAFAH1b2/3 function in mammalian biology. P11 showed excellent selectivity for PAFAH1b2/3 compared to other serine hydrolases, as measured by in vitro and in situ competitive ABPP studies; however, P11 could still interact with other proteins outside of the serine hydrolase class in cells. For instance, structurally related compounds have been shown to inhibit geranygeranyltransferase type I (GGTase-I),18, 32 although P11 itself displayed negligible activity against GGTase-1 in these past studies (< 20% inhibition at 50 μM). Further experiments are also needed to assess whether P11 can inhibit PAFAH1b2/3 in vivo. Confirmation of in vivo-target engagement for reversible inhibitors of enzymes can be challenging in the absence of bona fide substrate or product biomarkers, but has been achieved by competitive ABPP using kinetically attenuated probes33 or by performing ABPP experiments ex vivo in tissue lysates from inhibitor-treated animals.27 The pursuit of in vivo-active inhibitors and inactive control probes would also benefit from improvements in the physicochemical properties of P11 and its tetrahydropyridine analogues. The development of inhibitors that can selectively block either PAFAH1b2 or 1b3 is another important future goal to dissect the unique and overlapping functions of these enzymes. Our results have already shown that P11 exhibits ~10–20-fold greater potency for PAFAH1b2 over 1b3, indicating that further medicinal chemistry studies of the tetrahydropyridine scaffold could lead to PAFAH1b2-selective inhibitors. Finally, we are hopeful that pharmacological studies with P11, when combined with metabolomics and proteomic platforms to broadly assess the impact of PAFAH1b2/3 inhibition on cellular biochemistry, will lead to the discovery of endogenous substrate for these enzymes. This knowledge could in turn provide mechanistic insights into the functions played by PAFAH1b2/3 in cancer and other pathophysiological processes.
METHODS
Recombinant PAFAH1b2 Protein Expression and Purification
Mouse recombinant wild-type PAFAH1b2 was subcloned into pET-45b(+) (Novagen). BL21 (DE3) Escherichia coli containing this vector were grown in LB media containing 75 mg/L carbenicillin with shaking at 37 °C to an OD600 of 0.5. The cells were then induced with 1 mM IPTG and harvested 4 h later by centrifugation. Cells were lysed by stirring for 20 min at 4 °C in Tris buffer [50 mM Tris-HCl (pH 8.0) with 150 mM NaCl] and supplemented with 1 mg/mL lysozyme and 1 mg/mL DNase I. The lysate was then sonicated and centrifuged at 10,000 × g for 10 min. Talon cobalt affinity resin (Clontech; 400 μL of slurry / 10 g of cell paste) was added to the supernatant, and the mixture was rotated at room temperature for 1 h. Beads were collected by centrifugation at 1,400 × g for 3 min, washed twice with Tris buffer, and applied to a 1 cm gravity column. The column was washed twice with Tris buffer (10 mL / 400 μL of resin slurry) and Tris buffer with 500 mM NaCl once. The bound protein was eluted by the addition of 100 mM imidazole (2.5 mL). Imidazole was removed by passage over a Sephadex G-25M column (GE Healthcare). Protein concentration was determined using the BioRad DC Protein Assay kit. A S48A mutation was introduced into the mouse PAFAH1b2-pET-45b(+) construct using the Quikchange Site-Directed Mutagenesis Kit (Stratagene), and the resulting mutant protein was expressed following conditions described above.
PAFAH1b2 Fluopol-ABPP Assay
Prior to the start of the assay, 4.0 μL of Assay Buffer (0.01% Pluronic acid, 50 mM Tris HCl pH 8.0, 150 mM NaCl, 1 mM DTT) containing 1.25 μM of purified PAFAH1b2 protein were dispensed into 1536 microtiter plates. Next, 17 nL of test compound in DMSO (3.39 μM compound concentration) or DMSO alone (0.34% final concentration) were added to the appropriate wells and incubated for 120 min at 25 °C. The fluopol-ABPP assay was started by dispensing 1.0 μL of 375 nM FP-Rh probe in Assay Buffer to all wells. Plates were incubated for 90 min at 25 °C. Fluorescence polarization was read on a Viewlux microplate reader (PerkinElmer, Turku, Finland) using a BODIPY TMR FP filter set and a BODIPY dichroic mirror (excitation = 525 nm, emission = 598 nm) for 15 s for each polarization plane (parallel and perpendicular).
Details describing analysis and filtering of primary screening data can be found in Supporting Information.
Biochemical and Cell Biological Assays
See Supporting Information for details on preparation of proteomes and analysis by gel-based competitive ABPP, LC-MS PAF hydrolysis assays, gel filtration experiments, and cell survival assays.
Competitive ABPP-MudPIT Analysis
ABPP-MudPIT analyses were performed as previously described.23 Details can be found in Supporting Information.
In situ Competitive ABPP Assay for Reversible Inhibitors
Neuro2A cells cultured in isotopically heavy or light DMEM media were treated with compound or DMSO (2% final concentration), respectively, for 4 h at 37 °C in a 15 cm dish (20 mL total media volume). FP-alkyne (1 μM) was then added to both heavy and light cells for 30 min at 37 °C followed by removal of media and washing of cells with PBS (2x, 10 mL). Cells were scraped into 10 mL of PBS and isolated by centrifugation at 1,400 × g for 3 min. The cell pellet was resuspended in 1 mL of PBS, lysed, and membrane and soluble fractions isolated by centrifugation at 100,000 × g for 45 min. Total protein concentrations of each fraction were adjusted to 2 mg/mL in PBS (500 μL total reaction volume). Each fraction was subjected independently to click chemistry by addition of 200 μM Biotin-N3 (20 μL of a 50x stock in DMSO), 2 mM TCEP (40 μL of a 25x stock in DMSO), 200 μM TBTA (120 μL of a 17x stock in 4:1 t-butanol:DMSO), and 2 mM CuSO4 (40μL of a 25x stock in DMSO). For gel-based analysis, 25 μM of Rhodamine-N3 was used following the protocol described above. Please note that the click chemistry step quenches the FP-alkyne labeling reaction. Therefore, the steps prior to click chemistry should be performed in a rapid manner. High resolution LC-MS/MS analysis on a Thermo LTQ-Orbitrap mass spectrometer was performed as previously described.13 Additional details can be found in Supporting Information.
In vitro ABPP-SILAC analysis
PC3 cells were cultured in isotopically heavy and light RPMI media. Both heavy and light cells were harvested and separated into membrane and soluble fractions as described above. Total protein concentrations of each fraction were adjusted to 2 mg/mL in PBS (500 μL total reaction volume). Heavy and light proteomes were treated with P11 (10 μM) or DMSO (2% final concentration), respectively, for 30 min at 37 °C followed by treatment of proteomes with FP-biotin (1 μM) for 30 minutes at 37 °C. Samples were prepared for LC-MS/MS analysis as described above for Neuro2A proteomes. See Supporting Information for more details.
Compound Similarity Analysis
The MLSMR library (~330K compounds at the time of screening) was compared to the tetrahydropyridine active compounds using a fingerprint based approach. Extended connectivity fingerprints with a path length of 6 (ECFP-6) and Tanimoto similarities were calculated with Biovia Pipeline Pilot. The closest pairwise similarities between each of the MLSMR compounds and any of the tetrahydropyridines were then plotted with the histogram tool in Microsoft Excel’s Data Analysis Add-in.
Chemical Synthesis and Characterization
Tetrahydropyridines were synthesized following previously reported procedures.17 See Supporting Information for detailed synthetic procedures and chemical characterization.
Supplementary Material
Acknowledgments
We thank Kim Masuda and Chris Joslyn for technical assistance. This work was supported by the National Institutes of Health (DA033760, DA035864, MH084512 GM071779 and GM081282), the National Science Foundation (predoctoral fellowship to A.M.Z.), and the Skaggs Institute for Chemical Biology.
Footnotes
Detailed supplemental methods, synthetic procedures, and characterization of tetrahydropyridines, 1H-NMR spectra, HRMS data, and supplemental figures are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. The catalytic subunit of bovine brain platelet-activating factor acetylhydrolase is a novel type of serine esterase. J Biol Chem. 1994;269(37):23150–5. [PubMed] [Google Scholar]
- 2.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase [corrected] Nature. 1994;370(6486):216–8. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- 3.Hattori M, Adachi H, Aoki J, Tsujimoto M, Arai H, Inoue K. Cloning and expression of a cDNA encoding the beta-subunit (30-kDa subunit) of bovine brain platelet-activating factor acetylhydrolase. J Biol Chem. 1995;270(52):31345–52. doi: 10.1074/jbc.270.52.31345. [DOI] [PubMed] [Google Scholar]
- 4.Ho YS, Swenson L, Derewenda U, Serre L, Wei Y, Dauter Z, Hattori M, Adachi T, Aoki J, Arai H, Inoue K, Derewenda ZS. Brain acetylhydrolase that inactivates platelet-activating factor is a G-protein-like trimer. Nature. 1997;38(6611):89–93. doi: 10.1038/385089a0. [DOI] [PubMed] [Google Scholar]
- 5.Yan W, Assadi AH, Wynshaw-Boris A, Eichele G, Matzuk MM, Clark GD. Previously uncharacterized roles of platelet-activating factor acetylhydrolase 1b complex in mouse spermatogenesis. Proc Natl Acad Sci U S A. 2003;100(12):7189–94. doi: 10.1073/pnas.1236145100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sweeney KJ, Clark GD, Prokscha A, Dobyns WB, Eichele G. Lissencephaly associated mutations suggest a requirement for the PAFAH1B heterotrimeric complex in brain development. Mechanisms of development. 2000;92(2):263–71. doi: 10.1016/s0925-4773(00)00242-2. [DOI] [PubMed] [Google Scholar]
- 7.Page RM, Munch A, Horn T, Kuhn PH, Colombo A, Reiner O, Boutros M, Steiner H, Lichtenthaler SF, Haass C. Loss of PAFAH1B2 reduces amyloid-beta generation by promoting the degradation of amyloid precursor protein C-terminal fragments. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32(50):18204–14. doi: 10.1523/JNEUROSCI.2681-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hattori M, Arai H, Inoue K. Purification and characterization of bovine brain platelet-activating factor acetylhydrolase. J Biol Chem. 1993;268(25):18748–53. [PubMed] [Google Scholar]
- 9.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A. 2010;107(49):20941–6. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulvihill MM, Benjamin DI, Ji X, Le Scolan E, Louie SM, Shieh A, Green M, Narasimhalu T, Morris PJ, Luo K, Nomura DK. Metabolic profiling reveals PAFAH1B3 as a critical driver of breast cancer pathogenicity. Chem Biol. 2014;21(7):831–40. doi: 10.1016/j.chembiol.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon GM, Cravatt BF. Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J Biol Chem. 2010;285(15):11051–5. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niphakis MJ, Cravatt BF. Enzyme inhibitor discovery by activity-based protein profiling. Annu Rev Biochem. 2014;83:341–77. doi: 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- 13.Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat Chem Biol. 2011;7(7):469–78. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoover HS, Blankman JL, Niessen S, Cravatt BF. Selectivity of inhibitors of endocannabinoid biosynthesis evaluated by activity-based protein profiling. Bioorganic & medicinal chemistry letters. 2008;18(22):5838–41. doi: 10.1016/j.bmcl.2008.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat Biotechnol. 2009;27(4):387–94. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Direct visualization of serine hydrolase activities in complex proteome using fluorescent active site-directed probes. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Zhu XF, Lan J, Kwon O. An expedient phosphine-catalyzed [4 + 2] annulation: synthesis of highly functionalized tetrahydropyridines. J Am Chem Soc. 2003;125(16):4716–7. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]
- 18.Castellano S, Fiji HD, Kinderman SS, Watanabe M, Leon P, Tamanoi F, Kwon O. Small-molecule inhibitors of protein geranylgeranyltransferase type I. J Am Chem Soc. 2007;129(18):5843–5. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46(21):4477–86. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 20.Shoichet BK. Interpreting steep dose-response curves in early inhibitor discovery. J Med Chem. 2006;49(25):7274–7. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 21.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50(10):2385–90. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 22.Lone AM, Bachovchin DA, Westwood DB, Speers AE, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Rosen H, Cravatt BF, Saghatelian A. A Substrate-Free Activity-Based Protein Profiling Screen for the Discovery of Selective PREPL Inhibitors. J Am Chem Soc. 2011 doi: 10.1021/ja2036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, 3rd, Jeffrey SS, Cravatt BF. A streamlined platform for high-content functional proteomics of primary human specimens. Nat Methods. 2005;2(9):691–7. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 24.Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol. 2011;18:846–856. doi: 10.1016/j.chembiol.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon GM, Niphakis MJ, Cravatt BF. Determining target engagement in living systems. Nat Chem Biol. 2013;9(4):200–5. doi: 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagano JM, Hsu KL, Whitby LR, Niphakis MJ, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Hodder P, Srinivasan P, Gonzalez TD, Rosen H, Bahnson BJ, Cravatt BF. Selective inhibitors and tailored activity probes for lipoprotein-associated phospholipase A(2) Bioorganic & medicinal chemistry letters. 2013;23(3):839–43. doi: 10.1016/j.bmcl.2012.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi HG, Zhang J, Deng X, Hatcher JM, Patricelli MP, Zhao Z, Alessi DR, Gray NS. Brain Penetrant LRRK2 Inhibitor. ACS Med Chem Lett. 2012;3(8):658–662. doi: 10.1021/ml300123a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rostovtsev VV, Green JG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 29.Tornoe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. The Journal of organic chemistry. 2002;67(9):3057–64. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 30.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schurer SC, Nomura DK, Rosen H, Fu GC, Cravatt BF. Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc Natl Acad Sci U S A. 2011;108(17):6811–6. doi: 10.1073/pnas.1015248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe M, Fiji HD, Guo L, Chan L, Kinderman SS, Slamon DJ, Kwon O, Tamanoi F. Inhibitors of protein geranylgeranyltransferase I and Rab geranylgeranyltransferase identified from a library of allenoate-derived compounds. J Biol Chem. 2008;283(15):9571–9. doi: 10.1074/jbc.M706229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adibekian A, Martin BR, Chang JW, Hsu KL, Tsuboi K, Bachovchin DA, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Hodder PS, Rosen H, Cravatt BF. Confirming target engagement for reversible inhibitors in vivo by kinetically tuned activity-based probes. J Am Chem Soc. 2012;134(25):10345–8. doi: 10.1021/ja303400u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.