

Abstract
Dravet Syndrome (DS) is caused by heterozygous loss-of-function mutations in voltage-gated sodium channel NaV1.1. Our genetic mouse model of DS recapitulates its severe seizures and premature death. Sleep disturbance is common in DS, but its mechanism is unknown. Electroencephalographic studies revealed abnormal sleep in DS mice, including reduced delta wave power, reduced sleep spindles, increased brief wakes, and numerous interictal spikes in Non-Rapid-Eye-Movement sleep. Theta power was reduced in Rapid-Eye-Movement sleep. Mice with NaV1.1 deleted specifically in forebrain interneurons exhibited similar sleep pathology to DS mice, but without changes in circadian rhythm. Sleep architecture depends on oscillatory activity in the thalamocortical network generated by excitatory neurons in the ventrobasal nucleus (VBN) of the thalamus and inhibitory GABAergic neurons in the reticular nucleus of the thalamus (RNT). Whole-cell NaV current was reduced in GABAergic RNT neurons but not in VBN neurons. Rebound firing of action potentials following hyperpolarization, the signature firing pattern of RNT neurons during sleep, was also reduced. These results demonstrate imbalance of excitatory vs. inhibitory neurons in this circuit. As predicted from this functional impairment, we found substantial deficit in homeostatic rebound of slow wave activity following sleep deprivation. Although sleep disorders in epilepsies have been attributed to anti-epileptic drugs, our results show that sleep disorder in DS mice arises from loss of NaV1.1 channels in forebrain GABAergic interneurons without drug treatment. Impairment of NaV currents and excitability of GABAergic RNT neurons are correlated with impaired sleep quality and homeostasis in these mice.
Introduction
Dravet Syndrome (DS) is a debilitating, drug-resistant, and life-threatening childhood-onset epilepsy syndrome. Its manifestations begin with seizures induced by fever or hyperthermia at six-nine months, which progress to spontaneous myoclonic, tonic-clonic, absence, and partial seizures (Dravet et al., 2005; Oguni et al., 2001). During this period of frequent polymorphic seizures, children with DS develop several co-morbid conditions including psychomotor regression, ataxia, sleep disturbance, cognitive impairments, and many die prematurely (Dravet et al., 2005; Oguni et al., 2001). DS is caused by loss-of-function mutations in one allele of the SCN1A gene encoding the NaV1.1 sodium channel (Claes et al., 2003; Claes et al., 2001). Mouse models of DS develop its key phenotypic features including epilepsy with early (P21) onset, high susceptibility to hyperthermia-induced seizures, ataxia, spontaneous seizures, autistic-like behaviors, and premature death (Catterall et al., 2010; Han et al., 2012a; Kalume et al., 2013; Kalume et al., 2007; Oakley et al., 2009; Ogiwara et al., 2007; Yu et al., 2006). Deletion of NaV1.1 channels in DS mice preferentially reduces sodium current in inhibitory neurons in the hippocampus but not in excitatory neurons (Yu et al., 2006), suggesting that selective loss of excitability of inhibitory neurons is responsible for hyperexcitability in DS. Reduced NaV current and excitability in cerebellar Purkinje neurons, which are GABAergic inhibitory neurons, may cause ataxia (Kalume et al., 2007). These findings led to the unified hypothesis that reduced NaV current in GABAergic neurons in different brain regions underlies the multiple, seemingly unrelated co-morbidities of DS, such as sleep disturbance and cognitive impairment (Catterall et al., 2010; Yu et al., 2006). In support of this hypothesis, specific heterozygous deletion of NaV1.1 channels in forebrain GABAergic neurons reproduced seizures, comorbidities, and premature deaths analogous to those in DS mice (Cheah et al., 2012).
Sleep disturbances are common in epilepsies and are associated with poor seizure control and poor quality of life (Bazil, 2003; Steriade, 2005). Clinical evaluation of DS patients has revealed an abnormal sleep-wake cycle, with sleep-onset insomnia and difficulty maintaining sleep (Dravet et al., 2005; Kimura et al., 2005; Nolan et al., 2006). In a previous study, we examined sleep-wake cycle and found abnormal circadian rhythms in DS mice (Han et al., 2012b). In the studies reported here, we have examined sleep physiology in DS mice, uncovered abnormal sleep architecture, and correlated it with reduced sodium currents and action potential firing in the GABAergic neurons of the reticular nucleus of the thalamus (RNT). Our results show that, although sleep disorders in epilepsies are often attributed to side effects of antiepileptic drugs, sleep impairment in DS mice arises from mutation of NaV1.1 channels in forebrain GABAergic interneurons without involvement of drug treatment. This sleep impairment is correlated with cell-specific loss of sodium current and excitability of RNT GABAergic interneurons. Furthermore, our results suggest that both epilepsy and sleep impairment in DS may arise from impaired firing of GABAergic interneurons and therefore may be treatable by appropriate enhancement of GABAergic neurotransmission.
Materials and methods
All experiments with animals were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee of the University of Washington.
Generation of mutant mice
The DS mice used in this study were generated by targeted deletion of the last exon encoding domain IV from the S3 to S6 segment and the entire C-terminal tail of NaV1.1 channel as previously described (Yu et al., 2006). For electrophysiological experiments, mutant mice were generated by breeding pairs of Scn1a (+/−) of 50:50 C57BL6 and 129SvJ background and used at P13-14 as described (Yu et al., 2006), allowing direct comparison of these results to previous electrophysiological data on hippocampal and cerebellar Purkinje neurons. Use of P13-14 animals allowed studies of both heterozygote (HET) mice, which survive beyond weaning, and homozygote (HOMO) knockout mice, which all die on P15 (Yu et al., 2006). EEG-EMG-Video experiments required older HET mice, which were obtained by breeding Scn1a (+/−) males and Scn1a (+/+) females, both of C57BL6 genetic background, and used at P30-35. Scn1a (+/−) males and Scn1a (+/−) females breed poorly with each other and could not be used to generate sufficient HET or HOMO knockout animals for our experiments. Mice with forebrain interneuron-specific knock-out of NaV 1.1 were generated by the Cre-LoxP-mediated recombination as previously described (Cheah et al., 2012; Kalume et al., 2013). Briefly, these mutant mice were obtained by crossing homozygote Scn1a-floxed with Dlx-I12bCre mice, in which the intergenic region of the Dlx1 and Dlx2 homeo genes drives Cre recombinase expression in interneurons of forebrain and some other subcortical regions. Both mouse lines were of C57BL6 genetic background.
Genotyping of mutant mice
Genotypes were determined by PCR amplification of tail DNA samples using the following two sets of primer pairs: FHY209 (5′-CGAATCCAGATGGAAGAGCGGTTCATGGCT-3′) and FHY210 (5′-ACAAGCTGCTATGGACATTGTCAGGTCAGT-3′) for Wild type band (291 bp), and Neo5 (5′-AGGATCTCCTGTCATCTCACCTTGCTCCTG-3′) and Neo3 (5′-AAGAACTCGTCAAGAAGGCGATAGAAGGCG-3′) for mutant band (493 bp) (Yu et al., 2006). Genotypes of the mice with forebrain-interneuron-specific knock-out of NaV 1.1 were determined using tail DNA samples and the following Cre primers and flx primers: Cre-1 (5′-GGTTTCCCGCAGAACCTGAA-3′), Cre-2 (5′-CCATCGCTCGACCAGTTTAGT-3′), flx-311 (5′-CTTGATGGTTGAAATTCAC-3′), and flx-314 (5′-TATAGAGTGTTTAATCTCAAC-3′).
Cell dissociation
GABAergic RNT neurons were acutely dissociated from P13 - P14 mice as described (Huguenard and Prince, 1992). Animals were deeply anesthetized with halothane prior to decapitation. The brain was quickly removed and submerged in ice-cold dissection solution composed of 82 mM Na2SO4, 30 mM K2SO4, 5 mM MgCl2.6H2O, 10 mM HEPES, 10 mM D-Glucose, 0.001% Phenol Red, at pH 7.4 and bubbled with 95% O2/5% CO2. The brain was mounted on a chilled stage in the slicing chamber filled with ice-cold slush of dissection solution bubbled with O2/CO2. 400 μm thick coronal brain slices including the RNT were prepared on a Leica VT 1000S vibrating microtome and stored at room temperature for at least 1h in a holding chamber containing a solution composed of 126 mM NaCl, 2.5 mM KCl, 2 mM MgCl2.6H20, 2 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, 1 mM pyruvic acid, and saturated with O2/CO2. Typically, 3–4 slices were obtained from each brain. As needed, one or two slices were transferred to the enzyme chamber, treated with 1.5 mg/ml protease type XIV (Sigma) and rinsed in dissection solution containing trypsin inhibitor (1 mg/ml) and bovine serum albumin (1 mg/ml). The digested slice was then stored in Tyrode solution composed of 150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 10 mM Glucose, at pH 7.4. To isolate the RNT GABAergic neurons or ventrobasal thalamocortical neurons, the RNT or ventrobasal nucleus was visually identified and the desired nucleus was isolated using a razor blade. The isolated tissue was transferred to a 15 ml conical tube and triturated with a fire-polished pipette. Dissociated cells were plated on a poly-D-Lysine-coated cover slip on the bottom of a 35 mm dish and allowed to settle for at least 15 minutes.
Immunohistochemistry
Protocols previously described by Yu et al. (2006) and Kalume et al., (2007) were used to examine the distribution of voltage-gated sodium channels in thalamic neurons of P13–P14 mice. Briefly, 50 μm coronal brain sections were cut on a sliding frozen microtome and double labeled using either the polyclonal, affinity-purified, primary antibody anti-NaV 1.1, anti-NaV1.2, anti-NaV1.3 or anti-NaV1.6 (24–32 μg/ml; Alomone, Jerusalem, Israel) in combination with monoclonal anti-GAD 67 antibody (5 μg/ml; Millipore, Billerica, MA) for 36 h at 4 °C. After primary antibody incubation, the sections were washed in TBS for 1 h and immersed in goat anti-rabbit biotinylated secondary antibody and goat anti-mouse Alexa 555 for 1 h at 37 °C, rinsed, incubated in avidin D fluorescein and goat anti-mouse Alexa 555 for 1 h at 37 °C, rinsed, mounted onto gelatin-coated slides, coverslipped with Vectashield, and imaged using a Leica SL (Leica, Nussloch, Germany) confocal microscope located in the W. M. Keck Imaging Facility at the University of Washington. Tissues of animals of each genotype were processed identically and together in the same solutions. During imaging, the settings for gain, offset, and laser intensity were kept constant so that the staining intensity could be compared across genotypes. In control experiments, the primary antiserum was either omitted or replaced with normal rabbit serum during the staining protocol.
Electrophysiology
Isolated RNT or VBN cells were visually identified based on their large, pear-shaped soma. Whole-cell voltage- and current-clamp recordings were obtained using an Axopatch 200B amplifier (Molecular Devices, Union City, CA) and Pulse software (HEKA Electronics, Lambrecht/Pfalz, Germany). Recording glass pipettes had resistances of 2–4 MΩ In voltage clamp experiments, before compensation of whole cell capacitance, cells were held at −70 mV and a 10-mV hyperpolarizing voltage step was applied. Cell capacitance (Cm) was calculated as Cm=Q/V, where Q is the charge measured by integrating the capacitative current evoked with the voltage step. Whole cell capacitance was then compensated and series resistance was compensated by more than 90%. Whole cell sodium currents were recorded with an intracellular solution composed of: 112 mM CsCl, 5mM NaCl, 9 mM EGTA, 9 mM HEPES, 1.8 mM MgCl2, 14 mM Tris-creatine-PO4, 4 mM Mg-ATP, 0.3 mM Tris-GTP, at pH 7.4. The extracellular solution contained: 20 mM NaCl, 10 mM HEPES, 2 mM BaCl2, 300 μM CdCl2, 140 mM tetraethylammonium-Cl, at pH 7.4. Conductance-voltage (g-V) relationships were calculated from the current-voltage (I–V) relationships according to g = INa/(V-ENa), where INa represents the peak Na+ current measured at potential V and ENa the calculated equilibrium potential. Normalized activation and inactivation curves were fitted to Boltzmann relationships of the form: y = 1/(1 + exp [(V - V1/2)/k]) + A, where y is normalized gNa or INa, A the baseline, V the membrane potential, V1/2 the voltage of half-maximal activation, Va, or inactivation, Vh, and k is a slope factor. In fitting activation curves, A was fixed at 0. Analyses were performed using Igor Pro 5.0 (Wavemetrics Inc., OR). Statistical results are reported as mean ± SEM. Statistical significance for comparisons of the three genotypes were performed using one-way ANOVA followed by Tukey’s post test. Comparisons of effects of two genotypes were performed with unpaired Student’s t-test.
In current clamp experiments, firing patterns during rebound from a series of hyperpolarizations (800 ms duration, 10 pA increments) were recorded from cells. The internal solution contained: 117 mM K-methanesulfonate, 9 mM EGTA, 9 mM HEPES, 1.8 mM MgCl2.6H2O, 15 mM sucrose, 14 mM Tris-creatine PO4, 4 mM MgATP, 0.3 mM Tris-GTP, 5 mM KCl. The external solution was composed of 150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM MgCl2.6H2O, 10 mM HEPES, 10 mM glucose. The input-output relationship was defined as the relationship between the amplitude of current injected and the number of action potentials generated during the 500 ms following the end of hyperpolarization pulses.
Surgery
P30-33 mice used in these experiments were housed in the University of Washington Animal Facility under constant temperature and 12 hr light/12 hr dark cycle. They underwent survival surgery to implant EEG and EMG electrodes under ketamine/xylazine (130/8.8 mg/kg) anesthesia. Using aseptic technique, a midline incision was made anterior to posterior to expose the cranium. Fine (diameter: 130 μm bare; 180 μm coated) platinum wires were placed through small cranial holes created with a fine cutting needle and fixed in place with cyanoacrylite glue and dental cement. EEG electrodes were placed at visually identified locations - bilateral frontal and bilateral posterior. EMG electrodes were placed in back muscles. A reference electrode was placed at the midline cerebellum and a ground electrode was placed subcutaneously over the back. Electrode impedances were typically < 10 kΩ. After electrode placement, the skin was closed with sutures and the mice were allowed to recover overnight. EEG patterns were evaluated prior to recording to ensure complete recovery from anesthesia.
Video-EEG-EMG recording
A digital Video/EEG/EMG recording system (DEEG, Telefactor) was used to record EMG/EEGs from freely-behaving mice. The low frequency filter was set at 1 Hz, the high frequency filter was set at 70 Hz and a 70 Hz trap filter was used to reduce line noise if necessary. Simultaneous time-locked digital video recording was also obtained. For sleep architecture studies, data were collected from WT and DS mice for 8 continuous hours beginning at 8:00 AM. For sleep homeostasis studies, baseline and post-sleep deprivation data were collected from different groups of DS mice as described below under “Total sleep deprivation”.
Sleep architecture analysis
Data analysis was performed by an investigator blind to experimental groups. Mouse sleep states were classified as Wake, NREM, or REM based on video observations as well as EEG and EMG analysis. Wakefulness was characterized by stereotypic mouse exploration, low-amplitude, high frequency desynchronized EEG, and variable and high amplitude EMG. NREM sleep was characterized by behavioral arrest in the video record, low-frequency, high-amplitude synchronized EEG, and low amplitude EMG. REM sleep was accompanied by lack of movement and relaxed posture in the video record, faster EEG oscillations, and lower EMG amplitude. Average power spectral analyses were performed on the recordings in 5-s epochs, by Fast Fourier Transform, using custom designed IGOR macros (IGOR PRO 6, Wavemetrics Lake Oswego, OR). Spindles were identified by visual inspection of the EEG and by filtering the data in the frequency band between 10–15 Hz using a custom-designed IGOR macro. Epileptiform interictal spikes were identified using a threshold-crossing algorithm based on their large amplitude above baseline. The peak amplitude of the spike was marked and subsequently confirmed visually. Sleep EEG data are reported as mean ± SEM. Differences in sleep parameters between WT and DS mice were tested for statistical significance using unpaired Student’s t-test.
Total sleep deprivation
To permit control of circadian variations of sleep in these experiments, baseline (control) sleep data were recorded from mice a day before they weresubmitted to total sleep deprivation. Mice were allowed to sleep normally for 5 continuous hours beginning at light period onset (7:00 AM), and then baseline sleep video/EMG/EEG recordings were obtained continuously in the 3 subsequent hours. On the following day, beginning at light period onset, the same mice were kept awake for 5 consecutive hours by gentle handling and by introduction/removal of novel objects in the cage whenever they seemed drowsy or when delta waves appeared on the EEG. After sleep deprivation, mice were not disturbed and post-sleep deprivation recordings were obtained for 3 hours. To assess post-sleep deprivation rebound, sleep EEG activity in the 3 hours post-sleep deprivation and 3 hours post undisturbed sleep were compared. Mouse vigilance states were determined as described in the section above. Data are reported as mean ± SEM. Comparisons of sleep parameters after sleep deprivation and after normal sleep were assessed using a paired Student’s t-test. The threshold p value for statistical significance was 0.05.
Results
Sleep impairment in DS mice
Patients with DS have impaired sleep quality (Dravet et al., 2005; Nolan et al., 2008; Nolan et al., 2006). To investigate whether DS mice have impaired sleep quality, we carried out combined video, electroencephalographic (EEG), and electromyographic (EMG) recordings from 7 WT and 6 DS mice for 8 h during the light period, the predominant sleep phase for mice (Fig. 1A–C). We generated hypnograms to mark sleep and wake states in order to estimate total sleep time, which did not differ between DS mice and their WT littermates (80.5 ± 4 % in DS vs. 75.4 ± 3 % in WT mice; p > 0.05; Fig. 1D).
Fig. 1.

Unaltered total sleep duration in WT and DS mice. A, Examples of EEG-EMG recordings from a WT mouse and a hypnogram generated for sleep (S)/wake (W) scoring. EEG trace labels correspond to electrode positions illustrated in B. B, Drawing of a mouse skull illustrating the sites of EEG electrode placement. C, Magnified segments of wake and sleep EEG-EMG traces in the box in panel A. Scale bars: 1 s, 500 μV. D, Average total sleep duration for WT and DS mice.
The video-EEG-EMG records were visually inspected in 5-s epochs and scored for Wake, NREM sleep, and REM sleep states (Fig. 2). Wakefulness was indicated by low amplitude, mixed frequency EEG waves, high amplitude EMG activity, and normal mouse stereotypical behaviors including grooming and exploring (Fig. 2, top). NREM sleep was characterized by high amplitude, low frequency EEG waves in the delta band (1–5 Hz), lower amplitude EMG activity, and behavioral arrest with occasional small, brief body movements observed on video and EMG (Fig. 2, middle). REM sleep was characterized by low amplitude, mixed frequency EEG activities dominated by “sawtooth waves” in the theta band (5–10 Hz), lower EMG activity, and behavioral arrest with relaxed body observed on video indicating muscle hypotonia, a characteristic feature of REM sleep (Fig. 2, bottom). The durations of the sleep stages did not differ between WT and DS mice. The average NREM sleep duration was 91.7 ± 2.9 % of total sleep in DS mice vs. 91.8 ± 1.0 % in WT (p > 0.05), and the REM sleep duration was 8.2 ± 2.8 % of total sleep in DS mice vs. 8.2 ± 1.0 % in WT (p > 0.05). Initial visual inspection of our records from DS mice suggested more frequent wakes, less delta wave activity in NREM sleep, and less theta wave activity in REM sleep, when compared to WT mice at the same Zeitgeber Time to control for circadian regulation of sleep. These visual observations suggested that DS mice have impaired NREM and REM sleep patterns.
Fig. 2.

Less defined sleep EEG waveforms in DS mice than WT mice. Examples of wake, NREM sleep, and REM sleep EEG-EMG from WT (Left) and DS (Right) mice illustrating the less prominent and less sustained EEG delta waves during NREM sleep in a DS mouse, and the similarly less defined EEG theta waves during REM sleep of the same mouse. The stereotypic mouse behaviors for the EEG traces are characterized by activity in the associated EMG traces and depicted in adjacent photographs extracted from videos of experiments with the WT mouse. EEG trace labels correspond to electrode positions in Fig. 1B.
To quantify these observations, we computed the power spectral densities of the EEG during NREM and REM sleep between 1 and 20 Hz (Fig. 3A). For NREM sleep, we found that the average peak of power spectral density in the 1–5 Hz (delta) band was considerably lower in DS mice (153 ± 27 μV2/Hz, n=7) than in WT mice (421.6 ± 104 μV2/Hz, n=6; p= 0.002: Fig. 3B). Moreover, the power density was selectively reduced at lower frequencies in the delta range in DS mice (Fig. 3B, left), which is illustrated by the increase in the ratio of DS to WT spectral density at higher frequencies (Fig. 3B, right). For REM sleep, the power density spectral profiles in WT and DS peaked in the same frequency range, 5–10 Hz (theta), but the maximum power density was considerably lower in DS mice (149.8 ± 47.7 μV2/Hz, n=7 vs. 260 ± 50 μV2/Hz, n=6 in WT; p = 0.01; Fig. 3C). These findings demonstrate that DS mice have significantly lower EEG activity in frequencies that define the NREM and REM phases of sleep.
Fig. 3.

Decreased EEG power densities in DS mice during sleep. A, Unaltered mean power density profiles for combined wake and sleep states for WT and DS mice. B, Mean NREM sleep power density profiles and power ratios for DS and WT mice. (Left) Power density spectra for DS and WT mice. Inset shows mean ± s.e.m. of the integral of power in the delta frequency range. (Right) Ratio of DS/WT power density versus frequency. C, Mean REM sleep power density profiles and power ratios for DS and WT mice. (Left) Mean REM sleep power density profiles for DS and WT mice. (Right) Ratios of HET/WT power density in the theta frequency range.
Sleep spindles are one of the defining characteristic features of the EEG in NREM sleep. They are fast, synchronous, sinusoidal oscillations of EEG activity, in the 10 to 15 Hz (sigma) band, with a crescendo-decrescendo pattern. Spindles were identified during visual inspection of the EEG (Fig. 4A) and by automatically filtering the data in the frequency band between 10–15 Hz using a computer algorithm (Materials and Methods). On average, we identified approximately 3-fold more spindles in WT mice (217 ± 17, n=6) than DS mice (68.3 ± 15, n=7, p= 0.001) in eight-hour recording (Fig. 4B). This remarkable reduction in the number of sleep spindles would have a profound effect on the quality of sleep in DS.
Fig. 4.

Decreased incidence of sleep spindles during NREM sleep in DS mice. A, Examples of sleep spindles in WT and DS mice. EEG-EMG trace labels correspond to electrode positions in Fig. 1B. Spindles are marked by black bar above EEG traces. B, Average number of spindles per 8-h recording period for WT and DS mice.
Interictal spikes and brief waking episodes during NREM sleep in DS mice
NREM sleep is a marker of deep sleep, from which it is difficult to awaken a sleeping subject. Rodent NREM sleep is fragmented by numerous brief wakes (Tobler et al., 1997). Excessive sleep fragmentation is one of the hallmarks of sleep disturbance in mammals and is one of the specific symptoms of sleep disturbance in DS patients. The reduction in delta wave EEG power in DS mice during NREM sleep suggests that these mice may have a higher propensity for transitions from slow-wave sleep to wake state (McCormick and Bal, 1997; Steriade, 2005). To examine the extent of sleep fragmentation in DS mice, we recorded the incidence of brief (<5s) waking events in simultaneous EMG and EEG measurements (Fig. 5A). WT and DS mice experienced brief wakes exclusively during NREM sleep; however, DS mice had more than twice as many brief wakes as WT mice, indicating that their sleep was more fragmented (Fig. 5B).
Fig. 5.

Increased incidence of brief wakes and interictal spikes during sleep in DS mice. A, Examples of NREM sleep EEG illustrating brief wakes observed in both WT and DS records. EEG-EMG trace labels correspond to electrode positions in Fig. 1B. B, Mean ± s.e.m. of brief wakes in NREM and REM sleep per 8-h recording period. C, Mean ± s.e.m. of spikes in Wake, NREM, and REM sleep per 8-h recording period.
Visual evaluation of EEG records revealed numerous interictal spikes in DS mice (Fig. 5A). Interictal spikes are hallmarks of the epileptic brain, and in clinics they help to confirm the diagnosis of epilepsy and to characterize generalized vs. partial epilepsies (Fisher et al., 1997; Smith, 2005). Interictal spikes were defined as high amplitude, sharply contoured EEG waveforms with abrupt onset, brief interruption of background activity, and no association with movement on video (Oakley et al., 2009; Yu et al., 2006). The EEGs of WT mice did not have any spikes during sleep. In contrast, 100% of DS mice had interictal spikes during NREM sleep, with an average of 135 ± 33 spikes per eight hours of recording (Fig. 5C). No interictal spikes were observed during REM sleep or wake (Fig. 5C). Many interictal spikes preceded brief wakes in the EEG/EMG records (Fig. 5A). To investigate whether there was a correlation between the excessive incidence of brief wakes and the presence of spikes in DS mice, we counted the number of brief wakes with interictal spikes in the preceding 5 s in DS mice. Forty percent of the brief wakes were preceded by interictal spikes, and the frequency of interictal spikes in the 5 s preceding brief wakes was substantially higher (4.8 spikes/min) than in the 5 s following a brief wake (0.21 spikes/min), which is similar to the average number of spikes per min throughout the period of NREM sleep. These findings show that interictal spikes precede a fraction of brief wakes observed in DS mice and may contribute directly to the increase in brief waking episodes.
Altogether, our studies of sleep in DS mice reveal a substantial deficit in sleep quality. Delta wave power and the number of sleep spindles are profoundly reduced in NREM sleep, and the number of brief wakes is increased. Theta wave power is reduced more modestly in REM sleep. These deficits in sleep quality in DS mice correlate directly with poor quality of sleep in DS patients, suggesting that our mouse model recapitulates sleep impairment in DS accurately.
Whole-cell sodium current in thalamic neurons of NaV1.1 heterozygous and homozygous knockout mice
The RNT is a thin, crescent-shaped nucleus that lies between the cerebral cortex and thalamus. Its GABAergic neurons strongly modulate the bi-directional flow of information between the thalamus and cerebral cortex. A trisynaptic circuit connecting excitatory neurons of the ventrobasal nucleus (VBN) of the thalamus, pyramidal cells in the cerebral cortex, and RNT neurons is essential for generation of sleep spindles and for normal sleep architecture (Beenhakker and Huguenard, 2009; Fuentealba and Steriade, 2005; Steriade, 2005). GABAergic neurons of the RNT play a pivotal inhibitory role in the generation of oscillatory activity in this thalamocortical network, and this activity underlies sleep spindles and cortical delta-wave sleep EEG rhythms (Contreras and Steriade, 1995). To investigate the cellular basis for the sleep disturbance, we recorded NaV current from the inhibitory GABAergic neurons of the RNT and the excitatory TC relay neurons of the VBN. At postnatal day 14, RNT neurons express all four CNS subtypes of sodium channels--NaV1.1, NaV1.2, NaV1.3, and NaV1.6 (Fig. 6; (Lein et al., 2007; Yu et al., 2006)). In our mice low levels of NaV1.2 (Fig. 6, D1, E1, F1) and NaV1.3 (Fig. 6G1, H1 I1) channel protein were detected in the cell bodies of RNT neurons and levels were not noticeably changed in DS mice (HET) and in Nav1.1 homozygous knockout animals (HOMO). NaV1.6 channels were highly localized in the cell bodies and dendrites of these neurons (Fig. 6J1, K1, L1) but levels did not change dramatically with genotype. This contrasts with NaV1.1 channels which are also localized in neuronal cell bodies and proximal processes but levels are dramatically reduced in DS mice and are absent in HOMO mice (Fig. 6A1, B1, C1). The somatodendritic expression NaV1.1 channels indicates that they could play an important role in integration of incoming synaptic signals and initiation of action potentials. Our previous studies demonstrated that GABAergic interneurons in the hippocampus and the GABAergic Purkinje neurons in the cerebellar cortex of DS mice have greatly reduced NaV currents in their cell bodies (Kalume et al., 2007; Yu et al., 2006). To determine whether GABAergic neurons of the RNT are similarly impaired in DS mice, whole-cell sodium currents were recorded from RNT neurons acutely dissociated from WT and mutant mice (Fig. 7A). Sodium currents were evoked during step depolarizations from a holding potential of −90 mV to a series of test potentials. The currents were normalized to cell capacitance to correct for differences in cell size. The peak sodium current densities in neurons of NaV1.1 HET and HOMO mice were reduced compared to those of cells from WT mice (Fig. 7B). On average, the peak current density was – 86.7 ± 7 pA/pF in HET (n=16) and −60.8 ± 3.1 pA/pF in HOMO (n=11) compared to −112.3 ± 4 pA/pF in WT (n=9, p< 0.0001), corresponding to a reduction of peak sodium current of 22.5 ± 2 % in HET and 45.9 ± 2 % in HOMO (Fig. 7A, B). In contrast to the large effect on peak sodium currents, we observed little effect on the voltage dependence of sodium channel function (Fig. 7C, D). These findings indicate that deletion of NaV1.1 channels in the mutant mice reduces the density of sodium current with no detectable effects on voltage-dependent function of NaV channels in GABAergic RNT neurons.
Fig. 6.

Expression of NaV1.1, NaV1.2, NaV1.3, and NaV1.6 channels in the reticular nucleus of the thalamus of P14 WT, HET, and HOMO knockout NaV1.1 mice. A1, B1, C1, Staining with anti-NaV1.1 antibody showed labeling of cells bodies in the reticular nucleus of WT and HET but not HOMO knockout mice. A2, B2, C2, Corresponding GAD 67 staining and A3, B3, C3, merged images of NaV1.1 and GAD 67 staining. D1, E1, F1, Staining with anti-NaV1.2 antibody illustrating staining throughout the RNT region that did not differ between WT and HET or HOMO knockout animals. D2, E2, F2, Corresponding GAD 67 staining and D3, E3, F3, merged images of NaV1.2 and GAD 67 staining. G1, H1, I1, Staining with anti-NaV1.3 antibody demonstrating labeling of cell bodies in the reticular nucleus of WT, HET and HOMO knockout animals. G2, H2, I2, Corresponding GAD 67 staining and G3, H3, I3, merged images for NaV1.3 and GAD 67 staining. J1, K1, L1, Staining with anti-NaV1.6 showing expression of this channel in cell bodies and dendrites. J2, K2, L2, Corresponding GAD 67 staining and J3, K3, L3, merged images of NaV1.6 and GAD 67 staining.
Fig. 7.

Decreased sodium currents in inhibitory but not excitatory thalamic neurons. A, (Left) Micrograph of an RNT neuron. Scale bar: 20 μm. (Right) Representative sodium current traces, from a RNT neuron, evoked with 50-ms depolarizations from a holding potential of −90 mV to + 30 mV in 5-mV increments. For clarity, only the first 5 ms of the traces are illustrated. B, Mean current-voltage relationships recorded from RNT neurons for WT (filled circles), HET (DS, filled squares) and HOMO knockout (filled triangles) mice. C, Voltage-dependence of activation of the sodium conductance in RNT neurons of NaV1.1 WT, HET (DS), and HOMO knockout mice. The half-activation voltage for sodium current in cells of WT animals was −36.1.3 ± 0.6 mV (k= 4.4 ± 0.6 mV, n= 9, p> 0.05) compared to −34.0 ± 0.3 (k= 5.6 ± 0.3, n= 16) and −29.6 ± 1 (k=6.3 ± 0.6, n=11) in cells of HET and HOMO knockout animals, respectively. D, Voltage dependence of sodium current inactivation in GABAergic RNT neurons measured using a 20-ms test pulse to 0 mV following a 100-ms prepulse to a variable potential (5-mV increments). The half-inactivation voltages were −49.4 ± 0.5 mV (k= 5.3 ± 0.2, n=8, p> 0.05), −51.9 ± 0.2 (k= 5.4 ± 0.2, n= 16), and −50.8 ± 0.4 (k= 6.0 ± 0.4, n= 11) for cells of WT, HET (DS), and HOMO knockout mice, respectively. E, (Left) Micrograph of an excitatory thalamocortical neuron of the ventrobasal nucleus of the thalamus. Scale bar: 20 μm. (Right) Representative sodium current traces evoked in a thalamocortical neuron using a family of 50-ms depolarizations from −90 to 0 mV in 5 mV increments. F, Mean sodium current vs. voltage relationships from thalamocortical neurons of WT, HET (DS), and HOMO Knockout mice. G, Voltage-dependence of activation of the sodium conductance in thalamocortical neurons from NaV1.1 WT, HET (DS), and HOMO knock-out mice. H, Voltage-dependence of sodium current inactivation in thalamocortical neurons.
Excitatory relay neurons in the VBN are the projection neurons of the thalamus; they form reciprocal synapses with the GABAergic neurons of the RNT. Together these two thalamic neuronal types are essential cellular elements of the thalamocortical network that produces synchronized rhythmic activity characteristically observed during sleep (Timofeev and Steriade, 1998). Balanced excitability of these two neuronal types is likely to be required for normal circuit function. Previous studies demonstrated that inhibitory neurons are much more affected by Scn1a gene mutations than excitatory neurons in the hippocampus and cerebellar cortex (Kalume et al., 2007; Ogiwara et al., 2007; Yu et al., 2006). NaV1.1 channels are localized in cell bodies of VBN neurons (Lein et al., 2007; Yu et al., 2006). To determine the effects of Nav1.1 haploinsufficiency on NaV current in excitatory neurons of the thalamus, the VBN was separately dissected, cell bodies of excitatory thalamocortical relay neurons were dissociated, and their sodium currents were recorded and analyzed across genotypes. The peak current density was slightly reduced, but not significantly, in cells of DS HET mice and of KO mice. In addition, the voltage-dependence of activation and voltage-dependence of inactivation were not significantly different for cells of mutant mice compared to those of WT mice (Fig. 7E–H). The average peak current densities were: 117.9 ± 18 pA/pF (n=11) in WT, 110 ± 14 pA/pF (n=13) in HET, in 95.6 ± 13 pA/pF (n=15) in HOMO cells (Fig. 7E and F). The half-activation was −33.4 ± 0.2 (k= 5.2 ± 0.2) in WT, −35.1 ± 0.4 (k=5.8± 0.4) in HET, and −31.0 ± 0.5 (k=5.3 ± 0.5) in HOMO neurons. The inactivation was −54.6 ± 0.2 (k= 6.4 ± 0.2) in WT, −56.6 ± 0.2 (k= 6.2 ± 0.2) in HET, −57.0 ± 0.4 (k=5.5 ± 0.3) in HOMO (Fig. 7E–H). These findings indicate that deletion of NaV1.1 channels does not significantly affect the density or function of NaV channels in excitatory thalamocortical relay neurons and therefore would cause an imbalance of excitability between the excitatory VBN relay neurons and the inhibitory RNT neurons.
Reduced action potential firing in GABAergic RNT neurons
NaV current plays an important role in the generation of neuronal action potential firing. RNT neurons exhibit different firing patterns based on the sleep state (Llinas and Steriade, 2006). Rebound bursting of action potentials from hyperpolarized membrane potentials is the signature electrophysiological feature of these neurons during NREM sleep (Contreras and Steriade, 1995). Similar rebound bursts of action potentials can be generated in RNT neurons in vitro upon releasing the cell membrane potential from a hyperpolarized voltage (Contreras et al., 1992; Huguenard and Prince, 1992). To examine whether the decrease of NaV current in RNT neurons affects their rebound firing, we measured the number of action potentials generated in the 500 ms following termination of injection of hyperpolarizing currents for 800 ms. RNT neurons from WT mice exhibited the characteristic rebound bursts of action potentials after release of membrane potential from hyperpolarizing voltage (Fig. 8A). The number of action potentials during rebound activity was dependent on the holding potential and the intensity of the hyperpolarizing current injected. More negative holding potentials or stronger hyperpolarizing current generated more action potentials (Fig. 8B). At each level of injected hyperpolarizing current, RNT neurons from HET and HOMO mice fired fewer action potentials than WT, even though they were hyperpolarized to similar voltages (Fig. 8B). For example, at −100 pA of injected current, which hyperpolarized the cells to the same extent (WT, −111 ± 4 mV; HET, −110 ± 4 mV; HOMO, −107 ± 7 mV; p<0.05), the number of action potentials was decreased from 8.5 ± 1 in WT (n=14) to 4.0 ± 1 in HETs (n=15) and HOMO (n=9) (Fig. 8B; p<0.001). No changes were detected in other electrophysiological parameters: action potential threshold, width, amplitude, or input resistance (Table 1). This result demonstrates a substantial impairment of rebound burst firing of RNT neurons in DS mice.
Fig. 8.

Reduced rebound firing of RNT neurons in DS mice. A, Examples of rebound bursts of action potentials from WT, HET (DS), and HOMO knockout neurons for hyperpolarization with −80 pA current. B, Mean number of action potentials (+/− s.e.m) during the rebound plotted against the amount of hyperpolarizing current applied (bottom axis) and voltage reached at end of hyperpolarization (top axis).
Table 1.
Mean Electrophysiological Parameters
| Genotype | AP Threshold (mV) | AP Width (ms) | AP Peak Voltage (mV) | Input Resistance (MΩ) |
|---|---|---|---|---|
| WT | −35.1 ± 1.0 | 8.5 ± 0.4 | 40.6 ± 6.0 | 75.0 ± 9.0 |
| HET | −37.7 ± 1.0 | 8.6 ± 0.7 | 35.1 ± 6.0 | 69.8 ± 11 |
| KO | −38.1 ± 1.0 | 8.6 ± 0.4 | 33.2 ± 5.0 | 71.8 ± 9.0 |
Reduced rebound slow wave activity during homeostatic regulation of sleep
To ensure sleep homeostasis, sleep deficits elicit a compensatory increase in the intensity and/or duration of sleep, whereas excessive sleep reduces the propensity for sleep (Borbely, 1982). Post-deprivation rebound of slow wave activity in NREM sleep has been reported in many species, including mice (Huber et al., 2000; Larkin and Heller, 1998; Rechtschaffen et al., 1999). Because burst firing in RNT neurons plays an important role in the generation of the slow wave activity in NREM sleep (Steriade, 2005), our results presented above predict a decrease of slow wave activity in the EEG during rebound from total sleep deprivation. To control for circadian variations of sleep and examine this predicted impairment of sleep homeostasis in DS mice, we measured the extent of rebound slow wave activity by comparing sleep activity during 3 h immediately after 5 h of total sleep deprivation vs. after 5 h of normal sleep (Fig. 9, Control: see Materials and Methods). Following sleep deprivation, WT mice showed a robust increase in EEG power density during NREM sleep (Fig. 9A). In contrast, DS mice showed little effect of prior sleep deprivation on EEG power density during NREM sleep (Fig. 9B). The peak power density of the delta waves increased from 136.2 ± 20 after normal sleep to 309.5 ± 80 μV2/Hz after sleep deprivation in WT mice (n=5) but only from 132.1 ± 17 to 179.1 ± 15 μV2/Hz after sleep deprivation in DS mice (n=6; p<0.001). These findings correlate the loss RNT neuron excitability and action potential firing with impaired homeostatic rebound of NREM sleep.
Fig. 9.
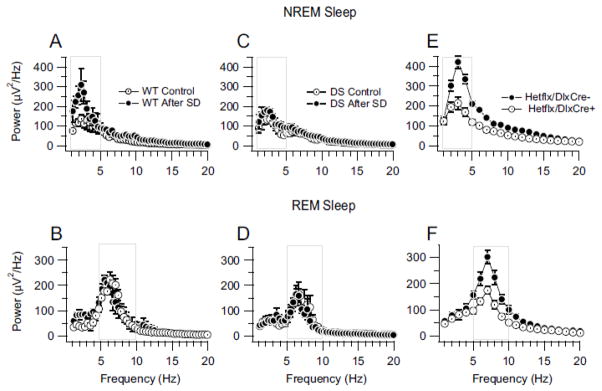
Reduced EEG power density during rebound from sleep deprivation in DS mice. A, Power density spectra for NREM sleep in WT mice after 5-h normal sleep (Control) and after 5-h sleep deprivation (SD). B, Power density spectra for REM sleep in DS mice after 5 h normal sleep (Control) and after 5-h sleep deprivation (SD). Boxes in A and B indicate the delta and theta frequency bands respectively. C, Rebound REM sleep EEG activity for DS mice after 5h of normal sleep (Control) and after 5h of sleep deprivation (SD). D, Rebound REM sleep EEG activity for DS mice after 5 h of sleep deprivation (SD). Boxes in C and D indicate the delta and theta frequency bands respectively. The power density measured under control conditions in these experiments is less than observed in Fig. 3 because here we recorded EEG for 3 h, beginning 5 h after the onset of the light period for homeostatic sleep regulation, whereas we recorded EEGs during 8 h of sleep from the onset of the light period for sleep architecture studies (See Materials and Methods). E and F, Decreased mean NREM (E) and REM (F) sleep EEG power density in conditional forebrain interneuron specific Scn1a KO mice. Boxes in E and F indicate the delta and theta frequency bands respectively
In contrast, as expected from previous studies (Endo et al., 1997; Rechtschaffen et al., 1999), the power density of the theta waves during REM sleep was not significantly increased by sleep deprivation in WT mice (221.7 ± 17 in control conditions vs. 222.8 ± 29 μV2/Hz after deprivation, n=5, p>0.05) or in DS mice (164.4 ± 22 in control conditions vs. 163.6 ± 34 μV2/Hz after deprivation, n=6, p>0.05) (Fig. 9C, D). These findings demonstrate a dramatic and selective loss of homeostatic regulation of slow wave activity following sleep deprivation in DS mice, as predicted from the deficit in rebound burst firing in RNT neurons that we have recorded. This loss of rebound firing of GABAergic RNT neurons likely contributes in a major way to the loss of EEG power density during both baseline and rebound NREM sleep and the substantial impairment of sleep quality in DS mice.
Sleep and circadian rhythms in mice with conditional deletion of Scn1a
DS mice carry a global heterozygous deletion mutation of Scn1a in both excitatory and inhibitory neurons. In order to directly test the role of NaV1.1 channels in forebrain GABAergic interneurons in DS, we generated conditional Scn1a heterozygous mice with deletion of one allele of Scn1a specifically in forebrain GABAergic interneurons by mating DlxI12b-cre and Scn1a floxed mouse lines (Cheah et al., 2012). These mice have reduced levels of NaV 1.1 channels in GABAergic interneurons of the hippocampus and cerebral cortex, but normal expression in excitatory neurons, and they develop seizures and die prematurely like DS mice (Cheah et al., 2012; Kalume et al., 2013). To investigate whether specific deletion of NaV 1.1 channels in forebrain GABAergic interneurons affects sleep rhythms, we recorded and examined the sleep video-EEG-EMG of these conditional HET mice. Sleep impairments similar to those of DS mice were observed. The delta power of the EEG was reduced in the NREM sleep (214.30 ± 30.3 μV2/Hz in Hetflx/DlxCre+, n=6 vs. 420.4 ± 31.4 μV2/Hz in Hetflx/DlxCre− mice, n= 5, p< 0.001) (Fig. 9E) and the theta EEG power was reduced in the REM sleep (175.1 ± 14.6 in Hetflx/DlxCre+ μV2/Hz, n=6 vs. 302.40 ± 25.7 Hetflx/DlxCre− mice, n=5, p< 0.001) (Fig. 9F). These findings indicate that heterozygous Scn1a deletion in forebrain GABAergic interneurons is sufficient to reproduce these sleep defects. Because DlxI12b-cre is expressed in the GABAergic RNT neurons (Potter et al., 2009), these findings further support the correlation between loss of NaV 1.1 channels in these neurons and impairment of sleep rhythms.
Our previous studies showed that DS mice have abnormal circadian rhythms characterized by longer circadian period, delayed daily onset of activity, and lack of light-induced phase shifts, as well as substantially reduced total wheel-running activity (Han et al., 2012b). To examine whether the specific knock-out of Scn1a in forebrain GABAergic interneurons causes similar abnormalities of circadian rhythms as in DS mice, we assessed wheel-running activity in the conditional HET mice and their control littermates. Conditional HET mice had slightly reduced total wheel-running activity during normal LD and DD test conditions (Fig. 10A and C). However, the length of circadian period (23.75 ± 0.03 h in Hetflx/DlxCre+, n=5 vs. 23.8 ± 0.05 h in Hetflx/DlxCre− mice, n=5, p>0.05), the delay of daily activity onset (5.0 ± 6.0 min in Hetflx/DlxCre+, n=5 vs. −7.5 ± 6.5 min in Hetflx/DlxCre− mice, n=5, p>0.05), and the effect of light pulse on activity phase shift (Light: −05.0 ± 4.0 min in Hetflx/DlxCre+, n=5 vs. −112.5 ± 5.0 min in Hetflx/DlxCre− mice, n=5, p>0.05; Dark: −8.5 ± 5.0 min in Hetflx/DlxCre+, n=5 vs. −0.1 ± 5.0 min in Hetflx/DlxCre− mice, n=5, p>0.05) did not significantly change in conditional HET mice (Fig. 10B and D). These findings demonstrate that the sleep impairment in DS mice illustrated in Figs. 1–5 and 9 is independent of effects of the DS mutation on circadian rhythm and thereby lend further support to the hypothesis that reduced excitability of RNT neurons is a major contributing factor to impairment of sleep quality in DS.
Fig. 10.
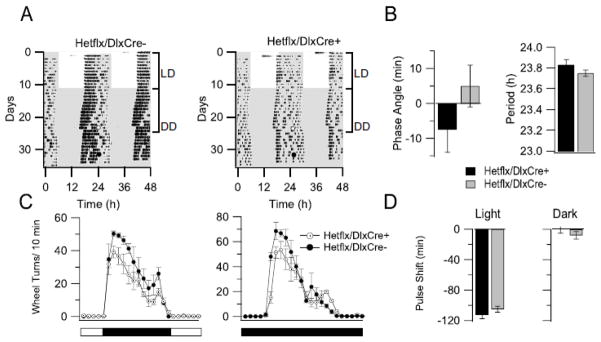
Mild abnormalities of circadian behavioral phenotypes in forebrain interneuron specific Scn1a KO mice. A, Wheel-running activity in a representative control mouse (left) and conditional HET mouse (right). Representative actograms of wheel-running activity display wheel revolutions as black bars on 48-h periods. White and gray areas represent the light and dark period, respectively. Light pulses of 30 min duration were applied to the mouse cage at CT 16 (white circle) and control pulses were similarly applied (dark circle). B, Phase angle and Period of the locomotor activity. C, Mild reduction of activity in the conditional KO mice compared to controls under LD condition (39.2 ± 3.6 in Hetflx/DlxCre+ wheel turns/10 min, n=5 vs. 50.3 ± 1.6 Hetflx/DlxCre− mice, n=5, p< 0.001) and DD condition (51.3 ± 4.5 in Hetflx/DlxCre+ wheel turns/10 min, n=5 vs. 68.6 ± 7.0 Hetflx/DlxCre− mice, n=5, p< 0.001). D, No significant effects of light pulses on conditional HET mice circadian phase shifting.
Discussion
Our results demonstrate substantial impairment of sleep in DS mice and provide unexpected insights into the key role of NaV1.1 channels in the function of RNT neurons and in integrative sleep physiology and homeostasis.
Functional and pathophysiological roles of NaV1.1 channels in RNT neurons
Our voltage-clamp recordings reveal decreased whole-cell NaV current in cell bodies of GABAergic neurons of the RNT, but not in cell bodies of neighboring excitatory neurons in the VBN of the thalamus. NaV1.1, NaV1.2, NaV1.3, and NaV1.6 proteins and mRNAs, encoded by Scn1a, Scn2a, Scn3a, and Scn8a genes respectively, are expressed in cell bodies of GABAergic interneurons of the RNT and excitatory thalamocortical neurons of the VBN (Lein et al., 2007; Yu et al., 2006). The preferential reduction of NaV current in RNT neurons indicates that the contribution of NaV1.1 current is greater in these GABAergic inhibitory neurons than in excitatory VBN relay neurons.
The decrease of NaV current in RNT neurons (22% in HET; 40% in HOMO) is less than the decrease in hippocampal GABAergic interneurons (53% in HET; 73% in HOMO) or cerebellar Purkinje neurons (40% in Het; 59% in HOMO) in DS mice (Kalume et al., 2007; Yu et al., 2006). Despite this comparatively small reduction of NaV current in RNT neurons rebound burst firing upon release from hyperpolarization is substantially impaired. Rebound burst firing is critical for generating EEG slow wave activity and sleep spindles and drives sleep behavior (Llinas and Steriade, 2006). Because rhythmic rebound bursts of action potentials are the characteristic firing pattern of RNT neurons during NREM sleep, our results predict that these cells are likely to be less active during this sleep stage in DS mice, and thereby cause impaired NREM sleep quality and homeostasis.
Our results with conditional HET mice where NaV1.1 channels in forebrain GABAergic interneurons eliminate reduction of NaV1.1 channels in excitatory neurons as a significant factor in the impairment of sleep quality in DS mice. However, interneurons in the cerebral cortex regulate firing of cortical pyramidal cells and may indirectly influence sleep rhythms by modulating the activity of these excitatory neurons. Surprisingly, heterozygous deletion of NaV1.1 channels does not have a detectable effect on sodium currents measured in whole-cell voltage clamp of cell bodies of dissociated interneurons from the cerebral cortex (Tai et al., 2014), suggesting a smaller defect in excitability of these neurons compared to RNT neurons. Nevertheless, parvalbumin-expressing, fast-spiking interneurons and somatostatin-expressing Martinotti cells in Layer V of the cerebral cortex do have significantly impaired action potential firing and synaptic boosting when intact neurons are studied in brain slices from DS mice (Tai et al., 2014). However, their impairment in action potential firing is less severe than we have observed for RNT neurons. Considering the lesser impairment of excitability of cortical interneurons and the more direct role of RNT neurons in generating sleep rhythms, it is likely that the loss of rebound firing of NRT neurons described here has a predominant role in sleep impairments in DS mice.
Loss of NaV1.1 channels impairs both baseline sleep and homeostatic regulation of sleep
Previous studies have shown that homozygous deletion of genes encoding voltage-gated ion channels, including CaV3.1, CaV3.3, CaV2.3, KV3.2, KV1.2, and SK2 channels, reduced the firing ability of RNT neurons and caused sleep abnormalities (Anderson et al., 2005; Cueni et al., 2008; Douglas et al., 2007; Espinosa et al., 2008; Lee et al., 2004; Vyazovskiy et al., 2002b; Zaman et al., 2011). Our results show that NaV1.1 channels are also required for normal firing of RNT neurons and that a dominant human disease mutation in NaV1.1 dramatically reduces sleep quality in heterozygous animals. In NREM sleep, delta wave activity and sleep spindles are reduced, and brief wakes are substantially increased in DS mice. The decreased delta wave activity in NREM sleep of the conditional HET mice shows that deletion of NaV 1.1 channels in forebrain GABAergic interneurons alone is sufficient to replicate the main sleep impairments observed in DS mice. Moreover, our results show that mutation of one allele of Scn1a also dramatically impairs sleep homeostasis in DS mice, as assessed from the large reduction in the rebound of slow wave activity after sleep deprivation. Given the crucial role of the RNT neurons in generating sleep rhythms, it is likely that impairment of burst firing by RNT neurons contributes substantially to the underlying mechanism that impairs the quality of baseline sleep and rebound sleep homeostasis in DS mice.
In contrast to NREM sleep, the reduced excitability of RNT neurons cannot account for the more subtle REM sleep abnormality in DS mice. Theta-band EEG activity during REM sleep is supported by rhythmic synchronized activity of the hippocampal network, accompanied by inhibition of thalamocortical and interhemispheric transmission (Cobb et al., 1995; Losonczy et al., 2010; Mann and Paulsen, 2007; Vyazovskiy et al., 2004; Vyazovskiy et al., 2002a). EEG rhythmic activity in REM sleep is highly modulated by activity of hippocampal GABAergic interneurons (Cobb et al., 1995; Mann and Paulsen, 2007), suggesting that decreased excitability in these cells would have a detrimental effect on EEG theta waves in REM sleep. Therefore, we postulate that this impairment may be related to the decrease of excitability in hippocampal GABAergic neurons observed in our previous studies (Yu et al., 2006). Thus, defects in NREM and REM sleep are both likely to arise from impaired firing of different classes of GABAergic interneurons.
The defects in sleep quality and homeostasis observed here are distinct mechanistically from the circadian defects in DS mice (Han et al., 2012b) because these circadian deficits were not observed when we deleted NaV1.1 channels only in forebrain GABAergic interneurons. The circadian defects are likely due to failure of firing of GABAergic neurons in the suprachiasmatic nucleus, the central circadian clock that governs daily rhythms of mammalian physiology. Together these findings suggest that the circadian and sleep physiology defects in DS mice may be caused by distinct functional impairments arising from the deletion of NaV 1.1 channels in different neuronal populations.
In a recent study, Papale and colleagues (2013) characterized sleep abnormality in a mouse model of GEFS+ expressing the R1649H mutation in one allele of Scn1a. The GEFS+ mice exhibited increased wakefulness and reduced NREM and REM sleep duration during periods of darkness, when mice are primarily awake, but not during the light phase, when mice are primarily sleeping. In contrast, the profound disruption of sleep that we observed occurred during the light phase, when mice sleep normally. Sleep rebound in the six hours following sleep deprivation was unaltered in R1648H mice, whereas DS mice studied here showed increased sleep activity following sleep deprivation. The severity of Scn1a-related seizure disorders correlates with the severity of reduction in sodium current and interneuron firing associated with the gene mutation (Catterall et al., 2010). Therefore, the milder disruption of sleep in these GEFS+ mice may reflect the partial loss of sodium channel function observed for the R1648H mutation compared to complete loss of function in DS mice.
Brief wakes, interictal spikes and sleep disturbance in DS mice
In addition to the reductions in slow wave activity and sleep spindles, the striking increase in the number of brief wakes during NREM sleep indicates that DS mice experience multiple sleep defects. The excessive brief wakes of DS mice, occurring exclusively during NREM sleep, illustrate both the abnormal fragmentation in NREM sleep and the resistance of REM sleep to brief wakes. The appearance of numerous interictal spikes exclusively during NREM sleep is consistent with results of several clinical and animal studies that lead to the idea that NREM sleep promotes the generation of epileptiform discharges via thalamocortical synchronous activity, whereas REM sleep is resistant to both brief wakes and interictal spikes (Asano et al., 2007; Kotagal and Yardi, 2008; Matos et al., 2010; Zhou et al., 2007). This result is also consistent with the idea that interictal spikes are a signature clinical feature of DS and suggests that evaluations of EEG interictal spikes during NREM sleep would increase the yield and accuracy of diagnostic exams (Arzimanoglou, 2009). The temporal association of a substantial portion (40%) of interictal spikes with brief wakes suggests that the excess brief wakes during NREM sleep in DS mice may be triggered primarily by these interictal spike discharges. Interictal spikes may also contribute to cognitive defects in DS patients by impairing memory consolidation during sleep. These findings lay the foundation for future studies of the intricate, reciprocal relationships among epilepsy, sleep, and cognition in DS mice.
Correlation of sleep impairment in DS mice and DS patients
Surveys of DS patients consistently report sleep problems, including disturbance of sleep time, impaired sleep quality, and increased incidence of nocturnal spikes, spike and wave discharges, and seizures (Nolan et al., 2008; Nolan et al., 2006; Skluzacek et al., 2009). Disturbance of sleep time correlates with circadian defects in DS mice (Han et al., 2012b). Reduction of slow wave activity and sleep spindles in DS mice correlate with poor quality of sleep in DS patients. High incidence of interictal spikes and brief wakes during NREM sleep in DS mice is consistent with increased nocturnal arousals and EEG abnormalities in patients. This close correlation between clinical findings in DS patients and our results on DS mice suggests that impairment of sleep rhythm, quality, and homeostasis in DS are caused by combined effects of impaired firing of GABAergic interneurons in suprachiasmatic nucleus and RNT.
A common mechanism for co-morbidities in DS
In this study, we have identified and characterized sleep architecture defects in DS mice and shown that they correlate with impaired rebound firing of bursts of action potentials by the GABAergic RNT neurons. In previous studies, we found that epilepsy, ataxia, circadian rhythm defects, cognitive impairment, autistic-like behaviors, and premature death all are caused by impaired action potential firing in GABAergic interneurons in different brain regions (Cheah et al., 2012; Han et al., 2012a; Kalume et al., 2013; Kalume et al., 2007; Yu et al., 2006). Thus, a clear pattern has emerged correlating the epilepsy and co-morbidities in DS with failure of action potential firing in different classes of GABAergic interneurons.
Highlights.
DS mice have abnormal sleep with increased brief wakes and reduced sleep spindles
Delta wave power is reduced in Non-Rapid-Eye-Movement sleep
Homeostatic sleep rebound after sleep deprivation is impaired
NaV current and rebound action potentials are reduced in inhibitory RNT neurons
Selective gene deletion in inhibitory neurons causes the same sleep impairment
Acknowledgments
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health under award number R01NS025704 to W.A.C., K01NS062862 to F. K., and NSF IOS0909716 to H.O.D. and by a grant from the McKnight Foundation to W. A. C.
Footnotes
Conflict of interest: None
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson MP, et al. Thalamic CaV3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A. 2005;102:1743–8. doi: 10.1073/pnas.0409644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzimanoglou A. Dravet syndrome: from electroclinical characteristics to molecular biology. Epilepsia. 2009;50(Suppl 8):3–9. doi: 10.1111/j.1528-1167.2009.02228.x. [DOI] [PubMed] [Google Scholar]
- Asano E, et al. Effect of sleep on interictal spikes and distribution of sleep spindles on electrocorticography in children with focal epilepsy. Clin Neurophysiol. 2007;118:1360–8. doi: 10.1016/j.clinph.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazil CW. Epilepsy and sleep disturbance. Epilepsy Behav. 2003;4(Suppl 2):S39–45. doi: 10.1016/j.yebeh.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–32. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- Catterall WA, et al. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849–59. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CS, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A. 2012;109:14646–51. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat. 2003;21:615–621. doi: 10.1002/humu.10217. [DOI] [PubMed] [Google Scholar]
- Claes L, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, et al. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–8. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Contreras D, et al. Bursting and tonic discharges in two classes of reticular thalamic neurons. J Neurophysiol. 1992;68:973–7. doi: 10.1152/jn.1992.68.3.973. [DOI] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–22. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L, et al. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci. 2008;11:683–92. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- Douglas CL, et al. Sleep in Kcna2 knockout mice. BMC Biol. 2007;5:42. doi: 10.1186/1741-7007-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravet C, et al. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol. 2005;95:71–102. [PubMed] [Google Scholar]
- Endo T, et al. Selective and total sleep deprivation: effect on the sleep EEG in the rat. Psychiatry Res. 1997;66:97–110. doi: 10.1016/s0165-1781(96)03029-6. [DOI] [PubMed] [Google Scholar]
- Espinosa F, et al. Ablation of KV3.1 and KV3.3 potassium channels disrupts thalamocortical oscillations in vitro and in vivo. J Neurosci. 2008;28:5570–81. doi: 10.1523/JNEUROSCI.0747-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, et al. Epilepsy for the neuroradiologist. Am J Neurorad. 1997;18:851–63. [PMC free article] [PubMed] [Google Scholar]
- Fuentealba P, Steriade M. The reticular nucleus revisited: intrinsic and network properties of a thalamic pacemaker. Prog Neurobiol. 2005;75:125–41. doi: 10.1016/j.pneurobio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Han S, et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012a;489:385–90. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, et al. NaV1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci U S A. 2012b;109:E368–77. doi: 10.1073/pnas.1115729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, et al. Topography of EEG dynamics after sleep deprivation in mice. J Neurophysiol. 2000;84:1888–93. doi: 10.1152/jn.2000.84.4.1888. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca2+-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–17. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–808. doi: 10.1172/JCI66220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, et al. Reduced sodium current in Purkinje neurons from NaV1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007;27:11065–74. doi: 10.1523/JNEUROSCI.2162-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, et al. A missense mutation in SCN1A in brothers with severe myoclonic epilepsy in infancy (SMEI) inherited from a father with febrile seizures. Brain Dev. 2005;27:424–30. doi: 10.1016/j.braindev.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Kotagal P, Yardi N. The relationship between sleep and epilepsy. Semin Pediatr Neurol. 2008;15:42–9. doi: 10.1016/j.spen.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Larkin JE, Heller CH. The disappearing slow wave activity of hibernators. Sleep Res Online. 1998;1:96–101. [PubMed] [Google Scholar]
- Lee J, et al. Lack of delta waves and sleep disturbances during non-rapid eye movement sleep in mice lacking alpha1G-subunit of T-type calcium channels. Proc Natl Acad Sci U S A. 2004;101:18195–9. doi: 10.1073/pnas.0408089101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–76. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Llinas RR, Steriade M. Bursting of thalamic neurons and states of vigilance. J Neurophysiol. 2006;95:3297–308. doi: 10.1152/jn.00166.2006. [DOI] [PubMed] [Google Scholar]
- Losonczy A, et al. Network mechanisms of theta related neuronal activity in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2010;13:967–72. doi: 10.1038/nn.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–9. doi: 10.1016/j.tins.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Matos G, et al. The relationship between sleep and epilepsy: evidence from clinical trials and animal models. J Neurol Sci. 2010;295:1–7. doi: 10.1016/j.jns.2010.05.003. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- Nolan K, et al. Coping with a child with Dravet syndrome: insights from families. J Child Neurol. 2008;23:690–4. doi: 10.1177/0883073808314162. [DOI] [PubMed] [Google Scholar]
- Nolan KJ, et al. Coping with Dravet syndrome: parental experiences with a catastrophic epilepsy. Dev Med Child Neurol. 2006;48:761–5. doi: 10.1017/S0012162206001629. [DOI] [PubMed] [Google Scholar]
- Oakley JC, et al. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci U S A. 2009;106:3994–9. doi: 10.1073/pnas.0813330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, et al. NaV1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguni H, et al. Severe myoclonic epilepsy in infants--a review based on the Tokyo Women’s Medical University series of 84 cases. Brain Dev. 2001;23:736–48. doi: 10.1016/s0387-7604(01)00276-5. [DOI] [PubMed] [Google Scholar]
- Papale LA, et al. Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+) Epilepsia. 2013;54:625–34. doi: 10.1111/epi.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter GB, et al. Generation of Cre-transgenic mice using Dlx1/Dlx2 enhancers and their characterization in GABAergic interneurons. Mol Cell Neurosci. 2009;40:167–86. doi: 10.1016/j.mcn.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtschaffen A, et al. Effects of method, duration, and sleep stage on rebounds from sleep deprivation in the rat. Sleep. 1999;22:11–31. doi: 10.1093/sleep/22.1.11. [DOI] [PubMed] [Google Scholar]
- Skluzacek JV, et al. Expanding the Clinical Spectrum: Updated Data on Comorbidities in Dravet Syndrome. International Workshop on Dravet Syndrome: Severe Myoclonic Epilepsy of Infancy- 30 Years Later; Verona, Italy. 2009. [Google Scholar]
- Smith SJ. EEG in the diagnosis, classification, and management of patients with epilepsy. J Neurol Neurosurg Psychiatry. 2005;76(Suppl 2):ii2–7. doi: 10.1136/jnnp.2005.069245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–24. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Tai C, et al. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet Syndrome. Proc Natl Acad Sci U S A. 2014;111:E3139–48. doi: 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Steriade M. Cellular mechanisms underlying intrathalamic augmenting responses of reticular and relay neurons. J Neurophysiol. 1998;79:2716–29. doi: 10.1152/jn.1998.79.5.2716. [DOI] [PubMed] [Google Scholar]
- Tobler I, et al. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–79. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy V, et al. Interhemispheric coherence of the sleep electroencephalogram in mice with congenital callosal dysgenesis. Neuroscience. 2004;124:481–8. doi: 10.1016/j.neuroscience.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, et al. Interhemispheric sleep EEG asymmetry in the rat is enhanced by sleep deprivation. J Neurophysiol. 2002a;88:2280–6. doi: 10.1152/jn.00304.2002. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, et al. Sleep EEG in mice that are deficient in the potassium channel subunit KV3.2. Brain Res. 2002b;947:204–11. doi: 10.1016/s0006-8993(02)02925-6. [DOI] [PubMed] [Google Scholar]
- Yu FH, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zaman T, et al. CaV2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron. 2011;70:95–108. doi: 10.1016/j.neuron.2011.02.042. [DOI] [PubMed] [Google Scholar]
- Zhou JL, et al. Effect of interictal spikes on single-cell firing patterns in the hippocampus. Epilepsia. 2007;48:720–31. doi: 10.1111/j.1528-1167.2006.00972.x. [DOI] [PubMed] [Google Scholar]