

Abstract
Occupational and environmental exposures to airborne asbestos and silica are associated with the development of lung fibrosis in the forms of asbestosis and silicosis, respectively. However, both diseases display distinct pathologic presentations, likely associated with differences in gene expression induced by different mineral structures, composition and bio-persistent properties. We hypothesized that effects of mineral exposure in the airway epithelium may dictate deviating molecular events that may explain the different pathologies of asbestosis versus silicosis. Using robust gene expression-profiling in conjunction with in-depth pathway analysis, we assessed early (24 h) alterations in gene expression associated with crocidolite asbestos or cristobalite silica exposures in primary human bronchial epithelial cells (NHBEs). Observations were confirmed in an immortalized line (BEAS-2B) by QRT-PCR and protein assays. Utilization of overall gene expression, unsupervised hierarchical cluster analysis and integrated pathway analysis revealed gene alterations that were common to both minerals or unique to either mineral. Our findings reveal that both minerals had potent effects on genes governing cell adhesion/migration, inflammation, and cellular stress, key features of fibrosis. Asbestos exposure was most specifically associated with aberrant cell proliferation and carcinogenesis, whereas silica exposure was highly associated with additional inflammatory responses, as well as pattern recognition, and fibrogenesis. These findings illustrate the use of gene-profiling as a means to determine early molecular events that may dictate pathological processes induced by exogenous cellular insults. In addition, it is a useful approach for predicting the pathogenicity of potentially harmful materials.
Background
Asbestosis and silicosis, the most common forms of pneumoconioses, are caused by inhalation of respirable asbestos fibers and crystalline silica particles, respectively. ‘Asbestos’ describes a group of silicate minerals comprising serpentine fibers (i.e. chrysotile) and rigid, highly pathogenic amphibole fibers (e.g. crocidolite, amosite, tremolite, actinolite, anthophyllite). Inhalation of asbestos causes major changes in parenchymal architecture, including diffuse interstitial collagen deposition and accumulation of dense connective tissue at specific loci (1–3). In addition, amphibole asbestos causes pleural fibrosis, and results in lung cancer and malignant mesotheliomas (3). The three naturally occurring polymorphs of crystalline silica of health concern are quartz, cristobalite and tridymite (2). Unlike asbestosis, silicosis is associated with the formation of concentric ‘whorled’ nodules of collagen-rich, dense fibrotic tissue and a prominent granulomatous response (1,3). Occupational exposures to silica, especially in smokers, have been associated with increased risks of lung cancer, but not mesotheliomas, in some cohorts, although this is a topic of debate (4–7).
Impingement of high concentrations of both fibers and particles on alveolar macrophages and epithelial cells lining the upper and lower respiratory tract is characterized by cell damage associated with the production of oxidants, a persistent inflammatory response, the generation of profibrotic mediators and scarring (8). Although exposure to each type of mineral can give rise to interstitial fibrosis, physicochemical differences between crystalline silica particles and asbestos fibers may lead to distinctive gene expression patterns resulting in the dissimilar pathological characteristics of silicosis and asbestosis. The role of the lung epithelium has until recently been viewed as a passive, physical barrier against inhaled particulates and other toxicants. However, studies show that lung epithelial cells play an active role in particulate uptake (9,10), immune cell recruitment (3), proliferation of epithelial and other cell types (11), and matrix remodeling (12). Since epithelial cells are target cells of lung carcinomas and contribute to the development of pulmonary fibrosis, understanding the molecular responses of human lung epithelial cells to pathogenic minerals is critical to unraveling the complex molecular events that initiate inflammatory responses, consequent fibrogenesis and other mechanisms intrinsic to these diseases. In this study, we therefore compared gene alterations induced by exposure to equitoxic doses of crocidolite asbestos and cristobalite silica in an isolate of normal human bronchial epithelial cells (NHBEs) (9). By gene-profiling, we furthermore explored expression signatures and their biological interactions. Selected common as well as mineral-specific gene alterations were confirmed in an immortalized bronchial epithelial cell line (BEAS-2B) by QRT-PCR. Additionally, further confirmation of selected findings at the protein level was obtained using various techniques. To our knowledge, these studies are the first to explore comparative gene expression-profiling in human airway epithelial cells using equitoxic doses of pathogenic minerals, i.e. an amphibole asbestos and a crystalline silica.
Results
Effects of mineral exposure on NHBE and BEAS-2B cell viability
We initially compared the effects of crocidolite asbestos and cristobalite silica on cell viability using surface area-based dosimetry, as the biological activity of these particulates is derived largely from their surface reactivity (13). Based on data from these experiments, a concentration of 15 × 106 μm2/cm2 dish was chosen for asbestos, and 15 and 75 × 106 μm2/cm2 dish for silica for NHBE gene expression studies (<LD50 concentrations) (Supplementary Material, Fig. S1). The viability of BEAS-2B cells exposed to minerals for 24 h was determined similarly (Supplementary Material, Fig. S2). As cells from the immortalized cell line were more resistant to equal surface area-based doses when compared with primary cell cultures, mineral concentrations of 75 and 150 × 106 µm2/cm2 dish were used for gene expression studies in BEAS-2B cells (9,14).
Strategies to detect gene expression alterations by minerals
Using equitoxic doses noted above, gene expression was assessed by Affymetrix GeneChip arrays analyzed with GeneSifter® and Bioconductor. An exposure period of 24 h was used as this is the time point for maximal gene expression changes by a number of pathogenic fibers and particles (9,10,15,16). In our previous studies, non-pathogenic glass beads and fine TiO2 did not induce any significant numbers of gene changes compared with unexposed cells (saline/vehicle control) (10,17), which were therefore used as controls here. First, to visualize the gene–sample relationship, an unsupervised average-linkage hierarchical clustering was applied to the 50 probe-sets with the highest variation across the 12 samples. This analysis clearly identified distinct clusters related to different minerals and doses (Fig. 1A). Cluster I comprises two subclusters: (a) including the unexposed control samples and the low dose (15 × 106 μm2/cm2) silica group and (b) containing the asbestos (15 × 106 μm2/cm2) samples. Cluster II contained the high dose (75 × 106 μm2/cm2) silica samples.
Figure 1.
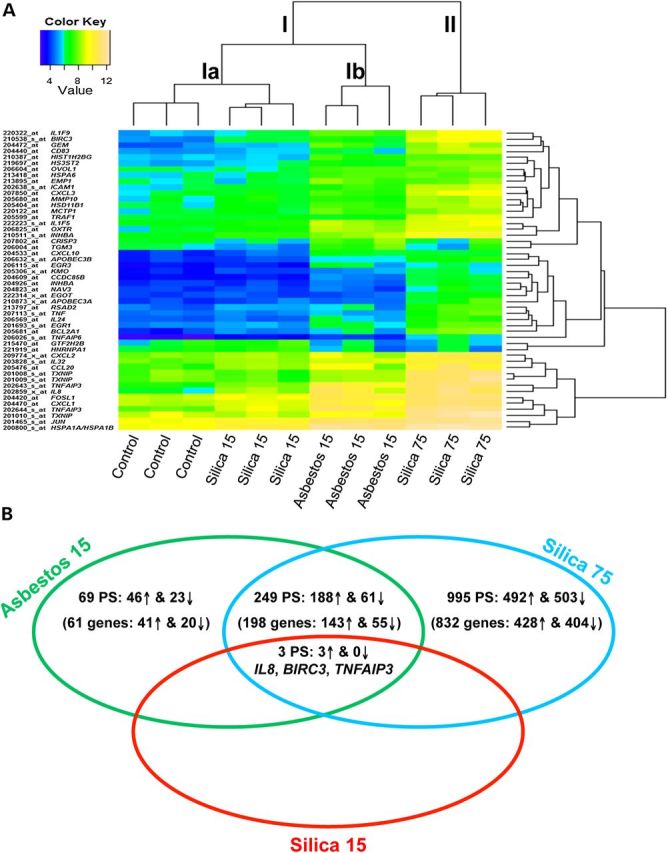
Unsupervised hierarchical cluster analysis and overall gene changes in mineral exposed NHBE cells. (A) Unsupervised hierarchical cluster analysis of gene expression profiles of unexposed control NHBE cells and cells exposed to crocidolite asbestos 15 × 106 µm2/cm2 (asbestos 15), cristobalite silica 15 × 106 µm2/cm2 (silica 15) or 75 × 106 µm2/cm2 (silica 75) (n = 3 samples/group) based on the log2 expression values of the top 50 most variable probe-sets. Individual samples are shown in columns and genes in rows. The log2 expression values for individual genes are indicated by color (yellow indicates a high and blue a low level of expression). (B) Venn diagram representation of the total number of probe-sets and genes (up-regulated and down-regulated) significantly altered by all three exposure groups compared. This includes those specific to each exposure as well as those commonly affected by different exposures [≥1.5-fold versus controls; adj. P < 0.05 (FDR < 0.05)].
To compare the magnitude of alterations in gene expression by each mineral and dose, we examined the total number of significant gene changes >1.5-fold versus unexposed controls, with an adj. P-value <0.05 (FDR < 0.05). Asbestos (15 × 106 μm2/cm2) altered levels of 321 probe-sets (262 genes) which was much more robust than silica, which altered only three probe-sets (three genes) at an equal concentration (15 × 106 μm2/cm2). However, at the higher amount, silica (75 × 106 μm2/cm2) exposures showed significant changes in 1247 probe-sets (1033 genes) (Table 1, Fig. 1B). A comprehensive list of significant gene changes for each group is described in Supplementary Material, Tables S1–S3.
Table 1.
Total number of gene alterations in NHBE cells (24 h)
| Mineral | Dose (S.A.), ×106 µm2/cm2 | Dose (mass), µg/cm2 | Probe-sets | Genes |
|---|---|---|---|---|
| Crocidolite Asbestos | 15 | 1.0 | 321 | 262 |
| Cristobalite silica | 15 | 2.94 | 3 | 3 |
| Cristobalite silica | 75 | 14.7 | 1247 | 1033 |
Total numbers of probe-sets and total numbers of genes significantly altered by each mineral and dose compared with unexposed controls. A threshold cut-off of 1.5-fold versus controls with an adj. P-value < 0.05 was used.
Comparative gene expression and divergent signaling pathways induced by asbestos and silica
To determine both common and divergent gene expression patterns and signaling pathways associated with mineral-induced gene expression changes, IPA® was used. Comparative analysis revealed that of 1316 probe-sets significantly altered across all three exposures (compared with controls), 252 were commonly altered by asbestos (15 × 106 μm2/cm2) and silica (75 × 106 μm2/cm2) (Supplementary Material, Table S4), 3 of which were also significantly altered by the low dose silica (15 × 106 μm2/cm2) (Fig. 1B). Conversely, 69 probe-sets were specifically altered by asbestos (15 × 106 μm2/cm2) (Supplementary Material, Table S5) and 995 by silica (75 × 106 μm2/cm2) (Supplementary Material, Table S6). These gene changes were analyzed to determine signaling pathways, biological and disease functions associated with mineral exposures. The top 10 canonical pathways and genes associated with these pathways were used to construct schematic representations of the major signaling events affected commonly or specifically by asbestos or silica (Figs 2, 5 and 7; Tables 2–4).
Figure 2.

Pathways commonly affected by asbestos and silica in NHBE cells. A schematic representation of 38 genes with altered expression by asbestos (15 × 106 µm2/cm2) and silica (75 × 106 µm2/cm2) at 24 h. Red: up-regulated, green: down-regulated [≥1.5-fold versus controls; adj. P < 0.05 (FDR < 0.05)]. Lines and arrows interconnecting molecules represent associations determined by IPA from the Ingenuity Knowledge Base, including both direct and indirect relationships. All gene names, fold-change values and adj. P-values are listed in Supplementary Material, Table S4.
Figure 5.

Pathways selectively affected by asbestos in NHBE cells. Graphical representation of the experimentally confirmed relationships between genes distinctively altered in expression by asbestos (15 × 106 µm2/cm2) using IPA software. Red and green colors indicate up- or down-regulation of gene expression levels respectively compared to unexposed control cells [≥1.5-fold versus controls; adj. P < 0.05 (FDR < 0.05)]. All gene names, fold-change values, and adj. P-values are listed in Supplementary Material, Table S5.
Figure 7.

Pathways selectively affected by crystalline silica in NHBE cells. Graphical representation of the experimentally confirmed relationships between genes distinctively altered in expression by silica (75 × 106 µm2/cm2) using IPA software. Red and green colors indicate up- or down-regulation of gene expression levels, respectively, compared with unexposed control cells [≥1.5-fold versus controls; adj. P < 0.05 (FDR < 0.05)]. All gene names, fold-change values, and adj. p-values are listed in Supplementary Material, Table S6.
Table 2.
Top 10 canonical pathways altered by both asbestos and silica in NHBE (24 h)
| Rank | Canonical pathway |
|---|---|
| 1 | Granulocyte adhesion and diapedesis |
| 2 | Agranulocyte adhesion and diapedesis |
| 3 | Glucocorticoid receptor signaling |
| 4 | TNFR2 signaling |
| 5 | Role of macrophages, fibroblasts and endothelial cells in rheumatoid arthritis |
| 6 | IL-6 signaling |
| 7 | Aldosterone signaling in epithelial cells |
| 8 | PPAR signaling |
| 9 | IL-17A Signaling in gastric cells |
| 10 | IL-10 Signaling |
Top 10 canonical pathways significantly enriched by alteration in gene expression by both asbestos and silica as determined by core analysis of commonly altered gene changes in IPA.
Table 3.
Top 10 canonical pathways altered by asbestos specifically in NHBE (24 h)
| Rank | Canonical pathway |
|---|---|
| 1 | Dermatan sulfate biosynthesis (late stages) |
| 2 | Production of nitric oxide and reactive oxygen species in macrophages |
| 3 | ERK/MAPK signaling |
| 4 | Chondroitin sulfate biosynthesis (late stages) |
| 5 | Estrogen biosynthesis |
| 6 | Heparan sulfate biosynthesis (late stages) |
| 7 | Melatonin degradation I |
| 8 | Chondroitin sulfate biosynthesis |
| 9 | FcγRIIB signaling in B lymphocytes |
| 10 | Dermatan sulfate biosynthesis |
Top 10 canonical pathways significantly enriched by alteration in gene expression by asbestos only as determined by core analysis of asbestos-specific gene changes in IPA.
Table 4.
Top 10 canonical pathways altered by silica-specifically in NHBE (24 h)
| Rank | Canonical pathway |
|---|---|
| 1 | Salvage pathways of pyrimidine deoxyribonucleotides |
| 2 | TREM1 signaling |
| 3 | Aryl hydrocarbon receptor signaling |
| 4 | Interferon signaling |
| 5 | Hepatic fibrosis/hepatic stellate cell activation |
| 6 | Role of JAK family kinases in IL-6-type cytokine signaling |
| 7 | Role of pattern recognition receptors in recognition of bacteria and viruses |
| 8 | Altered T- and B-cell signaling in rheumatoid arthritis |
| 9 | Role of IL-17A in arthritis |
| 10 | IL-6 signaling |
Top 10 canonical pathways significantly enriched by alteration in gene expression by silica only as determined by core analysis of silica-specific gene changes in IPA.
Common responses
Canonical pathways affected commonly by both minerals were associated with cell membrane receptors and downstream signaling [e.g. Caspase-1 (CASP1)], transcriptional regulators [i.e. early growth response (EGR) and activator protein-1 (AP-1) and nuclear factor kappa B (NF-κB)] and downstream biological functions. These biological functions included extracellular matrix (ECM) dissolution, leukocyte adhesion and migration, inflammation, fibrotic response, apoptosis regulation and cellular stress responses, as well as other biological activities such as mitogen-activated protein kinase (MAPK) inhibition and glucocorticoid signaling (Table 2, Fig. 2). It should be noted that all gene symbols are defined in Supplementary Material, Tables S4–S6. The up-regulation of interleukin-8 (IL8), matrix metallopeptidase 1 (MMP1) and baculoviral IAP-repeat-containing 3 (BIRC3) by both asbestos and silica exposures observed by microarray was confirmed in NHBE cells by QRT-PCR in a separate experiment (Supplementary Material, Fig. S3). These findings were also confirmed in BEAS-2B cells (Fig. 3A–C). Common up-regulation of the early-response gene, JUN, that encodes a member of the AP-1 transcription factor complex (Fig. 3D), and is known to have a pivotal role in asbestos- and silica-induced disease processes (18–20), was confirmed in BEAS-2B cells. In addition, up-regulation of a number of novel genes by both asbestos and silica exposure was confirmed in BEAS-2B cells including the transcriptional regulators EGR1 and TNFAIP3 (Fig. 3E and F), the inflammatory receptor IL1RL1 (Fig. 3G), the apoptosis inducer IL24 (Fig. 3H) and the cell stress response protein HSPA6 (Fig. 3I).
Figure 3.

Commonly altered genes by asbestos and silica exposure in BEAS-2B cells. Steady-state mRNA levels were measured by QRT-PCR in BEAS-2B cells exposed to asbestos and silica with two doses (75 and 150 × 106 µm2/cm2) for 24 h. Genes affected by both minerals were (A) IL8 (Interleukin-8), (B) MMP1 (Matrix metallopeptidase-1), (C) BIRC3 (Baculoviral IAP-repeat containing 3), (D) JUN (c-jun proto-oncogene), (E) EGR1 (Early growth response-1), (F) TNFAIP3 (TNFα-induced protein 3), (G) IL1RL1 (IL-1 receptor-like 1), (H) IL24 (Interleukin-24) and (I) HSPA6 (Heat shock protein A6). Gene expression levels were normalized to the housekeeping gene RPL13A (Ribosomal protein L13A). Values represent mean ± SEM. *P < 0.05 versus unexposed controls, n = 3, graphs are representative of two to three repeat experiments.
Additionally, some common changes observed at the mRNA level were investigated at the protein level in BEAS-2B cells (Fig. 4). In addition to the increased mRNA levels of CASP1 observed for both minerals in NHBE cells (Fig. 2), the enzymatic activity levels of this effector molecule of the NLRP3 inflammasome were found to be increased in BEAS-2B exposed to asbestos and silica for 24 h (Fig. 4A). Furthermore, we confirm that asbestos and silica both cause secretion of IL-8 (Fig. 4B), although comparing equal surface-area-based doses, asbestos induced higher levels of secreted IL-8 which is in agreement with our mRNA data (Fig. 3A). Moreover, both fibers and particles caused a marked increase in the protein expression of CLDN1 (Fig. 4C), a tight junction protein important in supporting the epithelium as a physical barrier and in regulating the inflammatory response (21,22).
Figure 4.
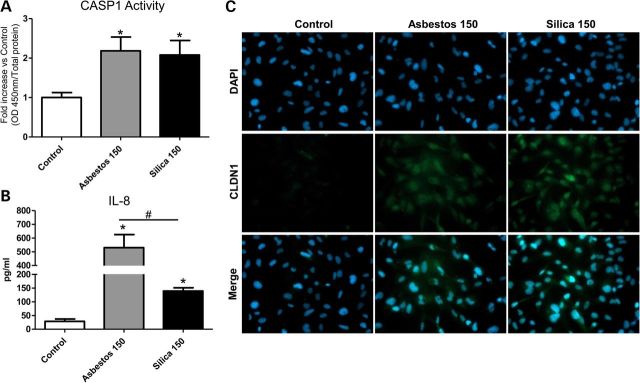
Commonly altered protein by asbestos and silica exposure in BEAS-2B cells. Confirmation of microarray findings at protein level in BEAS-2B cells after 24 h exposure to asbestos and silica (150 × 106 µm2/cm2). (A) Caspase-1 enzymatic activity levels relative to unexposed controls. (B) Secreted levels of IL-8 protein in conditioned medium of BEAS-2B (pg/ml). (C) Immunofluorescence images of BEAS-2B (left to right), unexposed controls, asbestos, silica and nuclear stain DAPI (top), CLDN1 in green (middle) and merged DAPI and CLDN1 (bottom). Negative controls (no primary antibody) were also included and all were negative for staining. Images were taken with ×40 objective lens.
Asbestos-specific responses
Genes selectively altered in expression after exposure to crocidolite asbestos (15 × 106 μm2/cm2) were grouped in a unique set of pathways which are listed in Table 3 and represented in Figure 5. This collection of experimentally confirmed relationships is associated with biological functions related to tissue damage (IL1RN) and iron homeostasis (TFRC) as well as small molecule biochemistry and cellular detoxification mechanisms (S100A12, CYP2C18 and SULT family members). Further downstream, asbestos-specific biological responses were linked to tumor morphology (MGEA5, CRNN, PLCG and TGMs) and cancer (CEACAM1, SEMA3A) as well as proliferation (FST, NCF1) and cell survival (ABCC10, RBCK1).
A selection of these findings was also investigated in the BEAS-2B epithelial cell line by QRT-PCR to confirm microarray-based findings in primary cells. Crocidolite-specific induction of SEMA3A expression was confirmed, and interestingly, silica exposure conversely decreased SEMA3A mRNA levels (Fig. 6A). In addition to the confirming of crocidolite-specific reduction in TFRC expression, we found that cristobalite silica also had an inverse effect on TFRC mRNA levels in BEAS-2B cells (Fig. 6B). Furthermore, exposure to asbestos, but not silica, caused a marked decrease (50%) in TFRC protein levels in BEAS-2B cells at 48 h post-exposure. Silica-induced increases in TFRC mRNA levels (24 h) were however not reflected at the protein level at 24 and 48 h, which were not significantly affected compared with controls (Fig. 6C and data not shown).
Figure 6.
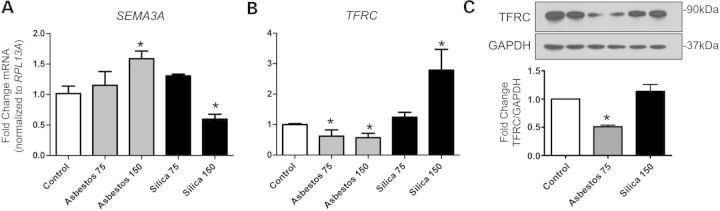
Asbestos-specific genes and protein altered in BEAS-2B cells. Steady-state mRNA levels were measured by QRT-PCR in BEAS-2B cells exposed to asbestos and silica at 2 doses (75 and 150 × 106 µm2/cm2) for 24 h. Genes significantly altered by asbestos only were (A) SEMA3A (Semaphorin 3A), (B) TFRC (Transferrin receptor C). Gene expression levels were normalized to the housekeeping gene RPL13A (Ribosomal protein L13A). Values represent mean ± SEM. *P < 0.05 versus unexposed controls (N = 3), (C) Western blot of TFRC protein levels (90 kDa) and house keeper GAPDH (37 kDa) after exposure to asbestos (75 × 106 µm2/cm2) or silica (150 × 106 µm2/cm2) for 48 h. Densitometry represents relative levels of TFRC normalized to GAPDH (N = 2) and graphs are representative of two to three repeat experiments.
Silica-specific responses
In NHBE cells, silica exposure at higher concentrations (75 × 106 μm2/cm2) specifically altered expression levels of various cell membrane receptors associated with pathogen-associated molecular pattern (PAMP) recognition (LY96, TLR1, CD14, TREM1), cell death (FAS) and inflammation (IFNGR1, TNFRSF9, TNFRSF10B, CXCR4, IL2RG). Additionally, silica exposure affected a number of transcriptional regulators (e.g. IRF, CREB and alternative NFκB family members) and a plethora of downstream effector molecules and biological functions, including pattern recognition and interferon responses, and additional inflammation, cell survival regulation and fibrogenesis pathways (Fig. 7, Table 4). Figure 8 depicts confirmation of findings in NHBE cells exposed to silica in BEAS-2B cells. Results demonstrate silica-specific up-regulation of the fibrogenic mediator FGF2 (Fig. 8A), the inflammatory regulator PTX3 (Fig. 8B), the transcriptional regulator CREB5 (Fig. 8C), as well as down-regulation of the SFRP1, a modulator of Wingless type (WNT) signaling (Fig. 8D), at the highest concentration (150 × 106 μm2/cm2). In addition, we show that silica selectively induced bFGF (FGF2) secretion from bronchial epithelial cells exposed for 24 h (Fig. 8E).
Figure 8.

Silica-specific genes and protein altered in BEAS-2B cells. Steady-state mRNA levels were measured by QRT-PCR in BEAS-2B cells exposed to asbestos and silica at two doses (75 and 150 × 106 µm2/cm2) for 24 h. Genes significantly altered by silica only were (A) FGF2 (fibroblast growth factor 2), (B) PTX3 (Pentraxin 3), (C) CREB5 (cAMP responsive element binding protein 5) and (D) SFRP1 (Secreted frizzled-related protein 1). Gene expression levels were normalized to the housekeeping gene RPL13A (Ribosomal protein L13A). Values represent the mean ± SEM. *P < 0.05 versus unexposed controls, n = 3. (E) Secreted levels of bFGF (FGF2) protein in conditioned medium of BEAS-2B cells exposed 24 h (pg/ml). Graphs are representative of two to three repeat experiments.
Discussion
Our studies demonstrate that robust gene-profiling was able to distinguish different mineral exposures based on global gene expression, as unsupervised average-linkage hierarchical cluster analysis accurately delineated samples based on both mineral exposure, and in the case of silica, the concentration used. Our studies also demonstrate unique features of crocidolite asbestos and cristobalite silica in patterns of toxicity and gene expression alterations. At equal doses (15 × 106 μm2/cm2), silica had minimal effects on gene expression, whereas the number of changes caused by asbestos was dramatically higher. However, at higher doses (75 × 106 μm2/cm2), silica induced markedly more gene alterations, mimicking dose–response effects observed in inhalation and intratracheal instillation studies (23,24).
Our data reveal gene changes that likely contribute to the mutual pathogenicity as well as divergent characteristics that may drive the increased carcinogenicity of amphibole asbestos and individual patterns of fibrogenicity. Both minerals altered mRNA levels of various membrane receptors, intracellular signaling molecules, transcriptional regulators and downstream effector molecules some linked to signaling pathways and biological functions that promote the development of mineral exposure-associated disease (3,25). In addition to identifying new genes that may be involved in asbestos and/or silica pathogenicity, our results confirm the highly active role of the epithelium as an initial player in these processes.
Common responses
Pathway analysis of genes altered by both minerals revealed that the canonical pathways most highly enriched represented disease mechanisms highly characteristic of the development of both pneumoconioses. These included not only genes well known to be associated with asbestos and silica-disease processes, but also revealed a number of novel genes intrinsic to complex signaling mechanisms that may contribute to disease. Common changes were observed in expression levels of documented membrane receptors associated with proteolysis (PLAUR) and the proinflammatory anaphylatoxin receptor (C5AR) as well as its ligand (C5) which has been shown to be activated by both minerals (26,27). Our data also revealed novel genes, associated with propagation of inflammation (IL6R, IL1RL1) and recognition of foreign materials (TLR2). The IL-33 receptor, IL1RL1 (also known as ST2), was up-regulated by both minerals and IL-33 signaling is a driving force in the development of asthma and allergic airways disease (28,29). The lung epithelium is a major source of IL-33 in response to lung injury, propagating signaling to other effector cells (e.g. TH2, mast cells, dendritic cells), which may be a feed-forward mechanism.
Both AP-1 and NFκB have been well established as key transcriptional regulators that drive inflammatory and fibrogenic disease (3,30,31). Moreover, both minerals induced increased levels of EGR1, which is induced by silica in type II alveolar epithelial cells (32) and is associated with epithelial apoptosis, protease expression and lung fibrosis (33,34). Imbalance of proteolysis and ECM dissolution are key mechanisms in the development of fibrosis (35,36), by dysregulation of key players such as various MMPs and protease inhibitors. Our study links both asbestos and silica exposure to increased expression of MMP1 in lung epithelial cells, which is also increased in human idiopathic pulmonary fibrosis (IPF) (37) and may contribute to further collagen breakdown, activation of cytokines and promotion of cell migration and proliferation. Increased expression of adherens and junction proteins upon injury to the epithelium is part of the restoration process of the physical barrier. For instance, CLDN1, which is increased in expression after exposure to asbestos and silica here, is also increased in the regenerative epithelium of human IPF and asbestosis lung tissues (21). CLDN1 also has a functional role in regulation of pro-fibrotic processes such as epithelial-mesenchymal-transition (EMT) and pro-inflammatory response (22,38). During this response to injury, expression of molecules such as ICAM1 and CYR61 promotes neutrophil extravasation and migration into the airways. This further perpetuates the potent inflammatory response, which is driven by various mediators including; acute phase proteins, cytokines and chemokines, which propagates the deregulated cycles of inflammation and injury seen in asbestosis and silicosis.
Activation of the intracellular danger sensing protein complex, the NLRP3 inflammasome, is a crucial step in propagation of the inflammatory response caused by exposure to occupational dusts (e.g. asbestos and silica) (39,40) as well as nanomaterials (e.g. carbon nanotubes) (41). Originally thought to be restricted to myeloid cells, activation has been demonstrated recently in epithelial (14,42,43) and mesothelial cells (44,45). Additionally, certain polymorphisms in the NLRP3 gene may be associated with IL-1β production and severe inflammation which may suggest a genetic predisposition for common inflammatory disorders (46–48).
IL-8, a potent neutrophil chemotactic factor, is a driving force in mineral-induced inflammation, and is shown to be elevated in asbestos (49) and silica-exposed subjects (50). Our data also reveal novel inflammatory molecules increased by both asbestos and silica exposures, including IL36G, IL36RN, IL32 and SAA2. Interestingly, IL-1 family members, IL36G (IL-36γ) and IL36RN (51), are highly expressed in the bronchial epithelium (52). IL-36γ is induced by toll-like receptor (TLR) ligands and TH17 cytokines and may promote neutrophilic airway inflammation (53). In conjunction with increased IL-36γ and IL-36Ra, our previous studies show increased expression of their receptors (IL1RL2 and IL1RAP) by crystalline but not amorphous, non-pathogenic silica (14). These seminal data suggest a prominent and uninvestigated role of IL-36 signaling in mineral exposure-induced inflammation.
Our data in concert suggest a scenario that may be common to pathogenic silicates. Owing to massive inflammation and tissue damage caused by exposures to fibrogenic agents, there is a consequent fibrotic response initiated in the epithelium in an attempt to repair damaged areas of the lung. For instance, both minerals increased expression of the profibrotic molecule IL13RA2, which has been shown to promote IL-13-driven induction of TGFβ in experimental fibrosis (54). Conversely, mineral exposures increased expression of the anti-fibrotic chemokine, CXCL10, which has been shown to alleviate fibrosis in bleomycin-induced experimental lung injury (55). This suggests a balance between anti-fibrotic and pro-fibrotic molecules that may be disrupted in susceptible individuals or at high overload concentrations of minerals. Cell proliferation in response to lung injury stimulates many biological activities and effector cell types including inflammatory cells and fibroblasts, but may also be compensatory in replenishing epithelial cells after injury and death of surrounding cells. The axis balancing cell death and proliferation is often disrupted in chronic disease, as regulation of apoptosis is key to controlling cell turnover of damaged cells. Genes associated with the regulation of apoptosis that were altered by both minerals include the anti-apoptotic molecule, BIRC3, which can be induced by TNFα-dependent mechanisms (56), and pro-apoptotic IL24, which is shown to drive apoptosis in various types of cancer cells by inhibition of survival and proliferation signaling and impedes lung cancer cell migration (57,58). In conjunction with exogenous stress, cell death can lead to the excessive release of damaging cellular contents, including oxidants, proteases and danger molecules, which further propagate cellular stress responses in surrounding tissue. A plethora of heat shock proteins are induced by both minerals, as well as the antioxidant, SOD2. SOD2 has been shown to play a pivotal role in mineral-induced disease development (59). Serum levels of SOD2 are elevated in silicotic patients (60) and polymorphisms in this gene are associated with increased asbestosis susceptibility (61).
Asbestos-specific responses
Many of the genes uniquely affected by crocidolite asbestos have not been described in lung epithelial cells previously, but may have key roles in lung injury and processes leading to carcinogenesis as indicated in Figure 5. Since we and others have linked asbestos exposures to cellular plasticity and cancer (62–64), it is not surprising that many genes affected by asbestos as opposed to silica are involved in processes related to cancer development. For example, ESPL1 (Separase) acts as an oncogene when overexpressed and plays an important role in the development of aneuploidy and tumorigenesis (65). ELF5 expression appears to be increased significantly in mouse mammary tumors relative to normal tissue (66). Previously, the up-regulation of SEMA3A has been linked to a feedback loop modulating growth-promoting signaling in mesothelial cells (67) and influencing apoptosis and proliferation in multiple cell types. SEMA3A is also essential for structural and functional abnormalities in tumor vascularization (68) and its induction by asbestos in NHBE and BEAS-2B cells is in line with our recent report on its up-regulation in human mesothelial cells exposed to crocidolite asbestos (69). We were also able to detect elevated CEACAM1 mRNA expression by asbestos. Recently, this gene was linked to the creation of a pro-angiogenic tumor microenvironment that supports tumor vessel maturation (70).
Exposure to foreign toxicants, like asbestos or other xenobiotic chemicals (e.g. those found in cigarette smoke), activates cellular detoxification mechanisms. For instance, sulfation is an important reaction in detoxification of xenobiotics, and sulfotransferases such as SULT1B1 enzymatically affect the bioactivation of pro-carcinogens to reactive electrophiles (71). In addition to sulfotransferases, asbestos induced increased mRNA levels of CYP2C18, a member of the cytochrome P450 family of detoxification enzymes. Together these data suggest that asbestos may activate and potentially disrupt xenobiotic detoxification mechanisms, which is suggested to contribute to asbestos-induced carcinogenic effects (72).
In the group of molecules involved in tumor morphology and remodeling, we observed increased expression levels of various enzymes, including transglutaminases (TGM3, and TGM5). Rigid amphibole fibers can penetrate and severely disrupt the cell membrane during or after uptake by cells. Up-regulation of transglutaminases, which are involved in protein cross-linking in molecular scaffolds and wound repair (73), may be part of a repair mechanism in an attempt to restore the compromised cell membrane.
The activin inhibitor FST has been shown to ameliorate inflammation and fibro-proliferative responses in a bleomycin model of pulmonary fibrosis in rats (74,75). It is possible that the up-regulation of FST, as observed here in the proliferation pathway in response to asbestos, is part of an endogenous defense mechanism of bronchial epithelial cells.
For amphibole asbestos, the surface chemistry, particularly bioavailable iron, is key in driving oxidant production and inflammation (76,77). Iron is mobilized and promotes oxidant production in lung epithelial cells exposed to crocidolite asbestos. Additionally, endogenous iron coats asbestos fibers in the lung, i.e. ferruginous bodies, one of the hallmarks of asbestosis (78,79). Iron sequestration is controlled by a number of molecules, including transferrin and the transferrin receptor (80). Our data indicate significant decreases in mRNA and protein levels of TFRC in human lung epithelial cells exposed to crocidolite asbestos. Crocidolite exposure causes intracellular sequestration of non-transferrin bound iron, potentially via the divalent metal ion transporter-1 (DMT1) (81). Accumulation of such intracellular iron stores likely leads to the down-regulation of TFRC, which, in turn, may increase the amount of extracellular iron. Broncho-alveolar lavage (BAL) fluids from asbestos-exposed subjects have higher levels of transferrin compared with unexposed subjects (82). Therefore, inhalation of asbestos can lead to severe disruption of normal iron homeostasis in the lungs, which may be a potential marker for exposure and/or disease development.
Silica-specific responses
The presence of massive inflammation is a driving force in the development and progression of silicosis. Our data show a robust inflammatory response to high-dose silica exposure with specifically increased expression of many potent inflammatory mediators. Silica induced, for instance, IL1A, IL1B, colony stimulating factors, CSF1, CSF2, as well as (C-X-C) and (C-C) motif chemokines. Interestingly, silica induced increased expression of TNFSF9 (CD137L/4–1BB) as well as its receptor TNFRSF9. TNFSF9 signaling is a key factor in the development of immune responses in T-cells, NK cells, macrophages and dendritic cells, and is also expressed in non-immune cells. Crosstalk between epithelial cells and inflammatory cells via TNFSF9-signaling drives neutrophil recruitment (83) which contributes to silica-induced inflammation. In keeping with these findings, there was also an evident interferon response. Type I interferon responses have been shown to play a role in silica-induced chronic inflammation (84); however, the mechanism by which they contribute to silica-induced inflammation is not well understood. In line with progressive inflammation, and interferon response, silica exposure was associated with pattern-recognition signaling pathways, with altered expression of membrane and intracellular pattern recognition-associated receptors (LY96, TLR1, CD14 and IFIH1/MDA-5). In addition, silica caused increases in antiviral-response proteins (OAS), and antigen-presentation molecules (CD83, HLADQB1). Silica also caused increased expression of the multifunctional pattern recognition molecule, PTX3 (85) which has been implicated in epithelial cell survival in co-culture models of silica exposure (86).
In response to silica particles, expression levels of a number of cell membrane receptors that mediate PAMP recognition, cell death and proinflammatory response were specifically altered. Silica exposure is associated with development of autoimmunity, where increased serum levels of (soluble) sIL2R are a marker of early immunological dysfunction in silica-exposed subjects (87). This correlates with our data, which reveal increased levels of IL2RG (i.e. the common gamma chain) mRNA. Furthermore, TNFα and the TNFR signaling pathway are crucial mediators in the development of silica-induced fibrosis (88), and soluble TNFR1 and soluble TNFR2 are increased in subjects exposed to silica (89). In conjunction with the up-regulated expression of a number of the TNF-receptor superfamily members, data reveal up-regulation of the adaptor protein TRAF1, which is crucial in dictating TNF-signaling via cascades including MAPK and NFκB. Canonical NFκB signaling is well known to drive silica-induced inflammation and fibrosis (90,91). However, our data are the first to reveal modulation of non-canonical (alternative) NFκB signaling, by up-regulation of NFKB2 (p52/p100) and RELB. Alternative NFκB signaling and auto-regulation are shown to modulate lung inflammation (92,93) and cross-talk between canonical and non-canonical NFκB regulates inflammatory responses in epithelial cells as well as airway inflammation and fibrosis in experimental models of allergic airways disease (94,95).
Owing to initial injury and the inability to clear silica particles, followed by cycles of inflammation that precede and continue during the development of silicosis. Exposure to silica promotes the production of various fibrogenic mediators, proteases, and proliferation and recruitment of fibroblasts in attempt to repair the damaged lung. Our data reveal silica-specific alteration of various fibrogenic mediators (e.g. VEGF, CTGF, FBRS, FGF2) and downstream signaling molecules associated with the fibrogenesis pathway. In particular, silica specifically induced mRNA levels and secretion of FGF2/bFGF. This potent fibrogenic factor (and others associated), which is induced by silica in lung epithelial cells (9,86) and potentially linked indirectly to fibroblast proliferation (14), may have a crucial impact on the intense fibrogenicity of silica. Furthermore, although WNT signaling is linked with aberrant proliferation and the development of fibrosis (96,97), it has not been associated with silica or silicosis yet. Interestingly we observed down-regulation of the WNT-signaling molecules SFRP1, WNT5A and up-regulation of DKK1 in response to silica. These findings suggest potential alteration of WNT signaling.
In summary, our study exemplifies how gene expression studies can predict acute responses of respiratory epithelial cells, the first cell type to encounter asbestos fibers or silica particles after inhalation, and link these events to biofunctional pathways leading to lung disease. Many of these alterations can be linked to biomarkers found in clinical samples of exposed subjects. Overall, our data reveal early alterations in both common gene expression that can be linked to inflammation and fibrosis by both pathogenic minerals and unique changes by silica promoting intense fibrogenicity and by asbestos that may be intrinsic to the development of lung cancers. Our analyses also highlight many novel molecules that can be pursued in functional studies, thus permitting their evaluation as new targets in detection, prevention and therapy of fibroproliferative diseases and cancers.
Materials and Methods
Cell culture
For primary cells, an isolate of normal primary NHBEs was purchased from Lonza, Clonetics®. NHBE cells were cultured and maintained in BEGM® with Reagentpack™ subculture reagents as described previously (9). For experiments with BEAS-2B cells, cells were cultured as recommended by ATCC as described previously (9).
Fiber/particle exposures
Respirable preparations of crocidolite asbestos fibers (NIEHS reference sample) and cristobalite silica particles (C&E Mineral Corp, King of Prussia, PA, USA) were sterilized and diluted to a concentration of 1.0 mg/ml prior to cell exposures at equal surface areas as described previously (9). Control samples were exposed to an equal volume of Hank's buffered saline solution (HBSS) alone (no-particle/fiber control). The fiber dimension data for this asbestos sample was previously reported (98), and particle characterization of cristobalite silica has previously been published (9,59).
RNA isolation
After exposure of cells, the medium was aspirated and total RNA was isolated using an RNeasy® Plus Mini Kit according to the manufacturer's protocol (Qiagen, Valencia, CA, USA) for NHBE cells. For BEAS-2B cell experiments, total RNA was isolated using the High Pure RNA Isolation Kit according to the manufacturer's protocol (Roche Applied Sciences, Indianapolis, IN, USA)
Quantitative real-time polymerase chain reaction (QRT-PCR)
For primary NHBE experiments, total RNA (1 μg) was reverse-transcribed with random primers using the AMV Reverse Transcriptase kit (Promega, Madison, WI, USA) according to the recommendations of the manufacturer, as described previously (15). All Taqman® qRT-PCR was performed using Assays-On-Demand™ primer and probe-sets (IL8, MMP1, BIRC3) used from Applied Biosystems (Foster City, CA, USA) and performed as described previously (15). For all BEAS-2B experiments QRT-PCR was performed as described previously (99). All forward and reverse primer sequences are described in Supplemental Material.
Affymetrix microarray gene expression-profiling
Microarray analysis was performed using Affymetrix GeneChip® Human Genome U133A 2.0 arrays (Affymetrix, Santa Clara, CA, USA). All procedures were performed by the Microarray Analysis Core Facility of the Vermont Genetics Network (VGN) using a standard Affymetrix protocol as described previously (100,101). GeneChip® Human Genome U133A 2.0 arrays (Affymetrix) targeting 18 400 genes were scanned twice (Hewlett-Packard GeneArray Scanner), the images overlaid and the average intensities of each probe cell compiled.
Microarray data were analyzed with Bioconductor tools in R with Bioconductor software (http://www.r-project.org/) (102), probe-level analysis was performed on the Affymetrix raw data (.cel files) with the robust multichip average (RMA) method to obtain a log2 expression value for each gene probe set (103,104). Unsupervised average-linkage hierarchical clustering, using Euclidian distance as metric, was performed to the RMA normalized data to visualize gene (probe set)/sample relationships of the top 50 most variable probe-set (103).
Genesifter analysis
Following data normalization using the RMA algorithm, the GeneSifter software (VizX Labs, Seattle, WA, USA) was used for supervised hierarchical cluster analysis to identify probe-sets/genes from log2-transformed data that significantly changed expression between the different groups (unexposed control, silica 15, asbestos 15 and silica 75; n = 3 per sample group) by pair-wise comparison analysis (t-test), and for the entire experimental group by multiple comparison analysis procedures (ANOVA). The raw P-values were corrected for multiple testing using the Benjamini and Hochberg false discovery rate method (FDR < 95%) or ‘adjusted P-value’ (105). A cut-off limit of 1.5-fold change versus controls (adj. P < 0.05) was used for analysis.
Ingenuity Pathway Analysis
Comparative analysis of pair-wise datasets (asbestos 15 versus controls; and silica 75 versus controls) revealed three groups of observations: (i) probe-sets significantly altered by both (asbestos 15 versus controls) and (silica 75 versus controls), (ii) probe-sets significantly altered by (asbestos 15 versus controls) only and (iii) probe-sets significantly altered by (silica 75 versus controls) only. These data sets were uploaded with gene ID (affymetrix), fold-change value, and adj. P-value in the Ingenuity Pathway Analysis program (IPA, Ingenuity Systems®, www.ingenuity.com) for core analysis. Core analysis of each dataset was performed (confidence level: experimentally observed). Genes associated with the top 10 canonical pathways significantly enriched by each comparison, in addition to a selection of genes with evident relevance to the canonical pathways and biological function, were used to construct schematic representations. The significance of the association between the input dataset and the canonical pathways was determined based on two parameters: (i) a ratio of the number of genes from the dataset that overlay to the pathway divided by the total number of genes that map to the canonical pathway and (ii) a P-value calculated determining the probability that the association between the genes in the dataset and the canonical pathway is not due to chance alone. Using the ‘connect’ tool, experimentally observed, direct and indirect connections were made for based on the Ingenuity Knowledge Base (Ingenuity Systems Inc., Redwood City, CA, USA). For clarity purposes, not all connections are shown, but only those pertinent to the biological scheme.
Immunofluorescence
For immunofluorescent staining of BEAS-2B cells, sterile glass coverslips (VWR 13 mm, Thickness No. 1) were placed in 24-well culture plates and coated with coating medium overnight at 37°C. Cells were seeded, maintained and exposed to minerals as described previously (9). Following exposure culture medium was aspirated, and cells washed three times with 1× PBS. Cells were then fixed in 4% phosphate-buffered formaldehyde solution, pH 7.0 (Klinipath 4078-9001) for 20 min at RT. Cells were washed three times with PBS and placed in blocking/permeabilization buffer (1.0% BSA, 0.1% Triton X-100 in PBS) for 1 h at RT. Cells were incubated in primary antibody (1:100 dilution) overnight at 4°C. Cells were washed three times with PBS, incubated with an Alexa Fluor®-conjugated secondary antibody (1:1000 in 1% BSA–PBS) for 1 h at RT (in dark). Cells were washed three times with PBS and incubated with DAPI solution (0.5 µg/ml) for 5 min at RT (in dark). Cells were washed two times with PBS and once with distilled water. Coverslips were then mounted on glass slides (Knittel Glass, Germany) using fluorescent mounting medium (DAKO). Cells were imaged on a Nikon Eclipse E800 (Nikon) equipped with an AxioCam MRc5 (Zeiss) using Zeiss ZEN (blue) 2012 software. Images were exported in .TIFF format.
Western blot
Cells were washed twice with cold HBSS, and lysed with 1× RIPA buffer [(RIPA buffer 10× Cell Signaling) with 1% Protease inhibitor cocktail (Sigma), 1% sodium orthovanadate, 1% PMSF and 1 mM DTT]. Total protein was determined using the Bio-Rad DC-protein determination kit (Bio-Rad, Hercules CA, USA). Twenty micrograms of protein were loaded onto pre-cast 12% (bis–tris) polyacrylamide gels (Bio-Rad). Gels were run, and transferred to nitrocellulose membranes. After blocking in 5% milk, membranes were incubated with primary antibodies (Anti-TFRC 1:5000, Anti-GAPDH 1:20 000) overnight at 4°C. Membranes were washed three times 15 min in TBS–Tween (0.1%), and then incubated in HRP-conjugated secondary antibody (1:5000 in 5% milk) for 1 h at RT. Membranes were washed three times in TBS–Tween (0.1%) and once in TBS, and then incubated in ECL substrate and developed. Densitometry was performed using QuantityOne software (Bio-Rad).
Caspase-1 activity assay
Caspase-1 activity was measured using a caspase-1 activity kit (R&D) following the manufacturer's protocol.
ELISA
To determine the levels of bFGF and IL-8 levels in cell culture medium, culture medium was centrifuged for 10 min at 14 000 rpm (4°C) to remove all cellular debris and minerals, and supernatant removed. Levels of bFGF and IL-8 in the supernatant were determined using a Human FGF-basic ELISA MAX Deluxe kit (Biolegend) and a Human IL-8 ELISA kit (Sanquin), respectively, following the manufacturerś protocols.
Antibodies
Primary antibodies: rabbit polyclonal Anti-Transferrin Receptor C (Sino Biological corp., #11020-RP02), Rabbit polyclonal anti-GAPDH (Cell Signaling), rabbit polyclonal anti-Claudin-1 (Cell Signaling #4933). Secondary Antibodies: Alexa Fluor® 488 conjugated anti-rabbit IgG (Life Technologies) and Peroxidase-conjugated anti-rabbit IgG (Vector Laboratories).
Statistical analysis
All statistical analyses for microarray and pathway analysis data are described above in their respective sections. For cell viability and QRT-PCR, data were analyzed by one-way analysis of variance (ANOVA) using the Student–Neuman–Keul's test to adjust for multiple pairwise comparisons between treatment groups, or the Student' t-test where appropriate. Data are expressed as mean values ± SEM. Differences with P-value <0.05 were considered statistically significant. Data presented are representative of multiple independent repeat experiments.
Supplementary Material
Funding
This work was supported by an unrestricted grant from the Weijerhorst Foundation.
Supplementary Material
Acknowledgements
The authors thank the Vermont Genetics Network Microarray Facility for gene expression-profiling (Grant Number P20 RR16462 from the INBRE Program of the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH)) and the Vermont Cancer Center DNA Analysis facility (supported by the Vermont Cancer Center, Lake Champlain Cancer Research Organization, and the UVM College of Medicine) at the University of Vermont (Burlington, VT) for assistance with QRT-PCR. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. We also thank Jennifer Diaz for help with preparation of the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Chong S., Lee K.S., Chung M.J., Han J., Kwon O.J., Kim T.S. Pneumoconiosis: comparison of imaging and pathologic findings. Radiographics. 2006;26:59–77. doi: 10.1148/rg.261055070. [DOI] [PubMed] [Google Scholar]
- 2.Guthrie G.D., Jr., Heaney P.J. Mineralogical characteristics of silica polymorphs in relation to their biological activities. Scand. J. Work Environ. Health. 1995;21(Suppl 2):5–8. [PubMed] [Google Scholar]
- 3.Mossman B.T., Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am. J. Respir. Crit. Care Med. 1998;157:1666–1680. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 4.Borm P.J., Tran L., Donaldson K. The carcinogenic action of crystalline silica: a review of the evidence supporting secondary inflammation-driven genotoxicity as a principal mechanism. Crit. Rev. Toxicol. 2011;41:756–770. doi: 10.3109/10408444.2011.576008. [DOI] [PubMed] [Google Scholar]
- 5.Gamble J.F. Crystalline silica and lung cancer: a critical review of the occupational epidemiology literature of exposure-response studies testing this hypothesis. Crit. Rev. Toxicol. 2011;41:404–465. doi: 10.3109/10408444.2010.541223. [DOI] [PubMed] [Google Scholar]
- 6.Guha N., Straif K., Benbrahim-Tallaa L. The IARC monographs on the carcinogenicity of crystalline silica. Med. Lav. 2011;102:310–320. [PubMed] [Google Scholar]
- 7.Leung C.C., Yu I.T., Chen W. Silicosis. Lancet. 2012;379:2008–2018. doi: 10.1016/S0140-6736(12)60235-9. [DOI] [PubMed] [Google Scholar]
- 8.Castranova V., Vallyathan V. Silicosis and coal workers’ pneumoconiosis. Environ. Health Perspect. 2000;108(Suppl 4):675–684. doi: 10.1289/ehp.00108s4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perkins T.N., Shukla A., Peeters P.M., Steinbacher J.L., Landry C.C., Lathrop S.A., Steele C., Reynaert N.L., Wouters E.F., Mossman B.T. Differences in gene expression and cytokine production by crystalline versus amorphous silica in human lung epithelial cells. Part. Fibre Toxicol. 2012;9:6. doi: 10.1186/1743-8977-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shukla A., MacPherson M.B., Hillegass J., Ramos-Nino M.E., Alexeeva V., Vacek P.M., Bond J.P., Pass H.I., Steele C., Mossman B.T. Alterations in gene expression in human mesothelial cells correlate with mineral pathogenicity. Am. J. Respir. Cell Mol. Biol. 2009;41:114–123. doi: 10.1165/rcmb.2008-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scapoli L., Ramos-Nino M.E., Martinelli M., Mossman B.T. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene. 2004;23:805–813. doi: 10.1038/sj.onc.1207163. [DOI] [PubMed] [Google Scholar]
- 12.Shukla A., Barrett T.F., Nakayama K.I., Nakayama K., Mossman B.T., Lounsbury K.M. Transcriptional up-regulation of MMP12 and MMP13 by asbestos occurs via a PKCdelta-dependent pathway in murine lung. FASEB J. 2006;20:997–999. doi: 10.1096/fj.05-4554fje. [DOI] [PubMed] [Google Scholar]
- 13.Fubini B., Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003;34:1507–1516. doi: 10.1016/s0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- 14.Peeters P.M., Perkins T.N., Wouters E.F., Mossman B.T., Reynaert N.L. Silica induces NLRP3 inflammasome activation in human lung epithelial cells. Part Fibre Toxicol. 2013;10:3. doi: 10.1186/1743-8977-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hillegass J.M., Shukla A., MacPherson M.B., Lathrop S.A., Alexeeva V., Perkins T.N., van der Vliet A., Vacek P.M., Gunter M.E., Mossman B.T. Mechanisms of oxidative stress and alterations in gene expression by Libby six-mix in human mesothelial cells. Part Fibre Toxicol. 2010;7:26. doi: 10.1186/1743-8977-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y.C., Karoly E.D., Dailey L.A., Schmitt M.T., Silbajoris R., Graff D.W., Devlin R.B. Comparison of gene expression profiles induced by coarse, fine, and ultrafine particulate matter. J. Toxicol. Environ. Health A. 2011;74:296–312. doi: 10.1080/15287394.2010.516238. [DOI] [PubMed] [Google Scholar]
- 17.Hillegass J.M., Shukla A., MacPherson M.B., Bond J.P., Steele C., Mossman B.T. Utilization of gene profiling and proteomics to determine mineral pathogenicity in a human mesothelial cell line (LP9/TERT-1) J. Toxicol. Environ. Health A. 2010;73:423–436. doi: 10.1080/15287390903486568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding M., Shi X., Dong Z., Chen F., Lu Y., Castranova V., Vallyathan V. Freshly fractured crystalline silica induces activator protein-1 activation through ERKs and p38 MAPK. J. Biol. Chem. 1999;274:30611–30616. doi: 10.1074/jbc.274.43.30611. [DOI] [PubMed] [Google Scholar]
- 19.Heintz N.H., Janssen Y.M., Mossman B.T. Persistent induction of c-fos and c-jun expression by asbestos. Proc. Natl Acad. Sci. USA. 1993;90:3299–3303. doi: 10.1073/pnas.90.8.3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heintz N.H., Janssen-Heininger Y.M., Mossman B.T. Asbestos, lung cancers, and mesotheliomas: from molecular approaches to targeting tumor survival pathways. Am. J. Respir. Cell Mol. Biol. 2010;42:133–139. doi: 10.1165/rcmb.2009-0206TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lappi-Blanco E., Lehtonen S.T., Sormunen R., Merikallio H.M., Soini Y., Kaarteenaho R.L. Divergence of tight and adherens junction factors in alveolar epithelium in pulmonary fibrosis. Hum. Pathol. 2013;44:895–907. doi: 10.1016/j.humpath.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Shiozaki A., Bai X.H., Shen-Tu G., Moodley S., Takeshita H., Fung S.Y., Wang Y., Keshavjee S., Liu M. Claudin 1 mediates TNFalpha-induced gene expression and cell migration in human lung carcinoma cells. PLoS One. 2012;7:e38049. doi: 10.1371/journal.pone.0038049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mossman B.T., Glenn R.E. Bioreactivity of the crystalline silica polymorphs, quartz and cristobalite, and implications for occupational exposure limits (OELs) Crit. Rev. Toxicol. 2013;43:632–660. doi: 10.3109/10408444.2013.818617. [DOI] [PubMed] [Google Scholar]
- 24.Mossman B.T., Lippmann M., Hesterberg T.W., Kelsey K.T., Barchowsky A., Bonner J.C. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J. Toxicol. Environ. Health B Crit. Rev. 2011;14:76–121. doi: 10.1080/10937404.2011.556047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallyathan V., Shi X., Castranova V. Reactive oxygen species: their relation to pneumoconiosis and carcinogenesis. Environ. Health Perspect. 1998;106(Suppl 5):1151–1155. doi: 10.1289/ehp.98106s51151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Governa M., Amati M., Valentino M., Visona I., Fubini B., Botta G.C., Volpe A.R., Carmignani M. In vitro cleavage by asbestos fibers of the fifth component of human complement through free-radical generation and kallikrein activation. J. Toxicol. Environ Health A. 2000;59:539–552. doi: 10.1080/009841000156664. [DOI] [PubMed] [Google Scholar]
- 27.Governa M., Fenoglio I., Amati M., Valentino M., Bolognini L., Coloccini S., Volpe A.R., Carmignani M., Fubini B. Cleavage of the fifth component of human complement and release of a split product with C5a-like activity by crystalline silica through free radical generation and kallikrein activation. Toxicol. Appl. Pharmacol. 2002;179:129–136. doi: 10.1006/taap.2002.9351. [DOI] [PubMed] [Google Scholar]
- 28.Kamekura R., Kojima T., Takano K., Go M., Sawada N., Himi T. The role of IL-33 and its receptor ST2 in human nasal epithelium with allergic rhinitis. Clin. Exp. Allergy. 2012;42:218–228. doi: 10.1111/j.1365-2222.2011.03867.x. [DOI] [PubMed] [Google Scholar]
- 29.Yagami A., Orihara K., Morita H., Futamura K., Hashimoto N., Matsumoto K., Saito H., Matsuda A. IL-33 mediates inflammatory responses in human lung tissue cells. J. Immunol. 2010;185:5743–5750. doi: 10.4049/jimmunol.0903818. [DOI] [PubMed] [Google Scholar]
- 30.Castranova V. Signaling pathways controlling the production of inflammatory mediators in response to crystalline silica exposure: role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 2004;37:916–925. doi: 10.1016/j.freeradbiomed.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 31.Rimal B., Greenberg A.K., Rom W.N. Basic pathogenetic mechanisms in silicosis: current understanding. Curr. Opin. Pulm. Med. 2005;11:169–173. doi: 10.1097/01.mcp.0000152998.11335.24. [DOI] [PubMed] [Google Scholar]
- 32.Chu L., Wang T., Hu Y., Gu Y., Su Z., Jiang H. Activation of Egr-1 in human lung epithelial cells exposed to silica through MAPKs signaling pathways. PLoS One. 2013;8:e68943. doi: 10.1371/journal.pone.0068943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee C.G., Cho S.J., Kang M.J., Chapoval S.P., Lee P.J., Noble P.W., Yehualaeshet T., Lu B., Flavell R.A., Milbrandt J., et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J. Exp. Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiang F., Bai M., Jin Y., Ma W., Xin J. Egr-1 mediates SiO(2)-driven transcription of membrane type I matrix metalloproteinase in macrophages. J. Huazhong Univ. Sci. Technol. Med. Sci. 2007;27:13–16. doi: 10.1007/s11596-007-0104-3. [DOI] [PubMed] [Google Scholar]
- 35.Scabilloni J.F., Wang L., Antonini J.M., Roberts J.R., Castranova V., Mercer R.R. Matrix metalloproteinase induction in fibrosis and fibrotic nodule formation due to silica inhalation. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;288:L709–L717. doi: 10.1152/ajplung.00034.2004. [DOI] [PubMed] [Google Scholar]
- 36.Tan R.J., Fattman C.L., Niehouse L.M., Tobolewski J.M., Hanford L.E., Li Q., Monzon F.A., Parks W.C., Oury T.D. Matrix metalloproteinases promote inflammation and fibrosis in asbestos-induced lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2006;35:289–297. doi: 10.1165/rcmb.2005-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herrera I., Cisneros J., Maldonado M., Ramirez R., Ortiz-Quintero B., Anso E., Chandel N.S., Selman M., Pardo A. Matrix metalloproteinase (MMP)-1 induces lung alveolar epithelial cell migration and proliferation, protects from apoptosis, and represses mitochondrial oxygen consumption. J. Biol. Chem. 2013;288:25964–25975. doi: 10.1074/jbc.M113.459784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suh Y., Yoon C.H., Kim R.K., Lim E.J., Oh Y.S., Hwang S.G., An S., Yoon G., Gye M.C., Yi J.M., et al. Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene. 2013;32:4873–4882. doi: 10.1038/onc.2012.505. [DOI] [PubMed] [Google Scholar]
- 39.Cassel S.L., Eisenbarth S.C., Iyer S.S., Sadler J.J., Colegio O.R., Tephly L.A., Carter A.B., Rothman P.B., Flavell R.A., Sutterwala F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl Acad. Sci. USA. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dostert C., Petrilli V., Van Bruggen R., Steele C., Mossman B.T., Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yazdi A.S., Guarda G., Riteau N., Drexler S.K., Tardivel A., Couillin I., Tschopp J. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1alpha and IL-1beta. Proc. Natl Acad. Sci. USA. 2010;107:19449–19454. doi: 10.1073/pnas.1008155107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hussain S., Sangtian S., Anderson S.M., Snyder R.J., Marshburn J.D., Rice A.B., Bonner J.C., Garantziotis S. Inflammasome activation in airway epithelial cells after multi-walled carbon nanotube exposure mediates a profibrotic response in lung fibroblasts. Part Fibre Toxicol. 2014;11:28. doi: 10.1186/1743-8977-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirota J.A., Hirota S.A., Warner S.M., Stefanowicz D., Shaheen F., Beck P.L., Macdonald J.A., Hackett T.L., Sin D.D., Van Eeden S., et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J. Allergy Clin. Immunol. 2012;129:1116–1125. doi: 10.1016/j.jaci.2011.11.033. e1116. [DOI] [PubMed] [Google Scholar]
- 44.Hillegass J.M., Miller J.M., MacPherson M.B., Westbom C.M., Sayan M., Thompson J.K., Macura S.L., Perkins T.N., Beuschel S.L., Alexeeva V., et al. Asbestos and erionite prime and activate the NLRP3 inflammasome that stimulates autocrine cytokine release in human mesothelial cells. Part Fibre Toxicol. 2013;10:39. doi: 10.1186/1743-8977-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson J.K., Westbom C.M., MacPherson M.B., Mossman B.T., Heintz N.H., Spiess P., Shukla A. Asbestos modulates thioredoxin-thioredoxin interacting protein interaction to regulate inflammasome activation. Part Fibre Toxicol. 2014;11:24. doi: 10.1186/1743-8977-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kukkonen M.K., Vehmas T., Piirila P., Hirvonen A. Genes involved in innate immunity associated with asbestos-related fibrotic changes. Occup. Environ. Med. 2014;71:48–54. doi: 10.1136/oemed-2013-101555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verma D., Lerm M., Blomgran Julinder R., Eriksson P., Soderkvist P., Sarndahl E. Gene polymorphisms in the NALP3 inflammasome are associated with interleukin-1 production and severe inflammation: relation to common inflammatory diseases? Arthritis Rheum. 2008;58:888–894. doi: 10.1002/art.23286. [DOI] [PubMed] [Google Scholar]
- 48.Ji X., Hou Z., Wang T., Jin K., Fan J., Luo C., Chen M., Han R., Ni C. Polymorphisms in inflammasome genes and risk of coal workers’ pneumoconiosis in a Chinese population. PLoS One. 2012;7:e47949. doi: 10.1371/journal.pone.0047949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ilavska S., Jahnova E., Tulinska J., Horvathova M., Dusinska M., Wsolova L., Kyrtopoulos S.A., Fuortes L. Immunological monitoring in workers occupationally exposed to asbestos. Toxicology. 2005;206:299–308. doi: 10.1016/j.tox.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 50.Lee J.S., Shin J.H., Lee J.O., Lee K.M., Kim J.H., Choi B.S. Serum levels of interleukin-8 and tumor necrosis factor-alpha in coal workers’ pneumoconiosis: one-year follow-up study. Saf. Health Work. 2010;1:69–79. doi: 10.5491/SHAW.2010.1.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Debets R., Timans J.C., Homey B., Zurawski S., Sana T.R., Lo S., Wagner J., Edwards G., Clifford T., Menon S., et al. Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J. Immunol. 2001;167:1440–1446. doi: 10.4049/jimmunol.167.3.1440. [DOI] [PubMed] [Google Scholar]
- 52.Gresnigt M.S., van de Veerdonk F.L. Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 2013;25:458–465. doi: 10.1016/j.smim.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 53.Chustz R.T., Nagarkar D.R., Poposki J.A., Favoreto S., Jr., Avila P.C., Schleimer R.P., Kato A. Regulation and function of the IL-1 family cytokine IL-1F9 in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011;45:145–153. doi: 10.1165/rcmb.2010-0075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fichtner-Feigl S., Strober W., Kawakami K., Puri R.K., Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat. Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 55.Jiang D., Liang J., Campanella G.S., Guo R., Yu S., Xie T., Liu N., Jung Y., Homer R., Meltzer E.B., et al. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J. Clin. Invest. 2010;120:2049–2057. doi: 10.1172/JCI38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pryhuber G.S., Huyck H.L., Staversky R.J., Finkelstein J.N., O'Reilly M.A. Tumor necrosis factor-alpha-induced lung cell expression of antiapoptotic genes TRAF1 and cIAP2. Am. J. Respir. Cell Mol. Biol. 2000;22:150–156. doi: 10.1165/ajrcmb.22.2.3783. [DOI] [PubMed] [Google Scholar]
- 57.Chada S., Bocangel D., Ramesh R., Grimm E.A., Mumm J.B., Mhashilkar A.M., Zheng M. mda-7/IL24 kills pancreatic cancer cells by inhibition of the Wnt/PI3 K signaling pathways: identification of IL-20 receptor-mediated bystander activity against pancreatic cancer. Mol. Ther. 2005;11:724–733. doi: 10.1016/j.ymthe.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 58.Sieger K.A., Mhashilkar A.M., Stewart A., Sutton R.B., Strube R.W., Chen S.Y., Pataer A., Swisher S.G., Grimm E.A., Ramesh R., et al. The tumor suppressor activity of MDA-7/IL-24 is mediated by intracellular protein expression in NSCLC cells. Mol. Ther. 2004;9:355–367. doi: 10.1016/j.ymthe.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 59.Janssen Y.M., Marsh J.P., Absher M.P., Hemenway D., Vacek P.M., Leslie K.O., Borm P.J., Mossman B.T. Expression of antioxidant enzymes in rat lungs after inhalation of asbestos or silica. J. Biol. Chem. 1992;267:10625–10630. [PubMed] [Google Scholar]
- 60.Zhang J.W., Lv G.C., Yao J.M., Hong X.P. Assessment of serum antioxidant status in patients with silicosis. J. Int. Med. Res. 2010;38:884–889. doi: 10.1177/147323001003800314. [DOI] [PubMed] [Google Scholar]
- 61.Franko A., Dodic-Fikfak M., Arneric N., Dolzan V. Manganese and extracellular superoxide dismutase polymorphisms and risk for asbestosis. J. Biomed. Biotechnol. 2009;2009:493083. doi: 10.1155/2009/493083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baldys A., Pande P., Mosleh T., Park S.H., Aust A.E. Apoptosis induced by crocidolite asbestos in human lung epithelial cells involves inactivation of Akt and MAPK pathways. Apoptosis. 2007;12:433–447. doi: 10.1007/s10495-006-0577-8. [DOI] [PubMed] [Google Scholar]
- 63.Mossman B.T., Lounsbury K.M., Reddy S.P. Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am. J. Respir. Cell Mol. Biol. 2006;34:666–669. doi: 10.1165/rcmb.2006-0047SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tamminen J.A., Myllarniemi M., Hyytiainen M., Keski-Oja J., Koli K. Asbestos exposure induces alveolar epithelial cell plasticity through MAPK/Erk signaling. J. Cell Biochem. 2012;113:2234–2247. doi: 10.1002/jcb.24094. [DOI] [PubMed] [Google Scholar]
- 65.Pati D. Oncogenic activity of separase. Cell Cycle. 2008;7:3481–3482. doi: 10.4161/cc.7.22.7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Galang C.K., Muller W.J., Foos G., Oshima R.G., Hauser C.A. Changes in the expression of many Ets family transcription factors and of potential target genes in normal mammary tissue and tumors. J. Biol. Chem. 2004;279:11281–11292. doi: 10.1074/jbc.M311887200. [DOI] [PubMed] [Google Scholar]
- 67.Catalano A., Caprari P., Rodilossi S., Betta P., Castellucci M., Casazza A., Tamagnone L., Procopio A. Cross-talk between vascular endothelial growth factor and semaphorin-3A pathway in the regulation of normal and malignant mesothelial cell proliferation. FASEB J. 2004;18:358–360. doi: 10.1096/fj.03-0513fje. [DOI] [PubMed] [Google Scholar]
- 68.Serini G., Maione F., Giraudo E., Bussolino F. Semaphorins and tumor angiogenesis. Angiogenesis. 2009;12:187–193. doi: 10.1007/s10456-009-9138-4. [DOI] [PubMed] [Google Scholar]
- 69.Shukla A., Bosenberg M.W., MacPherson M.B., Butnor K.J., Heintz N.H., Pass H.I., Carbone M., Testa J.R., Mossman B.T. Activated cAMP response element binding protein is overexpressed in human mesotheliomas and inhibits apoptosis. Am. J. Pathol. 2009;175:2197–2206. doi: 10.2353/ajpath.2009.090400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerstel D., Wegwitz F., Jannasch K., Ludewig P., Scheike K., Alves F., Beauchemin N., Deppert W., Wagener C., Horst A.K. CEACAM1 creates a pro-angiogenic tumor microenvironment that supports tumor vessel maturation. Oncogene. 2011;30:4275–4288. doi: 10.1038/onc.2011.146. [DOI] [PubMed] [Google Scholar]
- 71.Gamage N., Barnett A., Hempel N., Duggleby R.G., Windmill K.F., Martin J.L., McManus M.E. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 2006;90:5–22. doi: 10.1093/toxsci/kfj061. [DOI] [PubMed] [Google Scholar]
- 72.Anttila S., Raunio H., Hakkola J. Cytochrome P450-mediated pulmonary metabolism of carcinogens: regulation and cross-talk in lung carcinogenesis. Am. J. Respir. Cell Mol. Biol. 2011;44:583–590. doi: 10.1165/rcmb.2010-0189RT. [DOI] [PubMed] [Google Scholar]
- 73.Lorand L., Graham R.M. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 74.Aoki F., Kurabayashi M., Hasegawa Y., Kojima I. Attenuation of bleomycin-induced pulmonary fibrosis by follistatin. Am. J. Respir. Crit. Care Med. 2005;172:713–720. doi: 10.1164/rccm.200412-1620OC. [DOI] [PubMed] [Google Scholar]
- 75.de Kretser D.M., O'Hehir R.E., Hardy C.L., Hedger M.P. The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol. Cell Endocrinol. 2011;359:101–6. doi: 10.1016/j.mce.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 76.Aust A.E., Cook P.M., Dodson R.F. Morphological and chemical mechanisms of elongated mineral particle toxicities. J. Toxicol. Environ. Health B Crit. Rev. 2011;14:40–75. doi: 10.1080/10937404.2011.556046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghio A.J., Kennedy T.P., Whorton A.R., Crumbliss A.L., Hatch G.E., Hoidal J.R. Role of surface complexed iron in oxidant generation and lung inflammation induced by silicates. Am. J. Physiol. 1992;263:L511–L518. doi: 10.1152/ajplung.1992.263.5.L511. [DOI] [PubMed] [Google Scholar]
- 78.Chao C.C., Lund L.G., Zinn K.R., Aust A.E. Iron mobilization from crocidolite asbestos by human lung carcinoma cells. Arch. Biochem. Biophys. 1994;314:384–391. doi: 10.1006/abbi.1994.1457. [DOI] [PubMed] [Google Scholar]
- 79.Chao C.C., Park S.H., Aust A.E. Participation of nitric oxide and iron in the oxidation of DNA in asbestos-treated human lung epithelial cells. Arch. Biochem. Biophys. 1996;326:152–157. doi: 10.1006/abbi.1996.0059. [DOI] [PubMed] [Google Scholar]
- 80.Gkouvatsos K., Papanikolaou G., Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta. 2011;1820:188–202. doi: 10.1016/j.bbagen.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 81.Wang X., Wu Y., Stonehuerner J.G., Dailey L.A., Richards J.D., Jaspers I., Piantadosi C.A., Ghio A.J. Oxidant generation promotes iron sequestration in BEAS-2B cells exposed to asbestos. Am. J. Respir. Cell Mol. Biol. 2006;34:286–292. doi: 10.1165/rcmb.2004-0275OC. [DOI] [PubMed] [Google Scholar]
- 82.Lindahl M., Ekstrom T., Sorensen J., Tagesson C. Two dimensional protein patterns of bronchoalveolar lavage fluid from non-smokers, smokers, and subjects exposed to asbestos. Thorax. 1996;51:1028–1035. doi: 10.1136/thx.51.10.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim H.J., Lee J.S., Kim J.D., Cha H.J., Kim A., Lee S.K., Lee S.C., Kwon B.S., Mittler R.S., Cho H.R., et al. Reverse signaling through the costimulatory ligand CD137L in epithelial cells is essential for natural killer cell-mediated acute tissue inflammation. Proc. Natl Acad. Sci. USA. 2012;109:E13–E22. doi: 10.1073/pnas.1112256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giordano G., van den Brule S., Lo Re S., Triqueneaux P., Uwambayinema F., Yakoub Y., Couillin I., Ryffel B., Michiels T., Renauld J.C., et al. Type I interferon signaling contributes to chronic inflammation in a murine model of silicosis. Toxicol. Sci. 2010;116:682–692. doi: 10.1093/toxsci/kfq158. [DOI] [PubMed] [Google Scholar]
- 85.Mantovani A., Valentino S., Gentile S., Inforzato A., Bottazzi B., Garlanda C. The long pentraxin PTX3: a paradigm for humoral pattern recognition molecules. Ann. N. Y. Acad. Sci. 2013;1285:1–14. doi: 10.1111/nyas.12043. [DOI] [PubMed] [Google Scholar]
- 86.Herseth J.I., Volden V., Schwarze P.E., Lag M., Refsnes M. IL-1beta differently involved in IL-8 and FGF-2 release in crystalline silica-treated lung cell co-cultures. Part Fibre Toxicol. 2008;5:16. doi: 10.1186/1743-8977-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hayashi H., Maeda M., Murakami S., Kumagai N., Chen Y., Hatayama T., Katoh M., Miyahara N., Yamamoto S., Yoshida Y., et al. Soluble interleukin-2 receptor as an indicator of immunological disturbance found in silicosis patients. Int. J. Immunopathol. Pharmacol. 2009;22:53–62. doi: 10.1177/039463200902200107. [DOI] [PubMed] [Google Scholar]
- 88.Piguet P.F., Vesin C. Treatment by human recombinant soluble TNF receptor of pulmonary fibrosis induced by bleomycin or silica in mice. Eur. Respir. J. 1994;7:515–518. doi: 10.1183/09031936.94.07030515. [DOI] [PubMed] [Google Scholar]
- 89.Braz N.F., Carneiro A.P., Amorim M.R., de Oliveira Ferreira F., Lacerda A.C., Silva de Miranda A., Teixeira M.M., Teixeira A.L., Mendonca V.A. Association between inflammatory biomarkers in plasma, radiological severity, and duration of exposure in patients with silicosis. J. Occup. Environ. Med. 2014;56:493–497. doi: 10.1097/JOM.0000000000000164. [DOI] [PubMed] [Google Scholar]
- 90.Hubbard A.K., Timblin C.R., Shukla A., Rincon M., Mossman B.T. Activation of NF-kappaB-dependent gene expression by silica in lungs of luciferase reporter mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2002;282:L968–L975. doi: 10.1152/ajplung.00327.2001. [DOI] [PubMed] [Google Scholar]
- 91.van Berlo D., Knaapen A.M., van Schooten F.J., Schins R.P., Albrecht C. NF-kappaB dependent and independent mechanisms of quartz-induced proinflammatory activation of lung epithelial cells. Part Fibre Toxicol. 2010;7:13. doi: 10.1186/1743-8977-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morris G.F. An alternative to lung inflammation and fibrosis. Am. J. Pathol. 2010;176:2595–2598. doi: 10.2353/ajpath.2010.100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang L., Cui H., Wang Z., Zhang B., Ding J., Liu L., Ding H.F. Loss of negative feedback control of nuclear factor-kappaB2 activity in lymphocytes leads to fatal lung inflammation. Am. J. Pathol. 2010;176:2646–2657. doi: 10.2353/ajpath.2010.090751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tully J.E., Hoffman S.M., Lahue K.G., Nolin J.D., Anathy V., Lundblad L.K., Daphtary N., Aliyeva M., Black K.E., Dixon A.E., et al. Epithelial NF-kappaB orchestrates house dust mite-induced airway inflammation, hyperresponsiveness, and fibrotic remodeling. J. Immunol. 2013;191:5811–5821. doi: 10.4049/jimmunol.1301329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tully J.E., Nolin J.D., Guala A.S., Hoffman S.M., Roberson E.C., Lahue K.G., van der Velden J., Anathy V., Blackwell T.S., Janssen-Heininger Y.M. Cooperation between classical and alternative NF-kappaB pathways regulates proinflammatory responses in epithelial cells. Am. J. Respir. Cell Mol. Biol. 2012;47:497–508. doi: 10.1165/rcmb.2012-0014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guo Y., Xiao L., Sun L., Liu F. Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol. Res. 2012;61:337–346. doi: 10.33549/physiolres.932289. [DOI] [PubMed] [Google Scholar]
- 97.Konigshoff M. Lung cancer in pulmonary fibrosis: tales of epithelial cell plasticity. Respiration. 2011;81:353–358. doi: 10.1159/000326299. [DOI] [PubMed] [Google Scholar]
- 98.Campbell W.J., Huggins C.W., Wylie A.G. Bureau of Mines Report of Investigations. National Institute of Environmental Health Science; 1980. Chemical and physical characterization of amosite, chrysotile, crocidolite and nonfibrous tremolite for oral ingestion studies. ol. 8452, pp. 1–63. [Google Scholar]
- 99.Verhees K.J., Schols A.M., Kelders M.C., Op den Kamp C.M., van der Velden J.L., Langen R.C. Glycogen synthase kinase-3beta is required for the induction of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2011;301:C995–C1007. doi: 10.1152/ajpcell.00520.2010. [DOI] [PubMed] [Google Scholar]
- 100.Sabo-Attwood T., Ramos-Nino M., Bond J., Butnor K.J., Heintz N., Gruber A.D., Steele C., Taatjes D.J., Vacek P., Mossman B.T. Gene expression profiles reveal increased mClca3 (Gob5) expression and mucin production in a murine model of asbestos-induced fibrogenesis. Am. J. Pathol. 2005;167:1243–1256. doi: 10.1016/S0002-9440(10)61212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shukla A., Lounsbury K.M., Barrett T.F., Gell J., Rincon M., Butnor K.J., Taatjes D.J., Davis G.S., Vacek P., Nakayama K.I., et al. Asbestos-induced peribronchiolar cell proliferation and cytokine production are attenuated in lungs of protein kinase C-delta knockout mice. Am. J. Pathol. 2007;170:140–151. doi: 10.2353/ajpath.2007.060381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arijs I., Li K., Toedter G., Quintens R., Van Lommel L., Van Steen K., Leemans P., De Hertogh G., Lemaire K., Ferrante M., et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut. 2009;58:1612–1619. doi: 10.1136/gut.2009.178665. [DOI] [PubMed] [Google Scholar]
- 104.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 105.Benjamini Y., Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.