

Abstract
Nerve injury can lead to axonal regeneration, axonal degeneration, and/or neuronal cell death. Remarkably, the MAP3K dual leucine zipper kinase, DLK, promotes each of these responses, suggesting that DLK is a sensor of axon injury. In Drosophila, mutations in proteins that stabilize the actin and microtubule cytoskeletons activate the DLK pathway, suggesting that DLK may be activated by cytoskeletal disruption. Here we test this model in mammalian sensory neurons. We find that pharmacological agents designed to disrupt either the actin or microtubule cytoskeleton activate the DLK pathway, and that activation is independent of calcium influx or induction of the axon degeneration program. Moreover, activation of the DLK pathway by targeting the cytoskeleton induces a pro-regenerative state, enhancing axon regeneration in response to a subsequent injury in a process akin to preconditioning. This highlights the potential utility of activating the DLK pathway as a method to improve axon regeneration. Moreover, DLK is required for these responses to cytoskeletal perturbations, suggesting that DLK functions as a key neuronal sensor of cytoskeletal damage.
Keywords: DLK, axon regrowth, regeneration, cytoskeleton, neuron, DRG, neuropathy
Introduction
Axons are particularly vulnerable to injury and disease, and axon damage is a hallmark of many neurological disorders. Axon regeneration, axon degeneration, and neuronal cell death are three potential responses to axon injury. The MAP3K dual leucine zipper kinase, DLK, promotes all three of these responses, suggesting that DLK is a sensor of axon injury. Here we explore the activation mechanism for the DLK pathway.
DLK is a MAP3K in the mixed lineage kinase family that can activate the JNK and p38 families of stress MAP kinases (Hirai et al., 1996). In recent years, a series of studies have highlighted the essential role of DLK in promoting axonal regeneration after injury. DLK is required for axonal regeneration in worms, flies, and mice (Hammarlund et al., 2009; Shin et al., 2012; Xiong et al., 2010; Yan et al., 2009). In mice, DLK promotes the retrograde transport of injury signals that are necessary for activating a pro-regenerative program in sensory neurons and retinal ganglion cells (Shin et al., 2012; Watkins et al., 2013). Indeed, DLK is required for the preconditioning response in which a prior axon injury promotes more robust axonal regeneration in response to a subsequent injury (Shin et al., 2012). In mammals, DLK promotes axonal regeneration at least in part via activation of JNK and the subsequent phosphorylation and activation of the transcription factor cJun (Itoh et al., 2009; Shin et al., 2012; Watkins et al., 2013). Indeed, both JNK and Jun are also required for robust regeneration of PNS axons following injury (Rishal and Fainzilber, 2014).
DLK is the upstream kinase in the DLK/JNK/cJun regeneration pathway, and is postulated to be the sensor of axon injury. Hence, it is important to determine what aspect of axon injury induces DLK pathway activation. To date, four methods for activating DLK have been described. First, in Caenorhabditis elegans, an elegant Ca2+-dependent mechanism activates the DLK ortholog DLK-1 (Yan and Jin, 2012). Second, in C. elegans phosphatases act as negative regulators of DLK pathway activation (Baker et al., 2014; Tulgren et al., 2011). Third, DLK is activated by an increase in its protein levels via transgenic overexpression (Collins et al., 2006; Hammarlund et al., 2009; Yan et al., 2009), loss of the PHR ubiquitin ligase (Babetto et al., 2013; Collins et al., 2006; Nakata et al., 2005), or through a positive-feedback loop preventing its degradation (Huntwork-Rodriguez et al., 2013). Finally, we demonstrated in Drosophila that mutations in proteins that are required for a stable actin and microtubule cytoskeleton also activate DLK (Valakh et al., 2013). These findings lead us to hypothesize that cytoskeletal disruption activates the DLK pathway.
In this report we test the hypothesis that cytoskeletal disruption activates the DLK pathway by pharmacologically targeting the actin and microtubule networks in mammalian sensory neurons. Previously, we demonstrated that traumatic axonal injury leads to a DLK-dependent signal from the axon to the nucleus that triggers the phosphorylation of Jun and the induction of a pro-regenerative state in the neuron that improves axonal re-growth in response to a subsequent injury (Shin et al., 2012). Induction of this pro-regenerative state is essential for efficient axonal regeneration in vivo in the mammalian PNS (Tedeschi and Bradke, 2013). Here we find that compounds targeting the cytoskeleton also activate the DLK pathway, and this activation is sufficient to trigger a retrograde injury signal leading to phosphorylation of cJun and activation of a pro-regenerative state in adult sensory neurons. Pretreatment of neurons with cytoskeletal disrupting agents either in vitro or in vivo induces axonal regeneration in response to a subsequent injury. Hence, cytoskeletal perturbation is sufficient to induce the axonal preconditioning response. These findings support the model that cytoskeletal injury activates the DLK pathway. Moreover, DLK is required for the neuronal response to cytoskeletal injury, suggesting that DLK may function as a key sensor of cytoskeletal damage.
Results
Actin or microtubule destabilizing drugs activate the JNK pathway
A Drosophila mutant in the spectraplakin short stop activates the DLK/JNK pathway (Valakh et al., 2013). Short Stop functions as an actin-microtubule cross-linker to promote cytoskeletal stability (Applewhite et al., 2010; Suozzi et al., 2012). We observed a similar phenotype after RNAi knock-down of two subunits of the TCP1 complex, which is a chaperonin that folds actin and tubulin and that is also needed for proper cytoskeletal stability (Grantham et al., 2006; Ursic et al., 1994). These results led us to hypothesize that the DLK/JNK pathway is activated by cytoskeletal destabilization. Here we test this hypothesis by pharmacologically targeting the actin and microtubule cytoskeletons in mammalian sensory neurons.
In mouse, the JNK MAP kinase is the major downstream target of DLK. In DRG neurons, axon injury leads to DLK-dependent JNK phosphorylation of the transcription factor cJun in the nucleus (Ghosh et al., 2011; Shin et al., 2012; Watkins et al., 2013). We tested whether pharmacological manipulation of the cytoskeleton would activate this same pathway by analyzing accumulation of phosphorylated cJun (pcJun) in the nucleus of dissociated embryonic dorsal root ganglia (DRG) neurons. We used cytochalasin D and nocodazole to target the actin and microtubule cytoskeletons respectively, and chose low doses that affect filopodia formation without altering growth cone dynamics (cytochalasin D) or dampen microtubule dynamics while leaving polymer levels intact (nocodazole) (Dent et al., 2007; Gupton and Gertler, 2010; Jaworski et al., 2009; Perlson et al., 2013). Treatment of DRG neurons with cytochalasin D or nocodazole induces an approximately four-fold increase in the levels of pcJun compared to vehicle treatment (Figure 1). To test whether activation of pcJun was indeed through JNK, we treated with cytochalasin D or nocodazole in the presence of the JNK inhibitor SP600125 at doses that effectively block DLK-dependent responses to axon injury (Miller et al., 2009). JNK inhibition completely blocked drug-induced upregulation of pcJun levels in the neuronal cell bodies (Figure 1A,B), demonstrating that low doses of microtubule or actin destabilizers are sufficient to activate the JNK pathway in DRG neurons. This is consistent with findings in other cell types that cytoskeletal stress activates JNK kinases (Kaunas et al., 2006; Ren et al., 2009; Wang et al., 1998).
Figure 1. Actin and microtubule disrupting agents lead to the JNK-dependent phosphorylation of cJun.
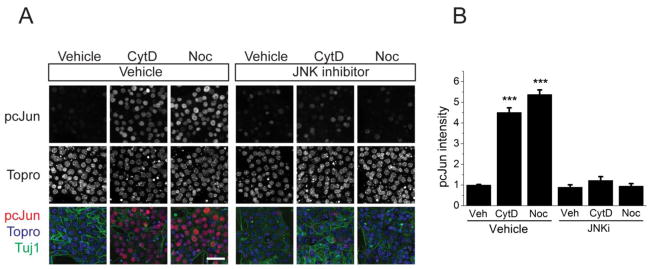
A. Application of either actin or microtubule disrupting drugs increased the levels of pcJun in a JNK-dependent manner. Sample confocal images of day 8 embryonic cultures stained for pcJun (red), β3 tubulin (green) and the nucleic acid dye Topro (blue) after 16-hour vehicle, nocodazole (Noc), or cytochalasin D (CytD) treatment. The pcJun increase was blocked by the JNK inhibitor SP600125 (15 μM). Scale bar = 50 μm.
B. Average pcJun intensity of the nuclei represented in A. ***p<0.001, compared to vehicle treated cells in each condition. n > 6 wells for each condition with more than 100 cells quantified for each well. Error bars indicate SEM.
Axonal cytoskeletal destabilization is sufficient to induce a cell-body response
Axonal injury in vivo induces a retrograde signal that originates at the axonal injury site and travels to the cell body where it triggers phosphorylation of cJun in the nucleus and induction of a pro-regenerative state (Shin et al., 2012). To investigate whether cytoskeletal destabilizing agents can activate a similar retrograde signal, we tested whether targeting the cytoskeleton exclusively in axons is sufficient to induce phosphorylation of cJun in the nucleus. To do this, we cultured embryonic DRG neurons in Campenot compartmentalized chambers, which provide fluid separation between the axonal and cell body compartments ((Campenot, 1982; Chan et al., 2004), Figure 2A). To target the cytoskeleton exclusively in the axon, we applied either nocodazole or cytochalasin D to the axonal compartment. Not all neurons send their axons into the axonal chamber, so we labeled those neurons with a retrograde tracer, wheat germ agglutinin conjugated to Alexa488 (WGA). Since the tracer only labels the cell bodies whose axons were exposed to drug, we were able to compare the levels of pcJun between drug treated and untreated cells in the same chamber. We find an approximately twofold increase in the intensity of pcJun staining in the cells whose axons were exposed to either cytochalasin D or nocodazole (Figure 2B,C). Thus, applying cytoskeletal destabilizing drugs exclusively in the axonal compartment is sufficient to induce an increase in pcJun levels in the cell body. Hence cytoskeletal disruption, like a nerve crush, is sufficient to activate a retrograde signal from axon to nucleus.
Figure 2. Cytoskeletal disrupting agents in the axon compartment induces the accumulation of pcJun in the cell body.

A. A schematic of a Campenot chamber used for compartmentalized cultures.
B. Sample confocal images of the cellular compartment of the Campenot chambers after addition of cytochalasin D (CytD) or nocodazole (Noc) along with the retrograde tracer WGA (green). The cell bodies are stained for pcJun (red) and a nuclear marker Topro dye (blue). Cells that are positive for WGA (green) show pcJun staining in their nuclei (arrows), indicating that treating the actin or microtubule cytoskeleton exclusively in the axon induces a retrograde signal leading to phosphorylation of cJun in the nucleus. Arrowheads point to cells that are negative for WGA, and hence were not exposed to cytochalasin D or nocodazole and so do not accumulate pcJun. Scale bar = 20μm.
C. Quantification of the pcJun intensity from the conditions in B. The cells positive for WGA are normalized to those negative for WGA from the same chamber. *p<0.05 compared to vehicle treated cells. Error bars represent SEM.
DLK is required for JNK pathway activation in response to cytoskeletal disruption
JNK is a target of many upstream kinases (Johnson and Nakamura, 2007), however in vivo DLK is the required MAP3K for JNK-dependent cJun phosphorylation in response to peripheral nerve injury (Shin et al., 2012). Here we tested whether DLK is also the required MAP3K for JNK-dependent cJun phosphorylation in response to microtubule and actin destabilizing drugs. We used a previously described conditional DLK allele (Miller et al., 2009) and delivered Cre recombinase to the cultured neurons via viral infection. In control neurons infected with an empty vector, nocodazole and cytochalasin D treatment robustly upregulates pcJun levels. However in the absence of DLK (Cre infected cells), the same drugs do not lead to an increase in pcJun levels (Figure 3). These data demonstrate that a single upstream MAP3K, DLK, is required for the JNK pathway activation following cytoskeletal disruption, and support the model that destabilizing either the actin or microtubule cytoskeleton activates the DLK pathway.
Figure 3. Phosphorylation of cJun in response to cytochalasin D and nocodazole treatment requires DLK.
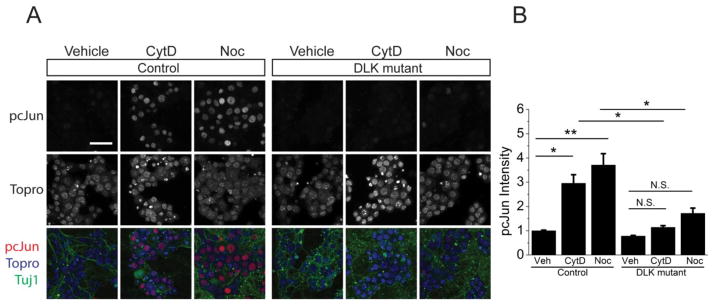
A. Sample confocal images of DLKF/F embryos infected either with an empty vector (control) or a vector with Cre recombinase (DLK mutant). The cells are stained for pcJun (red), β3 tubulin (green) and nucleic acid dye Topro (blue) after a 16-hour vehicle, cytochalasin D (CytD) and nocodazole (Noc) treatment. There is robust upregulation of pcJun in control but not DLK mutant neurons. Scale bar = 50 μm.
B. Normalized quantification of the pcJun intensity of the nuclei represented in A. *p<0.05, **p<0.01, compared to vehicle treated cells in each condition. n > 15 wells for each condition from 4 different mice with more than 100 cells quantified for each well. Error bars indicate SEM.
Calcium is not necessary to induce DLK pathway activation in mammalian neurons
While pharmacological targeting of actin and microtubules shares some features with traumatic nerve injury, there are additional consequences of axonal injury. When axons are cut there is a strong calcium influx that propagates throughout the cell (Avery et al., 2012; Cho and Cavalli, 2012). Indeed, in C. elegans calcium influx leads to an activation of DLK via the dissociation of DLK from an inhibitory isoform (Yan and Jin, 2012). Even though mammalian DLK lacks the hexapeptide involved in the calcium-dependent activation of worm DLK, calcium does play an important signaling role in the injured neuron and so we tested whether calcium influx functions as an intermediate between cytoskeletal disruption and DLK pathway activation.
To block calcium influx, we applied the cytoskeleton-destabilizing drugs in the presence of a cell-permeable fast calcium chelator BAPTA-AM (Figure 4). The presence of BAPTA did not affect the induction of pcJun in response to nocodazole or cytochalasin D treatment (Figure 4E,F). To test the efficacy of BAPTA-AM treatment for blocking calcium entry in these cells, we induced calcium influx with KCl depolarization (Crawford et al., 2011) or ionomycin treatment and visualized calcium with the cell-permeable calcium indicator dye fluo-4 AM. KCl depolarization as well as ionomycin treatment lead to a calcium influx that is effectively buffered by BAPTA-AM (Figure 4A,C). Taken together, these data demonstrate that calcium is not required for the DLK pathway activation in response to nocodazole or cytochalasin D.
Figure 4. Calcium is not necessary for pcJun upregulation.

A. Example fields of neurons filled with the calcium indicator fluo-4 after pretreated for 4 h with empty media (baseline) or 30 mM KCl (depolarized) in either calcium-containing media (vehicle) or media supplemented with 20 μM BAPTA-AM (BAPTA-AM).
B. Representative examples of neurons filled with the calcium indicator fluo-4 before (baseline) and after perfusion with 3μM ionomycin (ionomycin) under same conditions as in A.
C. Quantification of the conditions represented in A.
D. Quantification of the conditions represented in B.***p<0.001, **p<0.01.
E. pcJun upregulation was not affected by the presence of BAPTA-AM. Sample confocal images of day 7 wild-type embryonic cultures stained for pcJun (red), β3 tubulin (green) and nucleic acid dye Topro (blue) after 16-hour vehicle, cytochalasin D (CytD) or nocodazole (Noc) treatment in the presence of vehicle or 20μM BAPTA-AM. Scale bar = 50 μm.
F. Normalized quantification of the pcJun intensity of the nuclei represented in E. ***p<0.001, compared to vehicle treated cells in each condition. n > 8 wells. Error bars indicate SEM.
DLK pathway activation following cytoskeletal disruption does not require axonal degeneration
Axonal injury, in addition to causing calcium influx, can also result in axon degeneration (Wang et al., 2012). Hence, destabilization of the cytoskeleton could activate a degeneration program, which could secondarily activate the DLK pathway. First, we tested whether these doses of cytochalasin D or nocodazole trigger cell death or axon degeneration. Application of these drugs does not lead to cell toxicity as assessed by Ethidium Homodimer staining (vehicle: 4 ± 1 % EthD positive cells; 200nM CytD: 4 ± 2%; 200nM Noc: 3 ± 1%, p = 0.95 one-way ANOVA). To test for axon degeneration, we treated DRG neurons with vehicle as a negative control, axotomy as a positive control, cytochalasin D, and nocodazole, and then assessed axonal degeneration via calculation of a degeneration index that measures axonal fragmentation (Sasaki et al., 2009). While there is dramatic degeneration following axotomy, axonal fragmentation is not observed following drug treatment (degeneration index of vehicle-treated axons = 0.14 ± 0.02, nocodazole = 0.20 ± 0.02, cytochalasin D = 0.06 ± 0.02, axotomy = 0.89 ± 0.03; n=3 mice, one-way ANOVA p>0.5 between drug and vehicle treated groups). As a functional test of the role of the axonal degeneration program, we blocked axonal degeneration by overexpressing the potent axonal survival factor NMNAT2 (Gilley and Coleman, 2010). In this system NMNAT2 overexpression is sufficient to block axon degeneration for at least 24 hours after axotomy (Degeneration index of control infected cells = 0.64, NMNAT2 infected cells = 0.23, n=12, p<0.001, Figure 5C), however it has no effect on pcJun upregulation following nocodazole or cytochalasin D treatment (Figure 5A,B). Hence, in DRG neurons neither calcium influx nor the axon degeneration program is necessary for DLK pathway activation in response to cytoskeleton disrupting agents. These results are consistent with the model that cytoskeletal disruption during an injury is the trigger for the DLK pathway activation.
Figure 5. NMNAT2 overexpression does not block pcJun upregulation.

A. Application of cytoskeleton disrupting drugs does not cause axon degeneration in cultured neurons. Sample phase-contrast images of the axons of the embryonic DRG neurons after treatment with vehicle, nocodazole, cytochalasin D, or axotomy. Quantification is presented in the results.
B. pcJun upregulation is not affected by NMNAT2 overexpression. Sample confocal images of wild-type embryonic DRG neurons infected either with an empty vector (control) or a vector expressing the axoprotective molecule NMNAT2 (NMNAT2 o/e). The cells are stained for pcJun (red), β3 tubulin (green) and nucleic acid dye Topro (blue) after 16-hour hour vehicle, cytochalasin D (CytD) or nocodazole (Noc) treatment. Scale bar = 50 μm.
C. Normalized quantification of the pcJun intensity of the nuclei represented in A. ***p<0.001, compared to vehicle treated cells in each condition. n > 10 wells for each condition from 3 different mice with more than 100 cells quantified for each well. Error bars indicate SEM.
D. NMNAT2 overexpression suppresses axonal degeneration after axotomy. Bright field images of the axons distal to the cut site 24 hours after axotomy of day 7 embryonic cultures infected either with an empty vector (control) or a vector expressing NMNAT2.
Cytoskeletal destabilizing agents activate the JNK pathway and promote regeneration in adult DRG neurons
As animals mature, their neurons lose the ability to regenerate (Bates and Stelzner, 1993; Hasan et al., 1993). We wished to know whether cytoskeletal disruption activates the DLK/JNK pathway in adult neurons, and whether this activation is sufficient to promote axon regeneration. The dissection and plating of the neurons is an injury (Frey et al., 2015; Saijilafu et al., 2013; Zou et al., 2009), and we find that after one day in culture pcJun levels are high (Figure 6A–C). This is consistent with in vivo results demonstrating that a crush injury induces the phosphorylation of cJun in the DRGs (Shin et al., 2012). After 7 days in culture, pcJun in dissociated DRG neurons is nearly undetectable (Figure 6B–C), suggesting that cultured neurons have turned off the injury response program that was triggered by the initial dissection. Hence, we can now test whether the JNK pathway can be reactivated in adult cultured neurons following application of cytoskeleton destabilizing agents in the absence of traumatic injury. Indeed, treatment with nocodazole or cytochalasin D in these adult neurons restores high levels of pcJun (Figure 6A–C), demonstrating that cytoskeletal disruption can reactivate the JNK pathway in adult neurons. We next tested whether cytoskeletal destabilization induces regeneration-associated gene products. To this end, we measured the level of superior cervical ganglion 10 (SCG10), whose levels increase within hours of axonal injury in vivo (Shin et al., 2014). Treatment of adult DRG neurons with 200 nM nocodazole increased SCG10 levels 2 fold over vehicle treatment at day 7 in culture (DMSO treated: 1± 0.06; nocodazole treated: 1.96 ± 0.09, p<0.001). Hence, cytoskeletal disruption activates the DLK/Jun pathway and triggers the expression of pro-regenerative gene products.
Figure 6. Cytoskeletal disrupting agents induce pcJun upregulation and activate a pro-regeneration program in adult DRG neurons.

A. Timeline of the adult DRG neuron culture experiment. Cells were grown for 7 days in culture and their pcJun intensity and regrowth capacity was compared between Day 1 and Day 7 after vehicle or drug treatment.
B. Cytochalasin D and nocodazole treatment restores pcJun intensity. Sample confocal images of adult DRG neurons on day 1 and on Day 7 after 16-hour hour vehicle, cytochalasin D (CytD) or nocodazole (Noc) treatment. Cells are stained for pcJun (red) and β3 tubulin (green). Scale bar = 5 μm.
C. Normalized pcJun intensity from each condition in B. ***p<0.001 compared to day 7 vehicle.
D. Adult DRG neurons replated at the same time points as in B and allowed to grow for 18 hours in drug-free media to assess regeneration. Scale bar = 100 μm.
E. Cumulative distributions from a representative mouse corresponding to conditions in B and D with addition of naïve neurons.
F. Cumulative distributions from a representative mouse corresponding to conditions in B and D.
Since cytoskeletal destabilizing drugs activate the DLK/JNK pathway in adult neurons, we investigated whether they also induce a pro-regenerative functional response in adult neurons. We replated adult cultured DRG neurons at various times after initial dissection from the animal and measured how well neurons regrow neurites (Figure 6A). At DIV 1 replating of the neurons leads to robust axonal regrowth in the subsequent 18 hours, consistent with prior studies demonstrating that dissection from the animal acts as a preconditioning injury that promotes axon growth following replating of the neurons (Saijilafu et al., 2013; Zou et al., 2009). However by DIV 7, when the pcJun level is low, axonal growth in the 18 hours after replating is poor (Figure 6A,D–E). We then pretreated sensory neurons at DIV 6 with vehicle, cytochalasin D, or nocodazole for 24 hours, washed away the drug, and then replated the neurons and allowed them to extend axons for 18 hours in the absence of the drug. In contrast to untreated or vehicle-treated neurons, neurons pretreated with either cytochalasin D or nocodazole display robust axonal regrowth following replating (Figure 6A,D–F and Figure 7). These findings indicate that cytoskeletal disruption activates both the JNK pathway and a regenerative program in adult sensory neurons. We go on to quantify these responses and to test the DLK dependence of these effects.
Figure 7. Induction of pcJun and the pro-regenerative program by cytoskeletal disrupting agents requires DLK.

A. Schematic diagram outlining the adult DRG culture experiment. Drugs are present before the replating and are washed out for the regrowth phase.
B. Sample confocal images of adult DRG neurons cultured for 7 days. At DIV 6, neurons were treated for 24 hours with vehicle, cytochalasin D (CytD) or nocodazole (Noc). Cells are stained for pcJun (red) and β3 tubulin (green) at the end of the drug treatment. Littermate controls (DLKF/F); DLK mutant (DLKF/F, Adv-Cre). Scale bar = 10 μm.
C. Quantification of the pcJun intensity in the groups represented in B and normalized to vehicle-treated condition for each genotype. *p<0.05, N.S. = not significant. Over 150 neurons/condition derived from 3 different mice for each genotype were analyzed.
D. Pretreatment with cytochalasin D and nocodazole significantly increases neurite outgrowth in control but not in the DLK mutant mice. Representative cumulative distribution plots for the longest neurite per cell from one mouse of each genotype 18 hours after replating. Littermate controls (DLKF/F); DLK mutant (DLKF/F, Adv-Cre). ~100 neurons were measured for each condition.
E. Sample confocal images of replated DRG neurons on day 7 after pretreatment with vehicle, cytochalasin D or nocodazole. Neurons were replated and allowed to grow for 18 hours in the absence of drug in each condition. The relative paucity of neurites is due to the reduced ability of the neurons to regrow axons after replating on day 7 in culture. Littermate controls (DLKF/F); DLK mutant (DLKF/F, Adv-Cre). Scale bar = 100 μm.
F. Mean neurite length for each of the conditions represented in D and E. *p<0.05. n=3 mice for each genotype.
Cytoskeletal disrupting agents activate a DLK-dependent axon regeneration program
Following peripheral nerve injury in adult mice, DLK is required for both the accumulation of pcJun in DRG cell bodies and the activation of the pro-regenerative program induced by a preconditioning lesion (Shin et al., 2012). Here we test whether DLK is also required for the accumulation of pcJun in DRG cell bodies and the activation of the pro-regenerative program induced by pretreatment with drugs that destabilize the cytoskeleton in adult cultured DRG neurons. To this end, we mated a previously described conditional DLK allele (Miller et al., 2009) to AdvillinCre mice (Zurborg et al., 2011) to remove DLK selectively in sensory neurons. DRG neurons were cultured, and after 6 days in vitro were treated with nocodazole and cytochalasin D. This led to the accumulation of pcJun in littermate control but not DLK knockout sensory neurons (Figure 7A–C). Hence, cytoskeletal destabilization in cultured adult sensory neurons activates a DLK-dependent JNK pathway. To assess regrowth, we pretreated DIV 6 neurons with vehicle, cytochalasin D, or nocodazole, removed the drug, replated the neurons and allowed them to extend axons for 18 hours. For each neuron we measured the length of the longest neurite, plotted the cumulative probability of neurite length and measured the mean neurite length for each experiment. In sensory neurons from littermate controls, pretreatment with either cytochalasin D or nocodazole leads to a significant increase in neurite outgrowth (Figure 7D–F). This activation of the regenerative response requires DLK, as pretreatment with these concentrations of either cytochalasin D or nocodazole fails to promote neurite growth from DLK KO neurons (Figure 7D–F). Hence, cytoskeletal destabilization in vitro, like a preconditioning injury in vivo, activates a DLK-dependent pro-regenerative program that potentiates axonal regrowth. Moreover, this requirement for DLK shows that this single MAP3K functions as a key mediator of the cellular response to cytoskeletal injury in sensory neurons.
Targeting microtubules in vivo activates the pro-regenerative axon growth program
Having demonstrated that pretreatment with cytoskeletal destabilizing agents is sufficient to enhance axon regrowth from adult neurons in vitro, we next assessed whether targeting the cytoskeleton in vivo can, like a preconditioning nerve crush injury, induce the pro-regenerative state. In studies of preconditioning, three days after sciatic nerve crush there is a) accumulation of pcJun in sensory neuron cell bodies and b) improved axonal growth of neurons dissociated from these previously injured neurons (Shin et al., 2012). We performed these same two assays three days after treatment of the sciatic nerve with nocodazole or vehicle. Application of nocodazole to the nerve leads to accumulation of pcJun in the nucleus of significantly more sensory neurons compared to vehicle treatment (Figure 8A–C). Hence, targeting the microtubule network in axons is sufficient to activate retrograde JNK signaling in vivo. To assess the functional induction of the pro-regenerative state, we applied nocodazole or vehicle in vivo. After three days, we isolated and plated DRG neurons in the absence of nocodazole, and measured axonal regrowth following eighteen hours in culture. Neurons whose axons were pretreated with nocodazole in vivo grew axons that were approximately twice as long as those derived from vehicle-treated neurons (Figure 8D–F). Hence, microtubule destabilizing drug treatment in vivo, like a preconditioning nerve crush, is sufficient to activate the pro-regenerative program. These results suggest that cytoskeletal damage may be an important component of the injury signal that activates the regenerative response.
Figure 8. Nocodazole application in vivo induces pcJun upregulation and enhances axonal regrowth.

A. Schematic diagram outlining the in vivo experiment. Nocodazole was applied to the sciatic nerve for 3 days (blue bar) prior to dissection of DRGs for pcJun and regrowth assessment.
B. Sample confocal images of sections of adult DRGs stained for pcJun (red) and β3 tubulin (green). Application of nocodazole to the sciatic nerve significantly increases the number of neurons with pcJun positive nuclei. Boxed areas are shown at higher magnification in the adjacent panels. Scale bar = 100 μm.
C. Percentage of pcJun positive cells in the groups represented in B. **p<0.01, n = 3 mice.
D. In vivo application of nocodazole to the sciatic nerve prior to dissociating and plating neurons enhances the regrowth compared to vehicle treatment. Sample confocal images of dissociated DRG neurons 18 hours after plating stained for β3 tubulin. Scale bar = 100 μm.
E. Representative cumulative distribution plots for the longest neurite per cell from each mouse. Red lines represent mice treated with nocodazole and blue lines represent vehicle-treated animals. ~100 neurons were measured for each mouse.
F. Mean neurite length was measurement for each mouse represented in E. ***p<0.001.
Discussion
Our results establish that cytoskeletal disrupting agents activate the DLK/JNK pathway in sensory neurons, inducing a pro-regenerative state that enhances axonal regeneration in response to a subsequent injury. These findings support the hypothesis that DLK senses cytoskeletal damage following axon injury.
Cytoskeletal disruption activates the DLK pathway
DLK is a key player in the neuronal response to axon injury (Ferraris et al., 2013; Tedeschi and Bradke, 2013). In addition to its role in axon regeneration, DLK is also necessary for normal Wallerian degeneration in the PNS (Miller et al., 2009), and promotes degeneration of retinal ganglion cell axons in models of glaucoma (Watkins et al., 2013; Welsbie et al., 2013). Furthermore, DLK is required for programmed neuronal cell death during development, death of retinal ganglion cells in glaucoma models, and excitotoxic cell death in cortical neurons (Ghosh et al., 2011; Itoh et al., 2011; Pozniak et al., 2013; Welsbie et al., 2013). This central role for DLK in the response to axon injury has lead to the hypothesis that DLK is a sensor of axonal damage (Huntwork-Rodriguez et al., 2013; Watkins et al., 2013).
Despite the clear role of DLK in the response to axon injury, it is not known how axon damage leads to activation of the DLK pathway. Axonal injury results in many cellular changes that could activate the DLK pathway, including calcium influx, activation of signaling pathways, and damage to the axonal cytoskeleton. In C. elegans, calcium influx is an important activator of DLK (Yan and Jin, 2012). However Drosophila and mammalian DLK lack the calcium-sensitive hexapeptide present in worm DLK, suggesting that additional activation mechanisms exist. Following injury, DLK is part of a positive feedback loop in which its target, JNK, phosphorylates and stabilizes DLK to enhance pathway activation (Huntwork-Rodriguez et al., 2013). However since JNK is downstream of DLK, this elegant mechanism does not explain the initial activation of DLK following nerve injury. Recently, we demonstrated that in Drosophila DLK is activated by mutations in proteins that normally promote cytoskeletal stability (Valakh et al., 2013). Mutation of the spectraplakin short stop, an actin-microtubule cross-linker, or knockdown of subunits of TCP1, a chaperonin complex that helps fold both actin and tubulin, both lead to activation of the DLK pathway. In addition, mutations disrupting microtubules in C. elegans lead to DLK-dependent changes in levels of neuronal proteins and neuronal morphology (Bounoutas et al., 2011; Chen et al., 2014; Marcette et al., 2014). These findings lead us to hypothesize that cytoskeletal injury may be a signal that activates the DLK pathway.
Here we have tested this hypothesis by pharmacologically targeting both the actin and microtubule cytoskeletons. We find that low dose treatment with either cytochalasin D or nocodazole, to target the actin and microtubule cytoskeletons respectively, is sufficient to activate the DLK/JNK pathway in the absence of traumatic injury in mammalian sensory neurons. Neither calcium nor activation of the NMNAT-sensitive axon degeneration program is necessary for DLK pathway activation in response to cytoskeletal disruption, although either could be upstream of cytoskeletal damage and DLK activation following traumatic injury. Pharmacological targeting of the cytoskeleton and traumatic axon injury each trigger retrograde injury signals from axon to nucleus, each lead to DLK-dependent accumulation of pcJun in DRG cell bodies, and each induce a DLK-dependent pro-regenerative state that enhances axonal regeneration in response to subsequent injuries. These many parallels support the hypothesis that upon axon injury, cytoskeletal damage is an important signal activating the DLK pathway. Interestingly, the DLK/JNK pathway can in turn regulate microtubule stability (Hirai et al., 2011), suggesting that DLK may participate in a feedback mechanism that is activated by and responds to cytoskeletal damage.
Cytoskeletal disruption activates a DLK-dependent axon regeneration program
DLK is necessary for axon regeneration in worms, flies, and mice (Hammarlund et al., 2009; Itoh et al., 2009; Shin et al., 2012; Xiong et al., 2010; Yan et al., 2009). In mammals, DLK is required for regeneration of both peripheral sensory axons and central retinal ganglion cell axons, even in PTEN mutants that dramatically enhance the efficacy of regeneration (Shin et al., 2012; Watkins et al., 2013). This necessity for DLK raises an important question—is activation of the DLK pathway also sufficient to promote axon regeneration? In worms and flies, genetic overexpression of DLK does enhance regeneration (Hammarlund et al., 2009; Xiong et al., 2010). Our previous study demonstrated that mutations in short stop, which disrupts the cytoskeleton and activates the DLK pathway, is also sufficient to promote the regenerative response to nerve crush (Valakh et al., 2013). Here we asked whether pharmacological activation of the DLK pathway is sufficient to promote axon regeneration in mammalian sensory neurons.
In mammals, DLK is required for the preconditioning response, in which a prior nerve injury induces a pro-regenerative program that allows for more robust regeneration in response to a subsequent injury (Shin et al., 2012). This preconditioning paradigm highlights the potential therapeutic utility of activating this pro-regenerative program, but it is unlikely that nerve injury would be a clinically useful stimulus. Instead, it would be valuable to identify pharmacological manipulations that can substitute for the initial injury in the induction of the pro-regenerative state. Here we show that both cytochalasin D and nocodazole are capable of inducing this DLK-dependent regeneration program in the absence of traumatic nerve injury. The drugs are present only before injury and are removed during the regrowth phase and thus cannot affect the growth cone dynamics of regenerative axons. This is in contrast to the studies where cytoskeletal manipulation during the regrowth phase can either inhibit or enhance regeneration (Ertürk et al., 2007; Hellal et al., 2011; Sengottuvel et al., 2011) and highlights the importance of the activation of the transcriptional program to exploit the regenerative potential of neurons.
We have established an in vitro paradigm in which adult DRG neurons have recovered from the injury of being dissected from the animal (this new assay is characterized in detail in Frey et al., 2015). These neurons have low levels of the injury marker pcJun, and upon replating have limited capacity to regrow axons. However following addition of either cytochalasin D or nocodazole, there is an accumulation of pcJun, and upon replating in the absence of drug, robust axonal regrowth. Both of these effects are DLK dependent. Hence, pretreatment with these drugs behaves much like a prior nerve crush, activating injury signaling and allowing for improved regeneration upon a subsequent injury. We extended these findings in vivo, demonstrating that application of nocodazole to the sciatic nerve can mimic important effects of a preconditioning nerve lesion, including accumulation of pcJun and improved axonal outgrowth following subsequent neuronal culture. Previously it was demonstrated that application of colchicine to the sciatic nerve is sufficient to improve axon outgrowth following culture of the treated neurons (Smith and Skene, 1997). At the time, it was suggested that colchicine, which inhibits microtubule polymerization, blocked retrograde transport of factors that inhibited the growth potential of adult neurons. However, in light of our findings with nocodazole, we would suggest that colchicine, and likely other pharmacological agents that disrupt the axonal cytoskeleton, lead to activation of the pro-regenerative program. Future studies will investigate whether this new assay can identify drugs of other classes that are able to active the pro-regenerative program and enhance axonal regeneration.
DLK is a key sensor of cytoskeletal damage in DRG neurons
JNK MAP kinases respond to many cellular stresses, including cytoskeletal damage, and regulate a wide range of cellular processes (Dvorák et al., 2004; Hamel et al., 2006; Johnson and Nakamura, 2007). The specificity of JNK signaling is controlled in part via their activation by upstream kinases. DLK is a member of a large family of MAP kinase kinase kinases that activate JNK. Here we demonstrate that drugs that target either the actin or microtubule cytoskeletons activate JNK signaling in sensory neurons, and that this activation relies on a single MAP3K, DLK. We previously demonstrated that DLK is also the essential MAP3K for JNK pathway activation in vivo following nerve injury (Shin et al., 2012), an insult that also damages the cytoskeleton. These results suggest that in DRG sensory neurons DLK is the essential intermediate between cytoskeletal damage and JNK pathway activation. We note, however, that our studies used doses of cytochalasin D and nocodazole that triggered robust physiological responses but did not cause gross axonal damage. Indeed, we have found that higher doses of these drugs lead to effects that are not strictly DLK-dependent, indicating that additional kinases are activated upon a more catastrophic cytoskeletal insult. Nonetheless, our studies indicate that DLK is the most sensitive sensor of cytoskeletal damage triggering JNK activation in these neurons.
In this study we have highlighted the role of DLK in promoting axon regeneration, however DLK functions as a “double-edged sword,” (Tedeschi and Bradke, 2013) promoting axon degeneration and programmed neuronal cell death in some circumstances. Many genetic mutations that cause hereditary peripheral neuropathies as well as chemotherapy drugs that lead to painful neuropathies target components of the cytoskeleton (Edvardson et al., 2012; Gentil et al., 2013; Park et al., 2013; Solowska et al., 2014), and such cytoskeletal disruption can activate the DLK pathway. For example, the chemotherapeutic vincristine destabilizes microtubules and causes peripheral neuropathy, and in mouse models DLK is required for vincristine-induced axonal degeneration (Miller et al., 2009). We posit that DLK may act as a key intermediate between genetic and neurotoxic injuries to the cytoskeleton and neuropathic pathology in humans. If so, then the development of selective inhibitors of DLK (Ferraris et al., 2013) may open new therapeutic avenues for the treatment of neuropathy.
Materials and Methods
Mouse model
DLK conditional knock-out (DLKF/F) mice (Miller et al., 2009) and Advillin-Cre mice (Zurborg et al., 2011) were described previously. For embryonic and wild-type experiments CD-1 strain mice were used. Both male and female mice were used in the experiments. For DLK experiment controls were always sex-matched littermate mice. Mouse husbandry was under the supervision of Division of Comparative Medicine at Washington University.
Embryonic DRG culture preparation
Embryonic DRG culture was performed as described previously (Miller et al., 2009). Briefly, permanox plastic slides (Thermo Scientific) were coated with poly-d-lysine and laminin. DRGs were collected from embryonic day 12.5 mice, treated with trypsin-EDTA for 20 minutes at 37°C, and then dissociated into single cells. 2.5 μL of the cell suspension was placed as a spot on a dish at a density of approximately a half DRG per spot. To allow the cells to attach on the plastic, the plates were incubated for 15 min at 37°C before addition of Neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen), 25 ng/ml NGF, and 1 μM 5-fluoro-2′-deoxyuridine and 1 μM uridine (Sigma) that block cell division of non-neuronal cells. Cultures were maintained for 7 days before drug application. Compartmentalized cultures were assembled as previously described (Pazyra-Murphy and Segal, 2008).
Lentivirus preparation and infection
Lentivirus preparation and infection was performed as previously described (Babetto et al., 2013). Briefly, Cre and human NMNAT2 constructs were cloned into a FCIV-FM1 vector with an internal ribosome entry site (IRES)-enhanced YFP (Venus) cassette. Empty vector was used a control. The constructs were introduced into the embryonic DRG neurons on DIV2 via lentiviral transduction. The transduction efficiency was assayed via Venus fluorescence: ~ 100% of cells were expressing Venus for each construct tested.
Adult DRG culture preparation
Adult DRG cultures were prepared as described previously (Abe et al., 2010). Briefly, L4 - L6 DRGs were collected and incubated in DMEM with 0.7 mg/ml Liberase Blendzyme 3 (Roche), 0.6 mg/ml DNase (Sigma), and 10mg/ml bovine serum albumin for 15 min at 37°C. The DRGs were then incubated with trypsin-EDTA for 15 min at 37°C followed by trituration. Dissociated cells were plated on 12-well dishes (Costar) or on pemanox plastic slides (Thermo Scientific) coated with poly-d-lysine (Sigma) and laminin (Invitrogen) in DMEM with 10% FBS and 50 ng/ml NGF (Harlan Bioproducts).
Pharmacology
Nocodazole (Sigma) and cytochalasin D (Sigma) were used at the 100–200 nM final concentrations and prepared in the culture media from 10 mM stock in DMSO. JNK inhibitor SP600125 (Sigma) was used at 15 μM final concentration. BAPTA-AM (Sigma) was diluted from 20 mM stock solution in culture media to a final concentration of 20 μM and Ionomycin (Sigma) was diluted from 10mM stock solution to a final concentration of 3 μM in culture media. The drug treatments lasted for 16 hours in 37°C for embryonic cultures and for 24 hours with washout prior to replating in adult DRG cultures.
Quantifying DRG axon degeneration
DRG axon degeneration was quantified as previously described (Miller et al., 2009). Briefly, live DRG cultures were imaged using phase contrast microscopy with an inverted microscope and a 20X objective. Three non-overlapping images per well from 4 different wells were obtained per each condition. The degeneration index (DI) was calculated using a macro developed in ImageJ (National Institutes of Health, USA) (Sasaki et al., 2009). The mean DI of each condition was obtained by averaging the DIs of all images taken for that condition.
Calcium imaging
All calcium imaging experiments used embryonic spot cultures. For fluo-4 AM experiments, neurons were plated on glass-bottom 35mm Fluorodishes (World Precision Instruments Inc.). Cells were loaded with fluo-4 AM by 20 min incubation with 2 μM fluo-4 AM (Life Technologies) at 37°C. The cells were imaged on the Nikon D-Eclipse C1 confocal microscope using 40X water immersion objective. Imaging was performed at room temperature. To cause calcium influx, the cells were incubated in 30 mM KCl final concentration for 4 hours at 37°C. For the ionomycin experiments, 3 μM ionomycin was added to the cells in 100 μL of culture media 10 seconds into the imaging sequence. For BAPTA-AM experiments, the cell culture media was supplemented with 20 μM BAPTA-AM throughout the experiment. The experiments were repeated with changing the media to media made with 0 mM CaCl2 DMEM supplemented with 20 μM BAPTA-AM prior to imaging with the same results.
Analysis of cell death
Embryonic neurons were treated with 2 μM Ethidium Homodimer for 15–20 minutes. Images were collected with an Operetta High Content Imaging System (PerkinElmer) with 20X objective. Cell death was defined as neurons positive for ethidium homodimer. Percent cell death was quantified from at least 100 cells each from 3 independent experiments.
Immunohistochemistry
Embryonic spot cultures or dissociated adult DRG cultures were fixed in 4% paraformaldehyde in 1x PBS for 20 minutes at room temperature, washed in PBST and incubated in 1° antibody overnight. The cultures were washed and incubated in the secondary antibody for 1hr at room temperature, washed and mounted in VectaShield (Vector). The following primary antibodies were used: mouse anti-β3 tubulin antibodies, 1:1000 (Tuj1; Covance catalog number MMS-435P-250), rabbit anti-pcJun antibody, 1:300 (Cell Signaling, #9261), rabbit anti-SCG10, 1:5000 (Shin et al., 2014), Alexa Fluor 488-conjugated (Molecular probes, A11001) and Cy3-conjugated secondary antibodies (Jackson ImmunoResearch, Catalog # 111-165-144), 1:1000, and topro nucleic acid dye, 1:5,000 (Life technologies). SCG10 antibody was purified from Anti-SCG10 rabbit antiserum (Novus Biologicals) using the SulfoLink Kit (Thermo).
Imaging
Samples for quantification and representative images for the neuronal cultures were imaged using a Nikon D-Eclipse C1 confocal microscope using 20x (for embryonic cultures) or 40x (for adult cultures) objectives. All treatment groups for an individual experiment were imaged at the same gain and set such that signals from the brightest group for a given experiment were not saturating. Samples for the adult representative images were imaged using Leica TCS sP5 X confocal with 20x (for neurite length) or 40x (for pcJun intensity) objectives.
p-cJun and SCG10 quantification of dissociated DRG neurons
Mean intensity of p-cJun and SCG10 were measured in the nucleus and soma, respectively. For p-cJun confocal images were thresholded for topro dye fluorescence to select the nuclei by using the threshold tool of ImageJ. The regions were then transposed onto pcJun channel and mean fluorescent intensity was measured for the whole image. For SCG10, cell body outlines were performed manually using β3 tubulin. Then the outlines were transferred to the SCG10 channel to measure intensity of SCG10 in individual neurons. Typically ~100 cells were measured per well for embryonic cultures and 50 cells were measured per each condition in adult cultures. Counterstaining for neuron-specific β3 tubulin was used to confirm that the counted cells were neurons.
Adult replating and neurite outgrowth measurement
Adult DRG cultures were plated in 12 well-plates following DRG culture protocol at the density of 1 adult DRG/well and grown for 1–7 days. On the day of replating the media was aspirated and the cultures were rinsed with warm DMEM followed by the addition of 0.05% Trypsin-EDTA (Sigma) diluted in DMEM (0.025% final). The cells were incubated in trypsin for 5 mins at 37°C. Trypsin-EDTA was then removed and the cells were detached by gentle pipetting in 500 mls per well of adult culture medium and plated onto 4-well permanox slides (Thermo Scientific) for 18 hours to assess growth. To measure the neurite outgrowth the cells were fixed at the end of the 18 hour incubation in 4% PFA in 1x PBS for 20 minutes at RT and stained for β3 tubulin. The cells were imaged on a light microscope (Nikon eclipse 80i) using 10x air objective. Neurite length was quantified by tracing Tuj-1 positive axons with NeuronJ plugin of Image J. ~100 neurons were traced for each experiment. To get a representative measurement of the neurite length, 25–160 neurites per condition were averaged.
Surgeries, chemical treatments and sample preparations
All surgical procedures were approved by the Washington University in St Louis, School of Medicine Animal Studies Committee. For chemical treatment, sciatic nerve was exposed and soaked with DMSO-dissolved 10mM Nocodazole in Surgifoam (Johnson and Johnson) and the surgery site was sutured. For immunohistochemistry, DRGs were dissected, fixed for 1 h in 4% paraformaldehyde in PBS, incubated overnight in 30% sucrose, embedded in OCT solution (Tissue-Tek) and frozen. All surgeries and quantification was performed by experimenters blinded to the drug.
Statistical analysis
Student’s independent t-test or one-way ANOVA was used to test statistical significance. When ANOVA was used, Tukey post-test was performed for means comparison. p values greater than 0.05 were stated as not significant.
Highlights.
Cytoskeletal disruption activates the DLK injury response pathway in sensory neurons
Activation of the DLK pathway is sufficient to promote axon regrowth
Cytoskeletal disruption in vivo mimics a preconditioning injury
DLK is a key neuronal sensor of cytoskeletal damage
Acknowledgments
This work was supported by grants from the Craig H. Neilsen Foundation and the National Institutes of Health (DA020812, NS087562 and NS065053) to A.D., Muscular Dystrophy Association (MDA) career development award to E.B., and National Science Foundation Graduate Research Fellowship Grant No. DGE-1143954 to L.J.W. We thank members of the A.D. laboratory for helpful discussions and especially Jung Eun Shin for the help with setting up the mouse model and EJ Brace for comments on the manuscript.
Footnotes
Conflict of Interest
The authors declare no competing financial interest.
Author Contribution
A.D. and V.V. designed research. V.V., E.B., E.F., and L.W. performed experiments. V.V. and E.F. analyzed data. A.D. and V.V. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe N, Borson SH, Gambello MJ, Wang F, Cavalli V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem. 2010;285:28034–28043. doi: 10.1074/jbc.M110.125336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applewhite DA, Grode KD, Keller D, Zadeh AD, Slep KC, Rogers SL. The Spectraplakin Short Stop Is an Actin–Microtubule Cross-Linker That Contributes to Organization of the Microtubule Network. Mol Biol Cell. 2010;21:1714–1724. doi: 10.1091/mbc.E10-01-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, Freeman MR. WldS Prevents Axon Degeneration through Increased Mitochondrial Flux and Enhanced Mitochondrial Ca2+ Buffering. Curr Biol. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babetto E, Beirowski B, Russler EV, Milbrandt J, Diantonio A. The Phr1 Ubiquitin Ligase Promotes Injury-Induced Axon Self-Destruction. Cell Rep. 2013 doi: 10.1016/j.celrep.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker ST, Opperman KJ, Tulgren ED, Turgeon SM, Bienvenut W, Grill B. RPM-1 uses both ubiquitin ligase and phosphatase-based mechanisms to regulate DLK-1 during neuronal development. PLoS Genet. 2014;10:e1004297. doi: 10.1371/journal.pgen.1004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates CA, Stelzner DJ. Extension and regeneration of corticospinal axons after early spinal injury and the maintenance of corticospinal topography. Exp Neurol. 1993;123:106–117. doi: 10.1006/exnr.1993.1144. [DOI] [PubMed] [Google Scholar]
- Bounoutas A, Kratz J, Emtage L, Ma C, Nguyen KC, Chalfie M. Microtubule depolymerization in Caenorhabditis elegans touch receptor neurons reduces gene expression through a p38 MAPK pathway. Proc Natl Acad Sci. 2011;108:3982–3987. doi: 10.1073/pnas.1101360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB. Development of sympathetic neurons in compartmentalized cultures: II. Local control of neurite survival by nerve growth factor. Dev Biol. 1982;93:13–21. doi: 10.1016/0012-1606(82)90233-0. [DOI] [PubMed] [Google Scholar]
- Chan JR, Watkins TA, Cosgaya JM, Zhang C, Chen L, Reichardt LF, Shooter EM, Barres BA. NGF Controls Axonal Receptivity to Myelination by Schwann Cells or Oligodendrocytes. Neuron. 2004;43:183–191. doi: 10.1016/j.neuron.2004.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Lee A, Liao CP, Liu YW, Pan CL. RHGF-1/PDZ-RhoGEF and retrograde DLK-1 signaling drive neuronal remodeling on microtubule disassembly. Proc Natl Acad Sci. 2014;111:16568–16573. doi: 10.1073/pnas.1410263111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Cavalli V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012;31:3063–3078. doi: 10.1038/emboj.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire Restrains Synaptic Growth by Attenuating a MAP Kinase Signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Chang CY, Hyrc KL, Mennerick S. Calcium-Independent Inhibitory G-Protein Signaling Induces Persistent Presynaptic Muting of Hippocampal Synapses. J Neurosci. 2011;31:979–991. doi: 10.1523/JNEUROSCI.4960-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Kwiatkowski AV, Mebane LM, Philippar U, Barzik M, Rubinson DA, Gupton S, Van Veen JE, Furman C, Zhang J, et al. Filopodia are required for cortical neurite initiation. Nat Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- Dvorák Z, Maurel P, Ulrichová J, Modrianský M. Microtubule disarray in primary cultures of human hepatocytes inhibits transcriptional activity of the glucocorticoid receptor via activation of c-jun N-terminal kinase. Biomed Pap Med Fac Univ Palacký Olomouc Czechoslov. 2004;148:135–139. [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Jalas C, Shaag A, Maayan C, Axelrod FB, Elpeleg O. Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann Neurol. 2012;71:569–572. doi: 10.1002/ana.23524. [DOI] [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci Off J Soc Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris D, Yang Z, Welsbie D. Dual leucine zipper kinase as a therapeutic target for neurodegenerative conditions. Future Med Chem. 2013;5:1923–1934. doi: 10.4155/fmc.13.150. [DOI] [PubMed] [Google Scholar]
- Frey E, Valakh V, Karney-Grobe S, Shi Y, Milbrandt J, DiAntonio A. An in vitro assay to study induction of the regenerative state in sensory neurons. Exp Neurol. 2015;263:350–363. doi: 10.1016/j.expneurol.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentil BJ, Mushynski WE, Durham HD. Heterogeneity in the properties of NEFL mutants causing Charcot-Marie-Tooth disease results in differential effects on neurofilament assembly and susceptibility to intervention by the chaperone-inducer, celastrol. Int J Biochem Cell Biol. 2013;45:1499–1508. doi: 10.1016/j.biocel.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham J, Brackley KI, Willison KR. Substantial CCT activity is required for cell cycle progression and cytoskeletal organization in mammalian cells. Exp Cell Res. 2006;312:2309–2324. doi: 10.1016/j.yexcr.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Gertler FB. Integrin Signaling Switches the Cytoskeletal and Exocytic Machinery that Drives Neuritogenesis. Dev Cell. 2010;18:725–736. doi: 10.1016/j.devcel.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel M, Kanyi D, Cipolle MD, Lowe-Krentz L. Active stress kinases in proliferating endothelial cells associated with cytoskeletal structures. Endothel J Endothel Cell Res. 2006;13:157–170. doi: 10.1080/10623320600760191. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan SJ, Keirstead HS, Muir GD, Steeves JD. Axonal regeneration contributes to repair of injured brainstem-spinal neurons in embryonic chick. J Neurosci Off J Soc Neurosci. 1993;13:492–507. doi: 10.1523/JNEUROSCI.13-02-00492.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928–931. doi: 10.1126/science.1201148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Izawa M, Osada S, Spyrou G, Ohno S. Activation of the JNK pathway by distantly related protein kinases, MEKK and MUK. Oncogene. 1996;12:641–650. [PubMed] [Google Scholar]
- Hirai S, Banba Y, Satake T, Ohno S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-Jun N-terminal kinase pathway. J Neurosci Off J Soc Neurosci. 2011;31:6468–6480. doi: 10.1523/JNEUROSCI.5038-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J Cell Biol. 2013 doi: 10.1083/jcb.201303066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Commun. 2009;383:258–262. doi: 10.1016/j.bbrc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Wakayama K, Xu J, Bannerman P, Pleasure D, Itoh T. ZPK/DLK, a mitogen-activated protein kinase kinase kinase, is a critical mediator of programmed cell death of motoneurons. J Neurosci Off J Soc Neurosci. 2011;31:7223–7228. doi: 10.1523/JNEUROSCI.5947-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Kapitein LC, Gouveia SM, Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA, Di Stefano P, Demmers J, et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61:85–100. doi: 10.1016/j.neuron.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim Biophys Acta BBA - Mol Cell Res. 2007;1773:1341–1348. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaunas R, Usami S, Chien S. Regulation of stretch-induced JNK activation by stress fiber orientation. Cell Signal. 2006;18:1924–1931. doi: 10.1016/j.cellsig.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Marcette JD, Chen JJ, Nonet ML. The Caenorhabditis elegans microtubule minus-end binding homolog PTRN-1 stabilizes synapses and neurites. eLife. 2014;3:e01637. doi: 10.7554/eLife.01637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Park SB, Goldstein D, Krishnan AV, Lin CSY, Friedlander ML, Cassidy J, Koltzenburg M, Kiernan MC. Chemotherapy-induced peripheral neurotoxicity: A critical analysis. CA Cancer J Clin. 2013;63:419–437. doi: 10.3322/caac.21204. [DOI] [PubMed] [Google Scholar]
- Pazyra-Murphy MF, Segal RA. Preparation and maintenance of dorsal root ganglia neurons in compartmented cultures. J Vis Exp JoVE. 2008 doi: 10.3791/951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Hendricks AG, Lazarus JE, Ben-Yaakov K, Gradus T, Tokito M, Holzbaur ELF. Dynein Interacts with the Neural Cell Adhesion Molecule (NCAM180) to Tether Dynamic Microtubules and Maintain Synaptic Density in Cortical Neurons. J Biol Chem. 2013;288:27812–27824. doi: 10.1074/jbc.M113.465088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Ghosh AS, Gogineni A, Hanson JE, Lee SH, Larson JL, Solanoy H, Bustos D, Li H, Ngu H, et al. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J Exp Med. 2013;210:2553–2567. doi: 10.1084/jem.20122832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Jiang H, Yang F, Nakaso K, Feng J. Parkin Protects Dopaminergic Neurons against Microtubule-depolymerizing Toxins by Attenuating Microtubule-associated Protein Kinase Activation. J Biol Chem. 2009;284:4009–4017. doi: 10.1074/jbc.M806245200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishal I, Fainzilber M. Axon-soma communication in neuronal injury. Nat Rev Neurosci. 2014;15:32–42. doi: 10.1038/nrn3609. [DOI] [PubMed] [Google Scholar]
- Saijilafu, Hur E-M, Liu C-M, Jiao Z, Xu W-L, Zhou FQ. PI3K-GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nat Commun. 2013;4:2690. doi: 10.1038/ncomms3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BPS, Lund FE, Milbrandt J. Nicotinamide Mononucleotide Adenylyl Transferase-Mediated Axonal Protection Requires Enzymatic Activity But Not Increased Levels of Neuronal Nicotinamide Adenine Dinucleotide. J Neurosci Off J Soc Neurosci. 2009;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengottuvel V, Leibinger M, Pfreimer M, Andreadaki A, Fischer D. Taxol facilitates axon regeneration in the mature CNS. J Neurosci Off J Soc Neurosci. 2011;31:2688–2699. doi: 10.1523/JNEUROSCI.4885-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Geisler S, DiAntonio A. Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol. 2014;252:1–11. doi: 10.1016/j.expneurol.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci Off J Soc Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska JM, D’Rozario M, Jean DC, Davidson MW, Marenda DR, Baas PW. Pathogenic Mutation of Spastin Has Gain-of-Function Effects on Microtubule Dynamics. J Neurosci Off J Soc Neurosci. 2014;34:1856–1867. doi: 10.1523/JNEUROSCI.3309-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suozzi KC, Wu X, Fuchs E. Spectraplakins: master orchestrators of cytoskeletal dynamics. J Cell Biol. 2012;197:465–475. doi: 10.1083/jcb.201112034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Bradke F. The DLK signalling pathway—a double-edged sword in neural development and regeneration. EMBO Rep. 2013;14:605–614. doi: 10.1038/embor.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulgren ED, Baker ST, Rapp L, Gurney AM, Grill B. PPM-1, a PP2Cα/β phosphatase, regulates axon termination and synapse formation in Caenorhabditis elegans. Genetics. 2011;189:1297–1307. doi: 10.1534/genetics.111.134791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursic D, Sedbrook JC, Himmel KL, Culbertson MR. The essential yeast Tcp1 protein affects actin and microtubules. Mol Biol Cell. 1994;5:1065–1080. doi: 10.1091/mbc.5.10.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valakh V, Walker LJ, Skeath JB, DiAntonio A. Loss of the Spectraplakin Short Stop Activates the DLK Injury Response Pathway in Drosophila. J Neurosci. 2013;33:17863–17873. doi: 10.1523/JNEUROSCI.2196-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TH, Wang HS, Ichijo H, Giannakakou P, Foster JS, Fojo T, Wimalasena J. Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. J Biol Chem. 1998;273:4928–4936. doi: 10.1074/jbc.273.9.4928. [DOI] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L, Jr, Fuller J, Fu J, et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A. 2013;110:4045–4050. doi: 10.1073/pnas.1211284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, DiAntonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Jin Y. Regulation of DLK-1 kinase activity by calcium-mediated dissociation from an inhibitory isoform. Neuron. 2012;76:534–548. doi: 10.1016/j.neuron.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Ho C, Wong K, Tessier-Lavigne M. Axotomy-induced Smad1 Activation Promotes Axonal Growth in Adult Sensory Neurons. J Neurosci Off J Soc Neurosci. 2009;29:7116–7123. doi: 10.1523/JNEUROSCI.5397-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurborg S, Piszczek A, Martínez C, Hublitz P, Banchaabouchi MA, Moreira P, Perlas E, Heppenstall PA. Generation and characterization of an Advillin-Cre driver mouse line. Mol Pain. 2011;7:66. doi: 10.1186/1744-8069-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]