

Abstract
Daptomycin is a lipopeptide antibiotic that is used clinically against many gram-positive bacterial pathogens and is considered a key frontline bactericidal antibiotic to treat multidrug-resistant enterococci. Emergence of daptomycin resistance during therapy of serious enterococcal infections is a major clinical issue. In this work, we show that deletion of the gene encoding the response regulator, LiaR (a member of the LiaFSR system that controls cell envelope homeostasis), from daptomycin-resistant Enterococcus faecalis not only reversed resistance to 2 clinically available cell membrane–targeting antimicrobials (daptomycin and telavancin), but also resulted in hypersusceptibility to these antibiotics and to a variety of antimicrobial peptides of diverse origin and with different mechanisms of action. The changes in susceptibility to these antibiotics and antimicrobial peptides correlated with in vivo attenuation in a Caenorhabditis elegans model. Mechanistically, deletion of liaR altered the localization of cardiolipin microdomains in the cell membrane. Our findings suggest that LiaR is a master regulator of the enterococcal cell membrane response to diverse antimicrobial agents and peptides; as such, LiaR represents a novel target to restore the activity of clinically useful antimicrobials against these organisms and, potentially, increase susceptibility to endogenous antimicrobial peptides.
Keywords: LiaFSR, daptomycin, E. faecalis, antimicrobial peptides
The World Health Organization has named antimicrobial resistance as one of the greatest public health threats of the 21st century [1, 2]. Multidrug-resistant enterococci are among the most difficult microorganisms to treat in clinical settings because of resistance to all available antimicrobials. Moreover, infections caused by such multidrug-resistant bacteria often occur in patients who are critically ill and/or significantly immunocompromised, further complicating the treatment of these patients.
Daptomycin (DAP), a key front-line bactericidal agent for treating multidrug-resistant enterococci, is a cyclic lipopeptide that targets the bacterial cell membrane (CM) of gram-positive bacteria, becoming a de facto cationic antimicrobial peptide. The antibacterial mechanism of DAP action involves interactions with the CM in a calcium-dependent manner [3–5]. Calcium-complexed DAP seems to insert initially into the outer leaflet of the bacterial CM with preference for septal division planes [6]. Subsequently, DAP appears to oligomerize in a process that has been shown to depend on the presence of phosphatidylglycerol [4]. Oligomerization of DAP leads to disruption of the integrity of the CM and alterations of cell division that eventually lead to cell death [6]. We have previously shown that mutations in genes encoding the 3-component regulatory system LiaFSR (involved in the bacterial response to cell envelope–acting antibiotics and antimicrobial peptides [AMPs]), markedly reduced the activity of DAP [7–11]. The LiaFSR system [12–17], which is present in all gram-positive pathogens of clinical importance, including streptococci and Staphylococcus aureus [12–17], is composed of a transmembrane protein (LiaF/VraT), a membrane bound histidine-kinase (LiaS/VraS), and a response regulator (LiaR/VraR). In Bacillus subtilis, LiaR regulates the expression of a gene cluster (liaIHGFSR) that orchestrates the CM stress response against attack by antibiotics and AMPs [12, 18–20], although the mechanisms of this response are unknown. We previously showed that, in Enterococcus faecalis, adaptation against DAP-mediated CM disruption involves redistribution of cardiolipin microdomains in the CM, which are relocalized away from the septum to other CM areas [10]. We postulated that the phospholipid remodeling diverted DAP from the septum to other cardiolipin-rich areas in the CM, thus preventing the antibiotic from interacting with its main septal target [10] and, perhaps, from reaching the inner leaflet of the CM [3]. The remodeling of cardiolipin microdomains was associated with a deletion of isoleucine in position 177 of LiaF [10]. Moreover, our previous studies using quantitative experimental evolution revealed that development of DAP resistance in a clinical strain of E. faecalis follows a clear order and hierarchy of genetic changes [11]. We also showed that substitutions in LiaFSR appear to be the first pivotal event in the evolution of DAP resistance and, possibly, membrane adaptations [11]. In this work, we present data that the response regulator, LiaR, mediates CM adaptation in E. faecalis by controlling the remodeling of CM cardiolipin microdomains. Deletion of the liaR gene not only reversed resistance to DAP and AMPs but also produced hypersusceptibility to these molecules in vitro and resulted in attenuation of in vivo virulence. Thus, we postulate that interfering with the LiaFSR response may provide a novel strategy to restore and preserve the activity of antimicrobials (eg, DAP) and potentiate the innate immune clearance of resistant microorganisms. Such an approach may become a viable antimicrobial strategy against multidrug-resistant gram-positive organisms in the future.
MATERIALS AND METHODS
Bacterial Strains and Growth Conditions
Bacterial strains and their phenotypes are listed in Table 1. E. faecalis S613 is a vancomycin-resistant and DAP-susceptible clinical isolate and was recovered from the bloodstream of a patient with bacteremia in 2005 [22]. S613FΔliaF177gdpD170cls61 is a DAP-resistant derivative of S613 generated in vitro that harbors 3 alleles associated with DAP resistance (liaF, gdpD, and cls) [22]. All enterococci were routinely grown on brain-heart infusion agar or broth at 37°C with gentle agitation. Escherichia coli strains used for cloning and genetic manipulations were grown in Luria-Bertani (LB) broth or LB broth supplemented with chloramphenicol (10 µg/mL) when harboring derivatives of pHOU3.
Table 1.
Characteristics of Enterococcus faecalis Strains Used in This Study
| E. faecalis Strain | Relevant Characteristic(s) | DAP MIC, µg/mLa | Reference(s) |
|---|---|---|---|
| S613 | Vancomycin-resistant and daptomycin-susceptible bloodstream isolate exhibiting HLR resistance to both gentamicin and streptomycin | 0.5 | [7, 10] |
| S613F | Fusidic acid-resistant derivative of S613 | 0.5 | This study |
| S613FΔliaF177gdpD170cls61 | Daptomycin-resistant derivative of S613F harboring liaF177,b gdpD170,c and clsd61 resistance-associated alleles | 8 | [7, 10] |
| S613FΔliaF177gdpD170cls61ΔliaR | Derivative of S613FΔliaF177gdpD170cls61 harboring a nonpolar deletion of liaR | 0.094 | This study |
| S613FΔliaF177gdpD170cls61ΔliaR::liaR | S613FΔliaF177gdpD170cls61ΔliaR complementation of liaR in cis (native chromosomal location) | 8 | This study |
| OG1RF | Laboratory strain of E. faecalis | 2 | [21] |
| OG1RFΔliaR | OG1RF harboring a nonpolar deletion of liaR | 0.094 | This study |
| OG1RFΔliaR::liaR | OG1RFΔliaR complementation of liaR in cis (native chromosomal location) | 2 | This study |
| OG1RFΔliaR::pAT392liaR | OG1RFΔliaR complemented with liaR in trans, using plasmid pAT392 under the control of the constitutive P2 promoter | 2 | This study |
Abbreviation: HLR, high-level resistance.
a Daptomycin (DAP) minimum inhibitory concentrations (MICs) were determined by the use of Etest (bioMérieux) on Mueller-Hinton agar.
b Mutated allele encoding the transmembrane protein LiaF, resulting in deletion of isoleucine at position 177 of the putative protein.
c Mutated allele encoding a glycerophosphoryl diester phosphodiesterase, resulting in deletion of isoleucine at position 170 of the putative enzyme.
d Mutated allele encoding a cardiolipin synthase, resulting in deletion of lysine at position 61 of the putative protein.
Antimicrobial Susceptibility Testing
Minimum inhibitory concentrations (MICs) of DAP and telavancin were determined by the Etest, using the manufacturer's recommendations (Biomerieux), on Mueller-Hinton agar and incubated for 24 hours. Telavancin susceptibilities were assessed because of similarities in the mechanism of action of telavancin, compared with that of DAP (targeting CM). The results were interpreted using breakpoints issued by the Clinical and Laboratory Standards Institute (CLSI) [23, 24], except for telavancin (for which no breakpoint was available).
Mutant Construction and Genetic Manipulations
To create nonpolar deletions of liaR in S613FΔliaF177gdpD170cls61 (Table 1), we used the p-chloro-phenylalanine sensitivity counterselection (PheS*) system [25] with plasmid pHOU3 (which carries the chloramphenicol acetyltransferase gene), as described previously [7, 26, 27]. Briefly, fragments upstream and downstream of liaR (Supplementary Table 1) were amplified by crossover polymerase chain reaction (PCR), using the genomic DNA of E. faecalis S613 as a template. PCR products were cloned into pHOU3, using specific restriction enzymes (Supplementary Table 1). The plasmid constructs were electroporated into E. faecalis CK111 and, subsequently, delivered to E. faecalis S613FΔliaF177gdpD170cls61 and E. faecalis OG1RF by conjugation [7, 10, 26, 27]. First-recombination integrants were selected on agar containing chloramphenicol (10 µg/mL) and fusidic acid (25 µg/mL). To obtain the desired replacement, first-event integrants were subsequently grown on p-chloro-phenylalanine, and colonies were tested by replica plating in the presence of different DAP concentrations [7, 10, 26, 27]. DAP-susceptible mutants were characterized by pulsed-field gel electrophoresis and the open read frames of liaFSR, gdpD, and cls were sequenced in their entirety in both directions.
For complemented strains, the liaR gene was amplified by PCR with primers 1 and 4 (Supplementary Table 1), using S613 DNA as a template and the fragment (2113 bp) cloned into pHOU3. The recombinant plasmids were delivered into the E. faecalis S613FΔliaF177gdpD170cls61ΔliaR and E. faecalis OG1RFΔliaR mutants, using the same procedures specified above. The liaR gene was also cloned into plasmid pAT392 [28], using primers 9 and 10 (Supplementary Table 1), for trans complementation in E. faecalis OG1RF, and recombinant colonies were selected in the presence of gentamicin (125 µg/mL). The complemented strains were tested by replica plating in the presence of different DAP concentrations and characterized by pulsed-field gel electrophoresis.
Evaluation of Activity of AMPs
The in vitro bactericidal assays with AMPs were carried out as described previously [29–31] using a 2-hours microdilution method in Eagle minimal essential medium. We used an inoculum of 103 colony-forming units (CFU) of exponential phase cells at 2 concentrations of AMPs (Table 2). These AMPs concentrations were selected based on extensive pilot studies showing their inability to substantially reduce starting inocula of either parental strain over the 2-hour exposure period. The data were calculated and expressed as the relative percent of surviving colony-forming units of AMP-exposed versus AMP-unexposed cells. The percentage survival at 2 hours of E. faecalis S613FΔliaF177gdpD170cls61ΔliaR was compared to the survival of its parental strain (E. faecalis S613FΔliaF177gdpD170cls61) and complemented derivative (E. faecalis S613FΔliaF177gdpD170cls61ΔliaR::liaR). A minimum of 3 independent experiments was performed for each experiment.
Table 2.
Survival of Enterococcus faecalis Strains After 2 Hours of Exposure to Antimicrobial Peptides
| E. faecalis Strain | Survival, %, Mean ± SD |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LL37, µg/mL |
HBD-3, µg/mL |
Nisin, µg/mL |
GAL, µg/mL |
RP-1, µg/mL |
MER, µg/mL |
FRU, µg/mL |
||||||||
| 20 | 10 | 0.15 | 0.075 | 0.8 | 0.4 | 10 | 5 | 10 | 5 | 100 | 50 | 80 | 40 | |
| S613FΔliaF177gdpD170cls61 | 81 ± 6a | 83 ± 6a | 77 ± 15a | 93 ± 6 | 98 ± 7a | 98 ± 3a | 46 ± 12a | 59 ± 10a | 43 ± 14a | 93 ± 30a | 77 ± 3a | 89 ± 12a | 90 ± 3a | 89 ± 6 |
| S613FΔliaF177gdpD170cls61ΔliaR | 17 ± 21 | 46 ± 28 | 38 ± 21 | 85 ± 18 | 11 ± 12 | 21 ± 23 | 0 ± 0 | 0 ± 0 | 7 ± 5 | 36 ± 22 | 10 ± 4 | 16 ± 5 | 7 ± 6 | 68 ± 18 |
| S613FΔliaF177gdpD170cls61ΔliaR::liaR | 84 ± 14a | 94 ± 22a | 85 ± 22a | 95 ± 18 | 91 ± 20a | 105 ± 14a | 57 ± 15a | 59 ± 8a | 44 ± 34 | 82 ± 14a | 88 ± 10a | 104 ± 28a | 101 ± 14a | 96 ± 12a |
Abbreviations: FRU, friulimicin, a cationic lipopeptide; GAL, gallidermin, type A lantibiotic; HBD3, human β-defensin 3; LL-37, a cathelicidin neutrophil-derived antimicrobial peptide; MER, mersacidin, type B lantibiotic; nisin, type A lantibiotic; RP-1, synthetic antimicrobial peptide; SD, standard deviation.
a P < .05, compared with S613FΔliaF177gdpD170cls61ΔliaR survival at 2 h.
Caenorhabditis elegans Infection Model
The method used for the C. elegans infection model has been previously described [32]. Briefly, 60–90 synchronized young adult nematodes were infected with E. faecalis S613FΔliaF177gdpD170cls61, S613FΔliaF177gdpD170cls61ΔliaR, and S613FΔliaF177gdpD170cls61ΔliaR::liaR on brain-heart infusion agar medium containing gentamicin (10 µg/mL). The plates were incubated at 25°C, and worm death was scored daily. Worms were considered dead when they exhibited no response to a platinum wire pick. Kaplan–Meier log rank analysis was used to compare survival curves pairwise. P values of .05 were considered statistically significant. The software GraphPad Prism (version 5.0) was used for analyses.
10-N-nonyl Acridine Orange (NAO) Staining
Visualization of CM cardiolipin-rich domains was performed as described before [10], using the fluorescent dye N-nonyl acridine orange (NAO). NAO has been previously shown to bind preferentially to cardiolipin-arrays in the CM in several bacteria, including enterococci [33], and the association of NAO with cardiolipin molecules results in a green-to-red shift in fluorescence [34]. Fluorescence images were captured by an Olympus IX71 microscope with a PlanApo N 100X objective. At least 3 independent experiments were performed for each strain on different days. Fluorescence was viewed using the PicoQuant Imaging System. Excitation was at 490 nm, and emission was at either 528 or 617 nm at exposure times of 500 ms. Fluorescence areas identified in both wavelengths indicate a green-to-red shift, a property exhibited by NAO when bound to cardiolipin (Figure 3 and Supplementary Figure 2).
Figure 3.
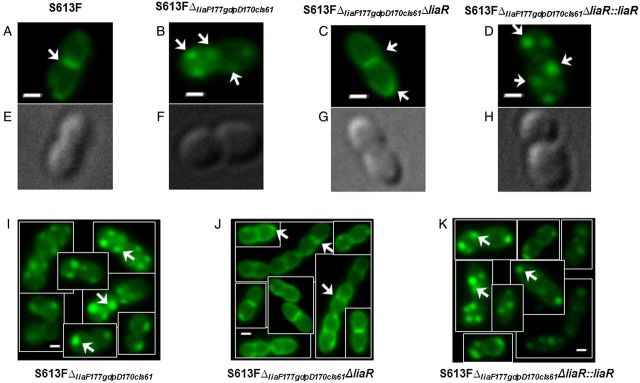
Staining of representative cells of Enterococcus faecalis S613F and derivatives with 10-N-nonyl acridine orange (500 nM). A–D and I–K, Fluorescence microscopy images of bacterial cells. E–H, Phase-contrast images of the same cells in panels A–D. A, S613F displays cardiolipin microdomains in septum and poles of the cell (white arrows). B and I, S613FΔliaF177gdpD170cls61 exhibits cardiolipin microdomains located away from the main septum and distributed throughout the cells. C and J, Deletion of liaR redistributes the cardiolipin microarrays back to the septum and poles, as shown in DAP-susceptible E. faecalis S613F. D and K, Reintroducing liaR in its native chromosomal location restored the remodeling of cardiolipin microdomains in the cell membrane. White arrows indicate the cardiolipin domains in the cell membrane, and bars denote lengths of 1 µm.
RESULTS
Deletion of liaR Restores Susceptibility to CM-Targeting Antibiotics and AMPs
To understand the role of LiaFSR in the enterococcal response to antimicrobials, we targeted the gene encoding the response regulator, LiaR, in E. faecalis strain S613FΔliaF177gdpD170cls61, a DAP- and fusidic acid–resistant derivative previously generated from E. faecalis S613 [22]. S613FΔliaF177gdpD170cls61 contains amino acid changes in 3 putative proteins associated with DAP resistance: (1) LiaFΔ177, a derivative of LiaF (a member of the LiaFSR system) lacking Ile177; (2) GdpDΔ170, a glycerophosphodiester phosphodiesterase with a deletion of Ile170 that is involved in CM phospholipid metabolism; and (3) ClsΔ61, a cardiolipin synthase harboring a deletion of Lys61 [7]. We generated a markerless nonpolar deletion of liaR (nucleotides 15–620) in E. faecalis S613FΔliaF177gdpD170cls61 (S613FΔliaF177gdpD170cls61ΔliaR), using the PheS* system, as described before [25]. Of note, deletion of liaR did not affect the growth of the mutant (Supplementary Figure 1A). Using the same PheS* system, we complemented the mutant (S613FΔliaF177gdpD170cls61ΔliaR::liaR) by delivering the original allele to the same chromosomal location. As shown in Figure 1A, deletion of liaR generated hypersusceptibility to DAP, fully reversing resistance to this antibiotic, with the MIC decreasing from 8 µg/mL to 0.094 µg/mL, far below the usual MIC of enterococci (breakpoint, 4 µg/mL). Complementation of the mutated strain with liaR restored the DAP resistance phenotype, indicating that liaR is necessary for DAP resistance in this E. faecalis strain. To confirm the role of liaR, we generated another liaR deletion in a laboratory strain of E. faecalis (OG1RF; DAP MIC, 2 µg/mL). Deletion of liaR in OG1RF also produced hypersusceptibility to DAP (reduction, 8 fold; MIC, 0.094), a phenomenon that was reversed by reintroducing liaR into its chromosomal location or on a multicopy plasmid (pAT392; Table 1), confirming liaR's role in the level of DAP susceptibility. Additionally, the liaR deletion also markedly increased the susceptibility to telavancin, resulting in an 8-fold MIC decrease (Figure 1A). All these MICs changes were reversed by reintroduction of liaR into its native chromosomal location (Figure 1A).
Figure 1.
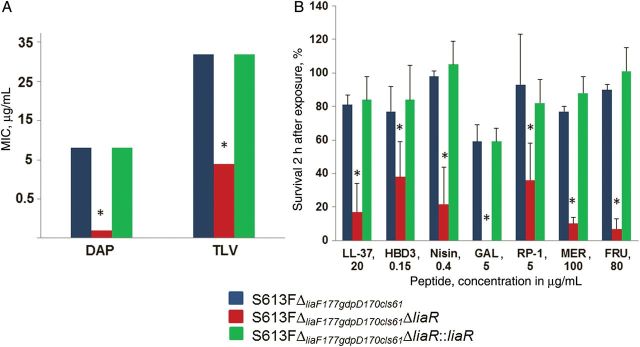
Minimum inhibitory concentrations (MICs) and susceptibility to antimicrobial peptides of Enterococcus faecalis derivatives. A, Change of daptomycin (DAP) and telavancin (TLV) MICs determined by the Etest (Biomerieux) on Mueller-Hinton agar. MIC determination was performed in triplicate for all strains, with the same result in each experiment. Of note, breakpoints for TLV are not available. B, Survival after exposure to antimicrobial peptides (AMPs). Results are from 3 independent experiments. *P < .001, compared with S613FΔliaF177gdpD170cls61. Abbreviations: FRU, friulimicin, naturally occurring lipopeptide; GAL, gallidermin, type A lantibiotic; HBD3, human β-defensin 3; LL-37, human cathelicidin; MER, mersacidin, type B lantibiotic; nisin, type A lantibiotic; RP-1, synthetic antimicrobial peptide.
AMPs are among the most ancient antibacterial mediators of the mammalian innate immune response [35]. Additionally, several prokaryotic organisms produce similar peptides that target competing bacteria. The main targets of prokaryotic- and eukaryotic-derived AMPs are the CM-associated precursors of peptidoglycan synthesis (lipid I and II), cell-wall teichoic acid and/or lipoteichoic acid precursors (lipid III and IV) [36], and the lipopolysaccharide (LPS) of gram-negative bacteria [35]. To determine whether liaR is also an important mediator of the E. faecalis response to AMPs (like calcium-complexed DAP), we tested the activity of a variety of AMPs against S613FΔliaF177gdpD170cls61, as well as its liaR deletion mutant and complemented derivatives. These peptides are from different origins and exhibit distinct mechanisms of action [35–40]. The peptides tested included (1) LL-37, a cathelicidin peptide produced by mammalian neutrophils and epithelium that has been shown to be active against both gram-positive and gram-negative pathogens (via binding of LPS) [35]; (2) human β-defensin 3, which is produced by epithelial cells and neutrophils and interacts with lipid II and LPS [37]; (3) nisin, a class I prokaryote bacteriocin and type A lantibiotic produced by Lactococcus lactis that interacts with lipid II, III, and IV [38]; (4) gallidermin, a nisin-related lantibiotic produced by Staphylococcus gallinarum that also targets lipid II, III, and IV [38]; (5) RP-1, a synthetic AMP mimicking the bactericidal domain of the PF-4 family of kinocidins [39, 41]; (6) mersacidin, a type B lantibiotic peptide produced by Bacillus species that targets lipid II [38]; and (7) friulimicin B, a naturally occurring cyclic lipopeptide produced by Actinoplanes friuliensis that interferes with peptidoglycan synthesis through the formation of a Ca(2+)-dependent complex with the bactoprenol phosphate carrier C(55)-P [38]. We evaluated survival of S613FΔliaF177gdpD170cls61ΔliaR, as well as its parental and complemented derivative, at 2 different concentrations of each AMP (Figure 1B and Table 2). Figure 1B shows that the liaR deletion had a profound and uniform impact on susceptibility to all AMPs tested, paralleling the effects seen with DAP and telavancin. Survival in the presence of AMPs was markedly reduced in the liaR deletion mutant, compared with its parental strain, and this effect was reversed by liaR complementation. Our results suggest that LiaR is a master regulator of the enterococcal CM response to both CM-acting antimicrobials and AMPs.
Deletion of liaR Also Affects In Vivo Virulence
To test whether the liaR deletion also affects the in vivo virulence of multidrug-resistant E. faecalis, we used the C. elegans model, which has been extensively used to examine the pathogenic properties of E. faecalis [32, 42]. AMPs are important mediators of the immune response in C. elegans, and this nematode possesses a complex signaling cascade that regulates the production of AMPs such as caenopores, lysozymes, and lectins [43–46] that behave as an important line of defense against bacterial pathogens. E. faecalis readily causes infection in the C. elegans model, colonizing the intestinal lumen and resulting in persistent infection leading to eventual death [32]. We tested the effect of the liaR deletion on the ability of S613FΔliaF177gdpD170cls61 to kill C. elegans. As shown in Figure 2, deletion of liaR significantly attenuated the virulence seen with the parental strain S613FΔliaF177gdpD170cls61 (P < .05), an effect that was initially evident 3 days after infection and continued over the 8-day observation period. Since the deletion did not affect the in vitro growth (Supplementary Figure 1), we postulate that the attenuation is likely due to increased susceptibility to AMPs.
Figure 2.

Killing of Caenorhabditis elegans by Enterococcus faecalis derivatives. Exposure of C. elegans to E. faecalis S613FΔliaF177gdpD170cls61ΔliaR results in significant attenuation, compared with the parental strain (S613FΔliaF177gdpD170cls61). The in vivo phenotype was restored by introducing liaR back into its native chromosomal location (S613FΔliaF177gdpD170cls61ΔliaR::liaR). The experiment was repeated 3 times with similar results (n = 60–90 worms). ***P < .0001.
LiaR Controls Localization of CM Cardiolipin Microdomains
We previously showed that emergence of DAP resistance in E. faecalis S613 was associated with important redistribution of cardiolipin microdomains in the CM, which relocalize away from the principle DAP target, the division septum [10]. This CM phospholipid remodeling was observed after we generated an Ile177 deletion in LiaF, suggesting that this mutation was likely to produce the CM effect via its response regulator, LiaR. Thus, to gain further insights into the mechanism of CM adaptation and the effect on susceptibility to DAP mediated by LiaR, we used the hydrophobic fluorescent dye NAO to assess localization of cardiolipin-enriched microdomains in the CM, using fluorescence microscopy, as previously described [33, 34]. As shown in Figure 3, NAO staining (500 nm) of DAP-resistant S613FΔliaF177gdpD170cls61 revealed the presence of cardiolipin domains that were localized away from the main division septum as previously reported [10]. Deletion of liaR produced a marked change in the cardiolipin-microdomain distribution so that, in the absence of LiaR, the CM cardiolipin-microdomains were redirected back to the septum, a pattern similar to that observed in the parental E. faecalis S613 strain (Figure 3 and Supplementary Figure 1). The pattern of cardiolipin-domain distribution in the CM was fully restored after complementation with liaR. This finding provided evidence that LiaR mediates CM phospholipid redistribution and that this structural phenomenon is mechanistically associated with resistance to a broad range of antibiotics and AMPs.
DISCUSSION
Our previous studies [7–10] focused on the mechanisms of DAP resistance in enterococci. These investigations provided insights into the molecular events associated with CM adaptation and the bacterial stress response in multidrug-resistant E. faecalis and suggested that interfering with such a process might be a novel antimicrobial strategy [7–10]. Indeed, we have recently shown that, in a clinical strain of E. faecalis, specific mutations triggered CM adaptation mechanisms to withstand the attack by AMPs. Using a pair of clinical DAP-susceptible (S613) and DAP-resistant E. faecalis (R712) strains obtained from a patient before and after DAP therapy, we demonstrated that development of DAP resistance involved initial diversion of the antibiotic molecule away from the septum. Importantly, this effect was associated with redistribution of cardiolipin microdomains in the CM [10]. We provided compelling evidence that the initial remodeling of cardiolipin microdomains was related to a deletion of Ile from position 177 of LiaF, one of the members of the LiaFSR system that, in B. subtilis, controls the bacterial CM stress response (LiaF appears to negatively regulate the histidine kinase LiaS) [18]. Moreover, by using an in vitro quantitative experimental evolution system, we showed that changes in the E. faecalis LiaFSR pathway were the initial changes associated with the adaptive response that led to successful selection of DAP-resistant strains [11]. The CM adaptive response in E. faecalis also involved alterations in phospholipid composition of the CM, that, among other effects, reduced the content of phosphatidylglycerol, which we presume interferes with the activity of DAP by reducing the ability to oligomerize inside of the CM [10]. The change in phospholipid composition was associated with alterations in enzymes involved in phospholipid metabolism (GdpD and Cls). However, the changes in phospholipid enzymes appeared late in the selection process, after exposure to serially increasing concentrations of DAP, and occurred in the presence of initial changes of the LiaFSR system.
Here, we targeted the LiaR response regulator, the effector of the LiaFSR system, to further dissect the role of this system in CM homeostasis in DAP resistance. Toward this end, we generated a nonpolar deletion of liaR from both a DAP-resistant clinical strain and from a laboratory isolate and then complemented the gene deletion. Our findings suggest that LiaR is a master regulator of the E. faecalis CM adaptive response to the challenge by a broad range of unrelated CM-targeting cationic antimicrobials of different origins, structures, and mechanisms of action. Indeed, our results further indicate that LiaR plays a major role in the development of resistance to CM-targeting compounds. Interestingly, deletion of liaR in DAP-resistant E. faecalis S613FΔliaF177gdpD170cls61 not only reversed resistance but also resulted in hypersusceptibility to the antibiotic, with MICs decreasing far below the values of the original parent strain. Thus, it is tempting to speculate that the LiaR regulon is crucial in phospholipid remodeling as part of the CM adaptive response and that, in its absence, membrane adaptation is weakened to such extent that CM disruptions from different origins have profound deleterious effects.
Importantly, the LiaR-mediated structural CM adaptations and changes in AMP susceptibilities in vitro were paralleled by changes in the in vivo virulence of this important pathogen. It should be emphasized that, although the nematode model used in our in vivo studies does not contain mammalian-type granulocytes, platelets, or epithelial cells, C. elegans possesses functional homologues therein [32]. Therefore, our finding of in vitro pan-hypersusceptibility of the liaR mutant to a wide-ranging array of such peptides suggests that the response to AMP homologues in C. elegans is likely playing a role in virulence attenuation. Our findings have important implications since they likely indicate that LiaR plays a major role in the overall CM homeostasis and stress response to such antimicrobial molecules and confirms that interfering with LiaR function would be an intriguing approach to not only restore antimicrobial susceptibility to current anti-enterococcal antimicrobials but also to increase the innate immune-mediated clearance of such pathogens. Also, since the LiaFSR system is present in all clinically important gram-positive bacteria [12–17, 47, 48], a potential small-molecule approach to interfere with LiaR function may have broader applicability. Indeed, deletion of liaR orthologs in other gram-positive pathogens has been associated with alterations of virulence and response to AMPs [16, 17, 20, 47, 48].
Additionally, in the current study, we show that one mechanism by which LiaR orchestrates the CM response in E. faecalis involve cardiolipin microdomain redistribution. Cardiolipin plays a crucial role in the organization of the bacterial CM with the formation of microarrays localized in septal and polar areas [49]. The cardiolipin domains appear to contribute to the function of multiprotein complexes that include CM transport, cell division, and metabolism [49]. Moreover, recent evidence suggests that increased cardiolipin content is associated with inhibition of DAP-mediated membrane permeabilization [3]. Interestingly, using a liposomal model, it was shown that when membranes containing phosphatidylglycerol were saturated with cardiolipin at molar fractions of 10% and 20%, DAP was unable to form pores or translocate to the inner leaflet of the CM despite the fact that oligomerization of the antibiotic molecule was not affected [3]. These findings are consistent with our working hypothesis of the DAP diversion mechanism, in which cardiolipin microarrays may trap DAP and prevent the antibiotic from reaching the critical targets at the septal division plane. Thus, this LiaR-mediated cardiolipin-microdomain redistribution appears to be the first step in preventing cationic AMP-mediated CM damage, a sophisticated evolutionary adaptive response devised by E. faecalis.
In conclusion, we provide evidence that LiaR is a master regulator of the multidrug-resistant E. faecalis response to antibiotics and AMPs and have identified the CM-associated mechanism for such regulation. LiaR (and its orthologs) emerges as a potential target for developing antiadaptation molecules as antimicrobials and may restore the clinical utility of current antibiotics, a novel strategy to overcome multidrug resistance in recalcitrant bacterial pathogens.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank William Miller, Isabel Reyes, and Karen Jacques-Palaz, for technical assistance in mutant construction; and Hans-Georg Sahl and Tanja Schneider for three peptides namely Gallidermin, Friulimicin and Mersacidin. Shaya Noorian, for technical assistance in AMPs assays.
Disclaimer. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (grants R01 AI093749 [to C. A. A.], R01 AI39108-15 [to A. S. B.], R01 AI076406 [to D. A. G.], R01 AI080714 [to Y. S.], and R01 AI047923 [to B. E. M.]; and the Department of Defense (grant W81XWH-12-2-0101 to M. R. Y.).
Potential conflict of interest. C. A. A. has received grant support from, consulted for, and/or provided lectures for Pfizer, Cubist, Bayer, Forest Pharmaceuticals, Novartis, and Theravance. B. E. M. has received grant support from, consulted for, and/or provided lectures for Theravance, Cubist, Forest, Pfizer, and The Medicines Company. A. S. B. has current research grants from ContraFect and Theravance Pharmaceuticals. He has received prior research grants from Cubist, University of Wurzberg, Trius pharmaceuticals, and the Medicines Company. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.World Health Organization (WHO). Antimicrobial resistance global report on surveillance 2014. Geneva, Switzerland: WHO, 2014. [Google Scholar]
- 2.Centers for Diseases Control and Prevention. Antibiotic resistance threats in the United States, 2013. Atlanta, GA: Department of Health and Human Services, CDC, 2013. [Google Scholar]
- 3.Zhang T, Muraih JK, Tishbi N, et al. Cardiolipin prevents membrane translocation and permeabilization by daptomycin. J Biol Chem 2014; 289:11584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muraih JK, Pearson A, Silverman J, Palmer M. Oligomerization of daptomycin on membranes. Biochim Biophys Acta 2011; 1808:1154–60. [DOI] [PubMed] [Google Scholar]
- 5.Bayer AS, Schneider T, Sahl HG. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 2013; 1277:139–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pogliano J, Pogliano N, Silverman JA. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J Bacteriol 2012; 194:4494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arias CA, Panesso D, McGrath DM, et al. Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med 2011; 365:892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munita JM, Panesso D, Diaz L, et al. Correlation between mutations in liaFSR of Enterococcus faecium and MIC of daptomycin: revisiting daptomycin breakpoints. Antimicrob Agents Chemother 2012; 56:4354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munita JM, Tran TT, Diaz L, et al. A liaF codon deletion abolishes daptomycin bactericidal activity against vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 2013; 57:2831–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran TT, Panesso D, Mishra NN, et al. Daptomycin resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. MBio 2013; 4:e00281-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller C, Kong J, Tran TT, Arias CA, Saxer G, Shamoo Y. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance [published correction appears in Antimicrob Agents Chemother 2014; 58:631]. Antimicrob Agents Chemother 2013; 57:5373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan S, Junker A, Helmann JD, Mascher T. Regulation of LiaRS-dependent gene expression in Bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J Bacteriol 2006; 188:5153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suntharalingam P, Senadheera MD, Mair RW, Lévesque CM, Cvitkovitch DG. The LiaFSR system regulates the cell envelope stress response in Streptococcus mutans. J Bacteriol 2009; 191:2973–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eldholm V, Gutt B, Johnsborg O, et al. The pneumococcal cell envelope stress-sensing system LiaFSR is activated by murein hydrolases and lipid II-interacting antibiotics. J Bacteriol 2010; 192:1761–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehta S, Cuirolo AX, Plata KB, et al. VraSR two-component regulatory system contributes to mprF-mediated decreased susceptibility to daptomycin in vivo selected clinical strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 2012; 56:92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klinzing DC, Ishmael N, Dunning-Hotopp JC, et al. The two-component response regulator LiaR regulates cell wall stress responses, pili expression and virulence in group B Streptococcus. Microbiology 2013; 159:1521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyle-Vavra S, Yin S, Jo DS, Montgomery CP, Daum RS. VraT/YvqF is required for methicillin resistance and activation of the VraSR regulon in Staphylococcus aureus. Antimicrob Agents Chemother 2013; 57:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf D, Kalamorz F, Wecke T, et al. In-depth profiling of the LiaR response of Bacillus subtilis. J Bacteriol 2010; 192:4680–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hachmann AB, Angert ER, Helmann JD. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob Agents Chemother 2009; 53:1598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kesel S, Mader A, Höfler C, Mascher T, Leisner MS. Immediate and heterogeneous response of the LiaFSR two-component system of Bacillus subtilis to the peptide antibiotic bacitracin. PLoS One 2013; 8:e53457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bourgogne A, Garsin DA, Qin X, et al. Large scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol 2008; 9:R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munoz-Price LS, Lolans K, Quinn JP. Emergence of resistance to daptomycin during treatment of vancomycin-resistant Enterococcus faecalis infection. Clin Infect Dis 2005; 41:565–6. [DOI] [PubMed] [Google Scholar]
- 23.Clinical Laboratory Standards Institute (CLSI). Performance standards for antimicrobial susceptibility testing: twenty-first informational supplement 2013. CLSI document MS100-S23 CLSI: Wayne, PA, 2013. [Google Scholar]
- 24.Pfaller MA, Mendes RE, Sader HS, Jones RN. Telavancin activity against Gram-positive bacteria isolated from respiratory tract specimens of patients with nosocomial pneumonia. J Antimicrob Chemother 2010; 65:2396–404. [DOI] [PubMed] [Google Scholar]
- 25.Kristich CJ, Chandler JR, Dunny GM. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 2007; 57:131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran TT, Panesso D, Gao H, et al. Whole-genome analysis of a daptomycin-susceptible Enterococcus faecium strain and its daptomycin-resistant variant arising during therapy. Antimicrob Agents Chemother 2012; 57:261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panesso D, Montealegre MC, Rincón S, et al. The hylEfm gene in pHylEfm of Enterococcus faecium is not required in pathogenesis of murine peritonitis. BMC microbiol 2011; 11:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arthur M, Depardieu F, Snaith HA, Reynolds PE, Courvalin P. Contribution of VanY D,D-carboxypeptidase to glycopeptide resistance in Enterococcus faecalis by hydrolysis of peptidoglycan precursors. Antimicrob Agents Chemother 1994; 38:1899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones T, Yeaman MR, Sakoulas G, et al. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob Agents Chemother 2008; 52:269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong YQ, Mukhopadhyay K, Yeaman MR, Adler-Moore J, Bayer AS. Functional interrelationships between cell membrane and cell wall in antimicrobial peptide-mediated killing of Staphylococcus aureus. Antimicrob Agents Chemother 2005; 49:3114–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeaman MR, Puentes SM, Norman DC, Bayer AS. Partial characterization and staphylocidal activity of thrombin-induced platelet microbicidal protein. Infect Immun 1992; 60:1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garsin DA, Sifri CD, Mylonakis E, et al. A simple model host for identifying Gram-positive virulence factors. Proc Natl Acad Sci U S A 2001; 98:10892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mileykovskaya E, Dowhan W, Birke RL, Zheng D, Lutterodt L, Haines TH. Cardiolipin binds nonyl acridine orange by aggregating the dye at exposed hydrophobic domains on bilayer surfaces. FEBS Lett 2001; 507:187–90. [DOI] [PubMed] [Google Scholar]
- 34.Petit JM, Maftah A, Ratinaud MH, Julien R. 10N-nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur J Biochem 1992; 209:267–73. [DOI] [PubMed] [Google Scholar]
- 35.Yount NY, Yeaman MR. Peptide antimicrobials: cell wall as a bacterial target. Ann N Y Acad Sci 2013; 1277:127–38. [DOI] [PubMed] [Google Scholar]
- 36.Vandamme D, Landuyt B, Luyten W, Schoofs L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol 2012; 280:2–35. [DOI] [PubMed] [Google Scholar]
- 37.Jarczak J, Kościuczuk EM, Lisowski P, et al. Defensins: natural component of human innate immunity. Hum Immunol 2013; 74:1069–79. [DOI] [PubMed] [Google Scholar]
- 38.Islam MR, Nagao J, Zendo T, Sonomato K. Antimicrobial mechanism of lantibiotics. Biochem Soc Trans 2012; 40:1528–33. [DOI] [PubMed] [Google Scholar]
- 39.Kilelee E, Pokorny A, Yeaman MR, Bayer AS. Lysyl-phosphatidylglycerol attenuates membrane perturbation rather than surface association of the cationic antimicrobial peptide 6W-RP-1 in a model membrane system: implications for daptomycin resistance. Antimicrob Agents Chemother 2010; 54:4476–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider T, Gries K, Josten M, et al. The lipopeptide antibiotic Friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob Agents Chemother 2009; 53:1610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yount NY, Cohen SE, Kupferwasser D, et al. Context mediates antimicrobial efficacy of kinocidin congener peptide RP-1. PLoS One 2011; 6:e26727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sifri CD, Mylonakis E, Singh KV, et al. Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice. Infect Immun 2002; 70:5647–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao P, Pinkston KL, Bourgogne A, et al. Library screen identifies Enterococcus faecalis CcpA, the catabolite control protein A, as an effector of Ace, a collagen adhesion protein linked to virulence. J Bacteriol 2013; 195:4761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balla KM, Troemel ER. Caenorhabditis elegans as a model for intracellular pathogen infection. Cell Microbiol 2013; 15:1313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simonsen KT, Gallego SF, Færgeman NJ, Kallipolitis BHK. Strength in numbers: “Omics” studies of C. elegansinnate immunity. Virulence 2012; 3:477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong D, Bazopoulou D, Pujol N, Tavernarakis N, Ewbank JJ. Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol 2007; 8:R194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tremblay YD, Lo H, Li YH, Halperin SA, Lee SF. Expression of the Streptococcus mutans essential two-component regulatory system VicRK is pH and growth-phase dependent and controlled by the LiaFSR three-component regulatory system. Microbiology 2009; 155:2856–65. [DOI] [PubMed] [Google Scholar]
- 48.Bergholz TM, Tang S, Wiedmann M, Boor KJ. Nisin resistance of Listeria monocytogenes is increased by exposure to salt stress and is mediated via LiaR. Appl Environ Microbiol 2013; 79:5682–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mileykovskaya E, Dowhan W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta 2009; 1788:2084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.