

Abstract
BACKGROUND
Animal studies suggest that the renin–angiotensin–aldosterone system is involved in neurocognitive function and the response to antihypertensive therapy. We investigated the impact of circulating aldosterone and renin activity on cognition and cerebral hemodynamics at baseline and after antihypertensive therapy for 1 year.
METHODS
Participants were older adults (n = 47; mean age = 71 years) enrolled in a clinical trial. Routine antihypertensive medications were replaced with the study regimen to achieve a blood pressure <140/90mm Hg. Executive function, memory, cerebral hemodynamics (blood flow velocity), CO2 vasoreactivity (measured using transcranial Doppler ultrasonography), plasma renin activity, and aldosterone were measured at baseline and at 6 and 12 months after the initiation of treatment.
RESULTS
At baseline, higher levels of circulating aldosterone were associated with lower blood flow velocity (β = −0.02; P = 0.03), lower CO2 vasoreactivity (β = −0.11; P = 0.007), and decreased autoregulation abilities (β = −0.09; P = 0.01). Those with higher levels of aldosterone at baseline demonstrated the greatest improvement in executive function (P = 0.014 for the aldosterone effect) and in CO2 vasoreactivity (P = 0.026 for the aldosterone effect) after 12 months of lowering blood pressure (<140/90mm Hg). Plasma renin activity was not associated with any of the measures.
CONCLUSIONS
Higher levels of aldosterone may be associated with decreased cerebrovascular function in hypertension. Those with higher aldosterone levels may benefit the most from lowering blood pressure. The role of aldosterone in brain health warrants further investigation in a larger trial.
Keywords: renin, aldosterone, vasoreactivity, cognition, blood flow velocity hypertension, blood pressure.
Animal studies suggest that the renin–angiotensin–aldosterone system (RAAS) is involved in many central nervous system processes in hypertension, including cognitive function and cerebral hemodynamic control.1–3 Angiotensin II infusion into rats’ brains is associated with impaired memory,4 neurovascular uncoupling, and hypoperfusion.5 In transgenic mice, activation of brain RAAS impairs cognitive function by decreasing cerebral blood flow.6 In humans, the evidence supporting a role of RAAS in brain function is derived indirectly from the observation that drugs that inhibit RAAS may be associated with cognitive preservation.7 To our knowledge, studies showing a relationship between RAAS activity and neurocognitive function in hypertensive older adults are nonexistent.
Aldosterone is the major contributor to the negative cardiovascular effects of RAAS. Higher circulating aldosterone predicts greater increase in blood pressure and incident hypertension with aging.8 In the Framingham cohort, higher aldosterone levels were associated with shorter leukocyte telomere length, suggesting a high inflammation and oxidative stress load.9 Aldosterone has multiple binding sites in the brain.10 However, its role in cognitive and cerebrovascular function has not been fully explored. On the vascular side, higher aldosterone levels are linked with endothelial dysfunction,11,12 which in turn may negatively affect cognitive function.13 While local brain endothelial function is difficult to assess, reactivity to changes in end-tidal CO2 may be an indirect marker of the central endothelium.14 Therefore, we hypothesized that in humans markers of elevated RAAS activity, higher aldosterone levels will be associated with lower scores on measures of cognition (memory and executive function) and impaired cerebral hemodynamics (autoregulation and vasoreactivity).
Prior studies suggest that there is a genetic basis for the individual heterogeneity of vascular response to antihypertensive therapy.15 We previously reported that polymorphisms in the angiotensinogen gene that are associated with higher RAAS activity16 predicted a better cognitive outcome with antihypertensive therapy.17 It is not known if these observations are related to the class of antihypertensive used, such as those that modulate RAAS, or are related to lowering blood pressure control. We therefore wanted to investigate this difference (class effect vs. blood pressure–lowering effect) in a recently completed clinical trial.
Our objective was to assess the association of circulating aldosterone and plasma renin activity (PRA) with cognitive function (memory and executive domains) and cerebral hemodynamics in hypertensive individuals off antihypertensive therapy. Our second objective was to investigate the impact of baseline RAAS markers on the cognitive/hemodynamic responses to antihypertensive therapy and/or controlling blood pressure to <140/90mm Hg.
METHODS
Study design
We analyzed of data from a clinical trial investigating the association between aldosterone and renin levels with cognitive and cerebrovascular measures at baseline while off antihypertensive medications and the impact of baseline aldosterone and renin (measured while off antihypertensive medication) on the response to antihypertensive therapy. The design of the original Antihypertensives and Vascular, Endothelial, and Cognitive Function trial (AVEC trial) was described previously.18 Briefly, this was a 12-month double-blind randomized controlled clinical trial to assess the differential effects of an angiotensin receptor blocker (ARB; candesartan), angiotensin-converting enzyme inhibitor (ACEI; lisinopril), and a diuretic (hydrochlorothiazide (HCTZ)) on cognitive function and cerebral hemodynamics. Inclusion criteria included the following: aged ≥60 years and hypertension (systolic blood pressure (SBP) ≥140mm Hg or diastolic blood pressure (DBP) ≥90mm Hg or receiving antihypertensive medications). Exclusion criteria included the following: dementia; intolerance to the study medications; SBP >200mm Hg or DBP >110mm Hg; elevated serum creatinine or serum potassium levels at baseline; receiving more than 2 antihypertensive medications; congestive heart failure; diabetes mellitus; stroke; and inability to perform the study procedures or unwilling to stop currently used antihypertensive medications. The goal of the intervention was to achieve blood pressure control defined as <140/90mm Hg.
Antihypertensive medications were tapered using a standard protocol described elsewhere.18 Participants were provided with a portable automated blood pressure monitor, and study personnel trained each participant on the use of this monitor. All participants received written instructions on tapering and discontinuation of antihypertensive medications as well as a description of symptoms associated with possible adverse events. They were asked to measure blood pressure 2 times a day (morning and before sleep) and record the readings in a diary. Contact by the study personnel was twice weekly for review of blood pressures. The timeline for medication taper was a 25%–50% decrease during week 1, 50%–75% decrease during week 2, and 100% decrease during week 3. Baseline measurements were performed afterward. Participants with significant blood pressure elevations (SBP >180/100mm Hg) for at least 2 consecutive readings or who developed hypertension-related symptoms were excluded (failed taper), asked to resume their usual dose of antihypertensive medications, and referred back to their primary care physician for chronic blood pressure management as usual. The Hebrew SeniorLife Institutional Review Board approved the study, and all participants provided written informed consent. The study was registered at ClinicalTrials.gov (NCT00605072).
Assessments
Baseline, 6-month, and 12-month assessments included questionnaires regarding social habits, family history, self-reported medical history, a medication inventory, height, and weight. Blood pressure was measured according to the American Heart Association guidelines.19 Two seated blood pressure readings were performed and averaged at each visit. The cognitive assessment included Trail Making Test (TMT) parts A and B, which assesses executive function, and Hopkins Verbal Learning Test, Revised (HVLT), which assesses memory. The cognitive scores that were used included TMT, parts B-A (in seconds), as measures of executive function, and the delayed and recognition scores of HVLT, as measures of memory.
RAAS markers
RAAS markers were measured at baseline (after antihypertensive taper) and at 6 and 12 months after 15–20 minutes of rest. Plasma aldosterone was measured using a radioimmunometric assay (RIA).20 The coat-a-count procedure is a solid phase radioimmunoassay that is based on an aldosterone-specific antibody immobilized to the wall of the polypropylene tube. The 125I-labelled aldosterone competes for a fixed time with aldosterone for antibody sites in the patient sample. Plasma renin activity was measured using competitive binding RIA using the GammaCoat plasma renin activity RIA kit (Stillwater, MN). The determination involves an initial incubation of plasma to generate angiotensin I, followed by quantification of angiotensin I by RIA. The lower limit of the assay was 2ng/dl.
Cerebral blood flow hemodynamics
Cerebral blood flow velocity (BFV) was measured in the middle cerebral artery using transcranial Doppler ultrasonography (2-MHz probe placed over the temporal bone; MultiDop X4, DWL-Transcranial Doppler Systems Inc., Sterling, VA). End-tidal CO2 was measured using a CO2 analyzer (Vacumed, Ventura, CA) attached to a nasal cannula. Mean BFV was measured at rest while sitting, during a 1-minute stand, and during a CO2 challenge (breathing a gas with 8% CO2 for 2 minutes and then mildly hyperventilated to an end-tidal CO2 level of approximately 25mm Hg for 2 minutes) at baseline and at 6 and 12 months. Beat-to-beat heart rate and blood pressure were simultaneously measured using continuous electrocardiogram recording and a Finometer (Finapres Measurement Systems, Arnhem, Netherlands). Data were analyzed offline using Matlab (Mathworks, Natik, MA). We used resting state for the BFV measure (averaged over 5 minutes). Autoregulation was assessed using the change in cerebrovascular resistance from sit-to-stand (Delta-CVR). Vasoreactivity was calculated as the slope for the change in BFV vs. end-tidal CO2. Because CO2 may also affect blood pressure,21 we derived the cerebrovascular conductance measure (cerebral blood flow/mean arterial blood pressure)22,23 and calculated CO2 vasoreactivity as the slope of the change in cerebrovascular conductance vs. the change in end-tidal CO2.23 This measure is a better marker of the vascular response to end-tidal CO2.24
Statistical analysis
Baseline associations between RAAS markers and cognitive or hemodynamic outcomes were explored using multiple linear regressions. Models were adjusted for age, gender, race, education level (for the cognitive outcomes), and body mass index (BMI). All models included both aldosterone and renin as independent variables to account for the feedback loops between aldosterone and renin. We elected to use this method instead of the ratio because it allowed us to compare individual associations of renin and aldosterone with our outcomes yet still adjust for the biological effects of each variable on the other. In contrast, using an independent variable that is a ratio of 2 independent variables in regression analysis may lead to incorrect inferences.25
We divided participants into 3 groups (low, medium, and high) according to their baseline tertiles of aldosterone levels. We then used linear mixed models to compare the changes between the 3 groups in cognitive and cerebral hemodynamic outcomes after 6 and 12 months. We specified an autoregressive covariance structure for the repeated measure due to the correlation between levels of aldosterone for each participant. Using the mixed models, we determined if the change in each tertile group was significant (within-group analysis) and if the changes in the 3 tertile groups were significantly different from each other (between-group analysis). We concluded that there is an effect of baseline aldosterone if the between-group analysis was significant, and we used the within-group analysis to identify the direction of the change in each group (increase or decrease). We performed a similar 3-group classification by baseline PRA. There were no differences in baseline aldosterone level (P = 0.31) or PRA (P = 0.55) by class of antihypertensive medication used (aldosterone: 7.5, 5.5, and 6.7 and PRA: 1.9, 4.4, 2.1 in the lisinopril, candesartan, and HCTZ groups, respectively). There were also no differences in aldosterone or PRA change during the study period (P = 0.06 for change of aldosterone and P = 0.64 for PRA). We therefore combined all 3 randomization groups to generate our results. We used the SAS statistical package (Version 9.2, Carey, NC) for these analyses.
RESULTS
Of the 63 eligible participants, 53 were randomized and 47 (89%) had all measures. Baseline characteristics of the sample are presented in Table 1. Of the 47 participants, 23 (49%) had controlled blood pressure (<140/90mm Hg) prior to entering the study, 46 (98%) had elevated blood pressure after tapering off the antihypertensive medications; 17 were randomized to lisinopril, 17 to candesartan, and 13 to HCTZ. The 3 randomized groups were similar in age, gender, race, education level, cognitive function, and cerebral hemodynamics as reported previously.26 Baseline blood pressure was similar in the 3 randomization groups (lisinopril: 153/85mm Hg; candesartan: 149/81mm Hg; and HCTZ: 155/83mm Hg; P = 0.60 for SBP and P = 0.41 for DBP). Further, SBP reductions were equivalent in all 3 groups (lisinopril: mean reduction ± standard error = 27±5mm Hg; candesartan: mean reduction ± standard error = 26±5mm Hg; and HCTZ: mean reduction ± standard error = 25±6mm Hg; P = 0.93). Blood pressure control (<140/90mm Hg) was 100% for the duration of the trial in the candesartan and HCTZ groups and 91% in the lisinopril group (P = 0.40 for the difference between the groups). Blood pressure control was achieved with only the study drug in the majority of participants (lisinopril for 14 participants, candesartan for 16 participants, and HCTZ for 12 participants). Add-on long-acting nifedipine was needed for 1 participant in the candesartan and HCTZ groups, and 2 participants needed add-on long-acting nifedipine and 1 add-on metoprolol in the lisinopril group.
Table 1.
Baseline characteristics of the study sample (n = 47)
| Measure | Mean (standard deviation) or % |
|---|---|
| Age, y | 71.3 (7.5) |
| Women, % | 60 |
| Black, % | 28 |
| High school education or higher,% | 79 |
| Body mass index, kg/m2 | 28.9 (5.8) |
| Blood pressure and heart rate sitting | |
| Systolic blood pressure, mm Hg | 151.9 (15.3) |
| Diastolic blood pressure, mm Hg | 82.6 (8.4) |
| Heart rate, beats/min | 65.2 (9.7) |
| Cognitive function | |
| Trail Making Test, part A (seconds to complete) | 38.3 (14.7) |
| Trail Making Test, part B (seconds to complete) | 102.1 (45.4) |
| Hopkins Verbal Learning Test, delayed recall | 7.9 (2.9) |
| Hopkins Verbal Learning Test, recognition | 22.6 (1.6) |
| Medical diagnosis | |
| Coronary heart disease, % | 46 |
| Past cancer, % | 19 |
| Osteoarthritis, % | 34 |
| Prestudy antihypertensive class | |
| Diuretics,% | 23 |
| Angiotensin-converting enzyme inhibitor, % | 38 |
| Angiotensin receptor blockers, % | 13 |
| Calcium channel blockers, % | 11 |
| Beta blockers, % | 29 |
| Laboratory | |
| Creatinine, mg/dl | 0.9 (0.3) |
| Estimated glomerular filtration rate,* (ml/min/1.73 m2) | 79.97 (18.43) |
| Serum sodium, mEq/l | 138.6 (2.9) |
| Serum potassium, mEq/ | 4.5 (0.4) |
| Plasma renin activity, ng/ml*hour | 2.8 (7.1) |
| Aldosterone, ng/dl | 6.8 (4.0) |
| Cerebral hemodynamics | |
| Sit-to-stand delta cerebrovascular resistance, mm Hg/cm/sec | −0.37 (0.61) |
| Blood flow velocity, cm/sec | 28.96 (7.37) |
| CO2 vasoreactivity, cm/sec/mm Hg per mm Hg PCO2 | 0.005 (0.003) |
*Using the Modification of Diet in Renal Disease study equation.
At baseline, higher aldosterone levels were associated with lower resting BFV (P = 0.021) and lower CO2 vasoreactivity (P = 0.002). These remained significant after adjusting for age, gender, race, and BMI, as shown in Table 2. Similarly, higher aldosterone levels were associated with smaller changes in CVR upon standing (P = 0.01). The contributions of aldosterone to the variance in hemodynamic measures, as reflected by the partial R2, were 0.16 for BFV, 0.24 for CO2 vasoreactivity, and 0.10 for change in CVR. Aldosterone levels were not associated with cognitive scores at baseline, and PRA was not associated with cognition or cerebral hemodynamics, as shown in Table 2. Plots of hemodynamic measures vs. aldosterone levels are shown in Figure 1.
Table 2.
Multiple regression analyses for the association of aldosterone and plasma renin activity with cognitive or cerebral hemodynamic measures
| Unadjusted | Adjusteda | |||
|---|---|---|---|---|
| Beta (SE) | P value | Beta (SE) | P value | |
| Aldosterone | ||||
| Blood flow velocity | –0.03(0.01) | 0.009 | –0.02(0.01) | 0.037 |
| DELTA-CVR | –0.06(0.03) | 0.046 | –0.09(0.03) | 0.01 |
| CO2 vasoreactivity | –0.11(0.03) | 0.002 | –0.11(0.04) | 0.007 |
| Trail Making Test (B-A) | 1.8(1.57) | 0.25 | 1.59(1.84) | 0.39 |
| HVLT | –0.11(0.12) | 0.37 | –0.11(0.14) | 0.44 |
| Plasma renin activity | ||||
| Blood flow velocity | –0.01(0.01) | 0.32 | –0.01(0.01) | 0.57 |
| DELTA-CVR | 0.03(0.04) | 0.53 | 0.04(0.04) | 0.33 |
| CO2 vasoreactivity | 0.01(0.08) | 0.93 | 0(0.08) | 0.97 |
| Trail Making Test (B-A) | 0.75(0.8) | 0.35 | 0.13(0.83) | 0.88 |
| HVLT | 0.04(0.06) | 0.5 | 0.06(0.06) | 0.36 |
aAdjusted for age, gender, race, and body mass index.
Abbreviations: DELTA-CVR, change in cerebrovascular resistance from sit to stand; HVLT, Hopkins Verbal Learning Test; SE, standard error.
Figure 1.

Scatter plots and fitted regression lines with the 95% confidence interval band of the cerebral hemodynamics vs. aldosterone levels measure at baseline while off routine antihypertensive therapy. Units of measurements are as follows: orthostatic change in cerebrovascular resistance (DEL-CVR), mm Hg/cm/sec; vasoreactivity, cm/sec/mm Hg per mm Hg PCO2; blood flow velocity (BFV), cm/sec. P values are testing the null hypothesis that the slope of the fitted line = 0 using the regression analyses.
Of the 53 randomized participants, 47 completed 6-month evaluations and 31 completed 12-month evaluations. At baseline, SBP and DBP were similar in the 3 aldosterone groups (mean = 153/85mm Hg in the lowest group, 150/81mm Hg in the medium group, and 156/83mm Hg in the high group; P = 0.24 for SBP and P = 0.32 for DBP). During follow-up, there were also no differences in blood pressure between the 3 groups (mean blood pressure at 12 months = 126/70mm Hg in the low group, 124/69mm Hg in the medium group, and 131/74mm Hg in the high group;, P value for SBP/DBP = 0.73/0.92). There was no difference in the blood pressure trends (decline) between the 3 groups (P = 0.93 for the change in SBP and P = 0.63 for the change in DBP between the aldosterone groups).
Participants with the highest level of aldosterone (≥8.2ng/dl; N = 11) at baseline demonstrated the greatest improvement in TMT, part B-A, at 12 months compared with the low (≤4ng/dl; N = 17) and medium (>4 and <8.2; N = 20) levels. This was significant after adjusting for age, gender, race, BMI, and renin levels (P = 0.01 for between-aldosterone group), as shown in Figure 2. Adding SBP to the model did not change the results (P = 0.026 for between-aldosterone group). Those with high levels of aldosterone at baseline also showed an improvement in CO2 vasoreactivity at 12 months; after adjusting for age, gender, race, BMI, and renin levels, P = 0.03 for between-aldosterone groups. Adding SBP to the model also did not alter the results (P = 0.01 for between-aldosterone group). There was no impact of baseline aldosterone levels on BFV (P = 0.47 for between-aldosterone groups) or delta-CVR (P = 0.38). There was no significant effect of baseline aldosterone on measures of memory (HVLT) or of baseline PRA on any cognitive or hemodynamic outcomes. To determine if these effects were related to the type of antihypertensive therapy or to the degree of change in aldosterone level during study period, we conducted an analysis with baseline aldosterone and change in aldosterone over the study period. Only the baseline aldosterone level was associated with the change in TMT, part B-A (P = 0.0097), and CO2 vasoreactivity (P = 0.026).
Figure 2.
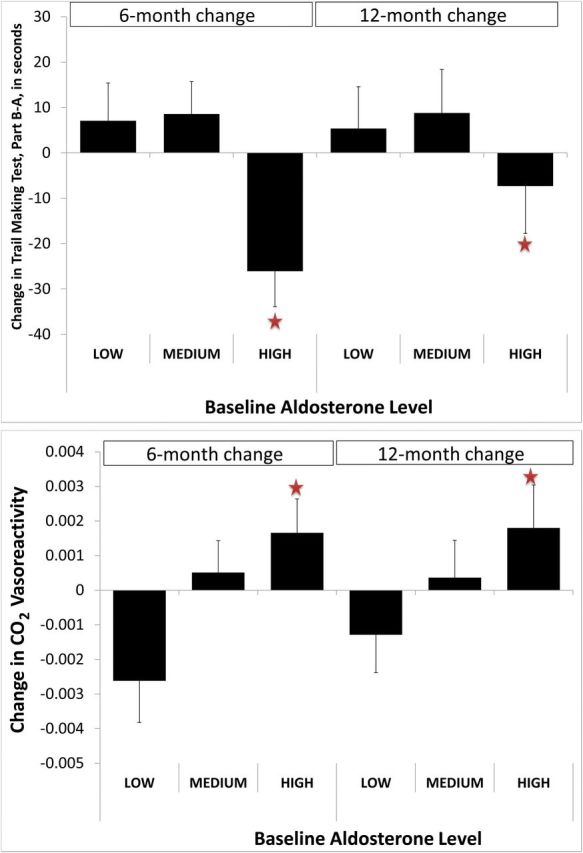
Change at 6 and 12 months relative to baseline in Trail Making Test, part B-A, in seconds, and CO2 vasoreactivity in cm/sec/mm Hg per mm Hg PCO2 by level of aldosterone at baseline while off antihypertensive therapy. Values are the least square adjusted for age, gender, race, body mass index, and renin levels. For Trail Making Test: between-group P value was 0.014; within-group P values were (low = 0.68, medium = 0.43, high = 0.001). For CO2 vasoreactivity: between-group P value was 0.026; within group P values were (low = 0.27, medium = 0.88, high = 0.01).
DISCUSSION
The results from this study suggest that higher levels of aldosterone are associated with lower cerebral BFV, CO2 vasoreactivity, and orthostatic change in CVR. They also suggest that those with high levels (>8ng/dl) of aldosterone before initiating antihypertensive therapy may benefit the most from lowering their blood pressure to <140/90mm Hg with regards to their executive function and CO2 vasoreactivity.
Aldosterone in the brain is scarce but is involved in central mechanisms of hypertension.27 In animals, central infusion of aldosterone elevated blood pressure,28 and infusion of angiotensin II led to impaired endothelial-dependent regulation of the cerebral microcirculation.29 Angiotensin II activates a central aldosterone-mineralocorticoid-receptor–mediated neuromodulation that leads to pressure dysregulation and initiation of hypertension.30 In recent years, a set of neurons in the nucleus tractus solitarii (NTS) in the brainstem was found to have an abundance of the mineralocorticoid receptors.31 NTS is involved in autonomic function including blood flow control via its connection to the carotid body. To our knowledge, our study is the first human study to show a relationship between plasma aldosterone and cerebral autoregulation and CO2 vasoreactivity, which may be explained mechanistically by these animal findings.
Aldosterone has relatively poorer penetration of the blood–brain barrier compared with other steroids.32 Nevertheless, higher expression of the mineralocorticoid receptor in the brain with hypertension increases the sensitivity to the relatively small amount of aldosterone that crosses into the brain.33 Our findings are based on circulating aldosterone and not central levels. This offers support to use of circulating aldosterone in future neurocognitive research, especially that related to vascular function. In addition, aldosterone has a variety of effects on the endothelium in all vascular beds.34 Our finding that higher aldosterone is linked to reduced CO2 vasoreactivity suggests that this is also true in the cerebrovascular bed.
Although we did not identify a cross-sectional association between aldosterone and cognition, we observed that controlling blood pressure to <140/90mm Hg was associated with better executive function and CO2 vasoreactivity changes in those with higher baseline aldosterone levels. This finding is similar to the observation that persons with genetic polymorphisms associated with higher RAAS activity derive greater cognitive improvement with antihypertensive therapy.17 If confirmed in future trials, it may be important clinically to identify patients with increased levels of aldosterone in hypertension, even in the nonpathological ranges, and to ensure that their hypertension is adequately controlled due to the possible advantage of cognitive and cerebrovascular protection. Our finding was specific to executive function, the domain that is highly susceptible to damage from hypertension.
We failed to demonstrate an association between renin and the cognitive and cerebral hemodynamic outcomes. This suggests that in the brain, aldosterone may play a more active role than proximal intermediary products of RAAS. One limitation of this study is the small sample size, and our findings should therefore be interpreted with caution. This is particularly important in the null results with regard to memory and renin. Larger studies are needed to further confirm our findings. We also did not have information about hypertension duration, degree of blood pressure control in our sample prior to study enrollment, and sodium chloride consumption as it may affect RAAS activity. However, our study is a first step toward understanding the role of aldosterone in the brains of hypertensive older adults. It offers translation of evidence for the role of aldosterone in cerebral hemodynamics and cognitive function observed in animal studies. Mineralocorticoid receptor antagonists are readily available. Hence, it is important to investigate the role of aldosterone in brain function as it may offer new modalities of treatment for cerebrovascular and cognitive disorders.
CONCLUSIONS
Higher levels of aldosterone may be associated with impaired cerebral autoregulation and declines in BFV and CO2 vasoreactivity. Higher baseline aldosterone levels may predict greater neurocognitive benefits from controlling blood pressure to below 140/90mm Hg, independently of the type of medication used or further changes in the RAAS markers. A larger study is needed to further investigate the role of aldosterone in brain health and is of critical importance in this arena.
DISCLOSURE
The author declared no conflict of interest.
ACKNOWLEDGMENTS
I.H. and the AVEC trial are supported by grant 1 K23 AG030057 from the National Institute on Aging. L.A.L. was supported by grants AG04390 and AG025037 from the National Institute on Aging. L.A.L. holds the Edyth and Irving S. Usen and Family Chair in Geriatric Medicine at Hebrew SeniorLife, Boston, MA. W.M.’s efforts were supported by SC CTSI (National Institutes of Health/National Center for Advancing Translational Sciences) through grant UL1TR000130.
REFERENCES
- 1. Diz DI, Kasper SO, Sakima A, Ferrario CM. Aging and the brain renin-angiotensin system: insights from studies in transgenic rats. Cleve Clin J Med 2007; 74 Suppl 1:S95–98. [DOI] [PubMed] [Google Scholar]
- 2. Savaskan E. The role of the brain renin-angiotensin system in neurodegenerative disorders. Curr Alzheimer Res 2005; 2:29–35. [DOI] [PubMed] [Google Scholar]
- 3. Wright JW, Harding JW. The brain renin-angiotensin system: a diversity of functions and implications for CNS diseases. Pflugers Arch 2013; 465:133–151. [DOI] [PubMed] [Google Scholar]
- 4. Bonini JS, Bevilaqua LR, Zinn CG, Kerr DS, Medina JH, Izquierdo I, Cammarota M. Angiotensin II disrupts inhibitory avoidance memory retrieval. Horm Behav 2006; 50:308–313. [DOI] [PubMed] [Google Scholar]
- 5. Kazama K, Wang G, Frys K, Anrather J, Iadecola C. Angiotensin II attenuates functional hyperemia in the mouse somatosensory cortex. Am J Physiol Heart Circ Physiol 2003; 285:H1890–H1899. [DOI] [PubMed] [Google Scholar]
- 6. Inaba S, Iwai M, Furuno M, Tomono Y, Kanno H, Senba I, Okayama H, Mogi M, Higaki J, Horiuchi M. Continuous activation of renin-angiotensin system impairs cognitive function in renin/angiotensinogen transgenic mice. Hypertension 2009; 53:356–362. [DOI] [PubMed] [Google Scholar]
- 7. Hajjar I, Keown M, Frost B. Antihypertensive agents for aging patients who are at risk for cognitive dysfunction. Curr Hypertens Rep 2005; 7:466–473. [DOI] [PubMed] [Google Scholar]
- 8. Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, Benjamin EJ, Levy D. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med 2004; 351:33–41. [DOI] [PubMed] [Google Scholar]
- 9. Vasan RS, Demissie S, Kimura M, Cupples LA, Rifai N, White C, Wang TJ, Gardner JP, Cao X, Benjamin EJ, Levy D, Aviv A. Association of leukocyte telomere length with circulating biomarkers of the renin-angiotensin-aldosterone system: the Framingham Heart Study. Circulation 2008; 117:1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geerling JC, Loewy AD. Aldosterone in the brain. Am J Physiol Renal Physiol 2009; 297:F559–F576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hashikabe Y, Suzuki K, Jojima T, Uchida K, Hattori Y. Aldosterone impairs vascular endothelial cell function. J Cardiovasc Pharmacol 2006; 47:609–613. [DOI] [PubMed] [Google Scholar]
- 12. Farquharson CA, Struthers AD. Aldosterone induces acute endothelial dysfunction in vivo in humans: evidence for an aldosterone-induced vasculopathy. Clin Sci (Lond) 2002; 103:425–431. [DOI] [PubMed] [Google Scholar]
- 13. Umemura T, Kawamura T, Umegaki H, Mashita S, Kanai A, Sakakibara T, Hotta N, Sobue G. Endothelial and inflammatory markers in relation to progression of ischaemic cerebral small-vessel disease and cognitive impairment: a 6-year longitudinal study in patients with type 2 diabetes mellitus. J Neurol Neurosurg Psychiatry 2011; 82:1186–1194. [DOI] [PubMed] [Google Scholar]
- 14. Ainslie PN, Murrell C, Peebles K, Swart M, Skinner MA, Williams MJ, Taylor RD. Early morning impairment in cerebral autoregulation and cerebrovascular CO2 reactivity in healthy humans: relation to endothelial function. Exp Physiol 2007; 92:769–777. [DOI] [PubMed] [Google Scholar]
- 15. Wu CK, Luo JL, Tsai CT, Huang YT, Cheng CL, Lee JK, Lin LY, Lin JW, Hwang JJ, Chiang FT. Demonstrating the pharmacogenetic effects of angiotensin-converting enzyme inhibitors on long-term prognosis of diastolic heart failure. Pharmacogenomics J 2010; 10:46–53. [DOI] [PubMed] [Google Scholar]
- 16. Bloem LJ, Manatunga AK, Tewksbury DA, Pratt JH. The serum angiotensinogen concentration and variants of the angiotensinogen gene in white and black children. J Clin Invest 1995; 95:948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hajjar I, Kritchevsky S, Newman AB, Li R, Yaffe K, Simonsick EM, Lipsitz LA. Renin angiotensin system gene polymorphisms modify angiotensin-converting enzyme inhibitors’ effect on cognitive function: the health, aging and body composition study. J Am Geriatr Soc 2010; 58:1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hajjar I, Hart M, Milberg W, Novak V, Lipsitz L. The rationale and design of the antihypertensives and vascular, endothelial, and cognitive function (AVEC) trial in elderly hypertensives with early cognitive impairment: role of the renin angiotensin system inhibition. BMC Geriatr 2009; 9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pickering TG, Hall JE, Appel LJ, Falkner BE, Graves J, Hill MN, Jones DW, Kurtz T, Sheps SG, Roccella EJ. Recommendations for blood pressure measurement in humans and experimental animals: Part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Hypertension 2005; 45:142–161. [DOI] [PubMed] [Google Scholar]
- 20. McKenzie JK, Clements JA. Simplified radioimmunoassay for serum aldosterone utilizing increased antibody specificity. J Clin Endocrinol Metab 1974; 38:622–627. [DOI] [PubMed] [Google Scholar]
- 21. Hetzel A, Braune S, Guschlbauer B, Dohms K. CO2 reactivity testing without blood pressure monitoring? Stroke 1999; 30:398–401. [DOI] [PubMed] [Google Scholar]
- 22. Ringelstein EB, Sievers C, Ecker S, Schneider PA, Otis SM. Noninvasive assessment of CO2-induced cerebral vasomotor response in normal individuals and patients with internal carotid artery occlusions. Stroke 1988; 19:963–969. [DOI] [PubMed] [Google Scholar]
- 23. Bishop CC, Powell S, Insall M, Rutt D, Browse NL. Effect of internal carotid artery occlusion on middle cerebral artery blood flow at rest and in response to hypercapnia. Lancet 1986; 1:710–712. [DOI] [PubMed] [Google Scholar]
- 24. Claassen JA, Zhang R, Fu Q, Witkowski S, Levine BD. Transcranial Doppler estimation of cerebral blood flow and cerebrovascular conductance during modified rebreathing. J Appl Physiol 2007; 102:870–877. [DOI] [PubMed] [Google Scholar]
- 25. Kronmal RA. Spurious correlation and the fallacy of the ratio standard revisited. J R Stat Soc Ser A Stat Soc 1993; 156:379–392. [Google Scholar]
- 26. Hajjar I, Hart M, Chen YL, Mack W, Novak V, C Chui H, Lipsitz L. Antihypertensive therapy and cerebral hemodynamics in executive mild cognitive impairment: results of a pilot randomized clinical trial. J Am Geriatr Soc 2013; 61:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomez-Sanchez EP, Gomez-Sanchez CM, Plonczynski M, Gomez-Sanchez CE. Aldosterone synthesis in the brain contributes to Dahl salt-sensitive rat hypertension. Exp Physiol 2010; 95:120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peysner K, Henry CA, Malvin RL. Central infusion of aldosterone increases blood pressure by mechanisms independent of Na retention. Clin Exp Hypertens A 1990; 12:399–414. [DOI] [PubMed] [Google Scholar]
- 29. Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol 2006; 26:826–832. [DOI] [PubMed] [Google Scholar]
- 30. Scheuer DA. Stimulation of aldosterone synthesis by angiotensin II in the brain: support for positive feedback in hypertension? Hypertension 2013; 62:459–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geerling JC, Kawata M, Loewy AD. Aldosterone-sensitive neurons in the rat central nervous system. J Comp Neurol 2006; 494:515–527. [DOI] [PubMed] [Google Scholar]
- 32. Pardridge WM, Mietus LJ. Regional blood-brain barrier transport of the steroid hormones. J Neurochem 1979; 33:579–581. [DOI] [PubMed] [Google Scholar]
- 33. Pietranera L, Brocca ME, Cymeryng C, Gomez-Sanchez E, Gomez-Sanchez CE, Roig P, Lima A, De Nicola AF. Increased expression of the mineralocorticoid receptor in the brain of spontaneously hypertensive rats. J Neuroendocrinol 2012; 24:1249–1258. [DOI] [PubMed] [Google Scholar]
- 34. Oberleithner H. Aldosterone makes human endothelium stiff and vulnerable. Kidney Int 2005; 67:1680–1682. [DOI] [PubMed] [Google Scholar]