

Abstract
Plasmodium falciparum parasites that are resistant to artemisinins have been detected in Southeast Asia. Resistance is associated with several polymorphisms in the parasite's K13-propeller gene. The molecular epidemiology of these artemisinin resistance genotypes in African parasite populations is unknown. We developed an assay to quantify rare polymorphisms in parasite populations that uses a pooled deep-sequencing approach to score allele frequencies, validated it by evaluating mixtures of laboratory parasite strains, and then used it to screen P. falciparum parasites from >1100 African infections collected since 2002 from 14 sites across sub-Saharan Africa. We found no mutations in African parasite populations that are associated with artemisinin resistance in Southeast Asian parasites. However, we observed 15 coding mutations, including 12 novel mutations, and limited allele sharing between parasite populations, consistent with a large reservoir of naturally occurring K13-propeller variation. Although polymorphisms associated with artemisinin resistance in P. falciparum in Southeast Asia are not prevalent in sub-Saharan Africa, numerous K13-propeller coding polymorphisms circulate in Africa. Although their distributions do not support a widespread selective sweep for an artemisinin-resistant phenotype, the impact of these mutations on artemisinin susceptibility is unknown and will require further characterization. Rapid, scalable molecular surveillance offers a useful adjunct in tracking and containing artemisinin resistance.
Keywords: falciparum malaria, artemisinin resistance, drug resistance, molecular epidemiology
(See the editorial commentary by Sibley on pages 667–9, and the major article by Takala-Harrison et al on pages 670–9.)
Global control of falciparum malaria relies upon the high efficacy of artemisinin combination therapies (ACTs) [1]. ACTs are recommended in nearly all malaria-endemic countries [2]. However, in Southeast Asia, ACT efficacy is threatened by reduced Plasmodium falciparum susceptibility to artemisinins, which clinically manifests as delayed parasite clearance [3]. While thus far restricted to Southeast Asia, this delayed-clearance phenotype is increasingly common [4] and correlates with increases in ACT failures [5]. Delayed clearance is therefore widely considered to be associated with clinically significant P. falciparum artemisinin resistance. The spread of these artemisinin-resistant parasites could undermine artemisinin-based therapies and imperil global malaria control.
The artemisinin-resistant phenotype reported in Southeast Asia has recently been ascribed to mutations in the kelch propeller domains of the parasite gene PF3D7_1343700 (hereafter, the “K13-propeller gene”) [6]. K13-propeller polymorphisms are associated with in vitro parasite survival in the presence of dihydroartemisinin and with delayed clearance in vivo after artemisinin therapy. In Cambodia, where this delayed parasite clearance was first reported, 3 polymorphisms—C580Y, R539T, and Y493H—are prevalent and associated with prolonged parasite half-life after treatment. An additional polymorphism, M476I, was selected in a Tanzanian parasite by cyclic in vitro artemisinin pressure. Recent allelic exchange experiments, in which C580Y was introduced into an artemisinin-susceptible parasite, resulted in in vitro artemisinin resistance [7]. These data support the use of K13-propeller mutant genotypes as a marker for reduced parasite susceptibility to artemisinins [8].
Rapid surveillance of parasite populations for drug resistance can help to inform the selection of drugs by control programs. Across sub-Saharan Africa, ACT for uncomplicated malaria has had high cure rates [9, 10], and parasite clearance is rapid in in vivo studies [11, 12]. As an adjunct to in vivo drug efficacy studies, we recently described a protocol suitable for large-scale molecular surveillance of drug resistance markers that uses deep sequencing pools of parasite isolates by second generation sequencing to quantify P. falciparum alleles [13]. Herein, we report the adaptation and rapid application of this protocol to screen for P. falciparum K13-propeller mutations in >1100 infections from across sub-Saharan Africa. To our knowledge, this is the first survey of K13-propeller polymorphisms in African parasites.
METHODS
Design of Pooled Sequencing Protocol
We first sequenced a fragment of the P. falciparum K13-propeller gene in 22 parasites from Anlong Veng, Cambodia [5], and from the laboratory P. falciparum strain 3D7 (ATCC, Manassas, VA). We used a hemi-nested protocol to amplify a 751-bp region (nucleotides 1279–2030, representing codons 427–676) of PF3D7_1343700, which included described single-nucleotide polymorphisms recently associated with delayed parasite clearance [6]. In the 25-µL first-round reaction, we used 2.5 µL of the Roche FastStart Hi-Fidelity Buffer, 0.5 µL of the Roche FastStart Hi-Fidelity Enzyme, 400 nM of the primers ArtinnerF (GCCTTGTTGAAAGAAGCAGAA) and ArtouterR (CGCCATTTTCTCCTCCTGTA), 10 nM of dNTPs, and 5 µL of template. Cycling conditions were 95°C for 15 minutes, followed by 35 cycles at 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes, with an extension at 72°C for 5 minutes. The second round consisted of the primers ArtinnerF and ArtinnerR (GTGGCAGCTCCAAAATTCAT) and 3 µL of the first-round product as template, with otherwise identical constituents and cycling conditions. Products were sequenced using ABI BigDye Terminator chemistry, aligned to reference PF3D7_1343700 (http://www.plasmodb.org; accessed 12 January 2014), and scored for polymorphisms by using Sequencher (Gene Codes, Ann Arbor, MI). After identification of an isolate (Cam91) containing only the C580Y mutation, we confirmed its monoclonality by genotyping at 8 neutral microsatellite loci [14] and deep sequencing of a polymorphic segment of the P. falciparum circumsporozoite protein [15].
Control Mixture Amplification and Sequencing
We investigated both the sensitivity and precision of minor allele frequency (MAF) estimation by amplifying the K13-propeller gene from mixtures of 3D7 (wild type) and Cam91 (mutant) genomic DNA (gDNA). We first quantified parasite DNA in each specimen, using a real-time polymerase chain reaction (PCR) assay [16], and then prepared 6 dilutions of 3D7:Cam91 gDNA in ratios of 7:1, 15:1, 31:1, 63:1, 127:1, and 255:1; in these dilutions, the C580Y mutation would be expected to be present in proportions of 12.5%, 6.25%, 3.13%, 1.56%, 0.78%, and 0.39%, respectively. From these dilutions, we amplified the target in duplicate reactions and prepared these products for sequencing by acoustic shearing to produce 300-bp fragments. Library preparation was done using the Ion Plus Fragment Library Kit and Ion Xpress Barcode Adaptors (Life Technologies). The final sequencing library consisted of equimolar amounts of the 12 bar-coded libraries and was sequenced on an Ion Torrent 316 chip, using the 400-bp sequencing kit.
Determination of False-Discovery Rate and Setting Minor Allele Frequency Cutoffs
Because both PCR and Ion Torrent sequencing are error prone, mitigating the risk of false discovery is critical. Therefore, we optimized the specificity of minor allele detection by enforcing a suite of nonsequential quality controls on the reads and their constituent bases. First, reads were mapped against the reference sequence by using bowtie2 with default unpaired read settings [17]. Custom python scripts created using pysam were used to generate a pileup (base-wise multiple alignment), filter each base for inclusion, and calculate the final frequency of any detected minor alleles (Figure 1A) [18]. During filtering, we minimized common known artifacts from sequencing or misalignment. Therefore, bases were censored if they were within (1) poorly mapped reads (q < 10); (2) reads of <200 bp; (3) 10 bp of the end of a read, where false mismatches are common; or (4) reads with poor alignment scores to the reference sequence (≤80). Additionally, any minor alleles had to occur on both forward and reverse strands.
Figure 1.
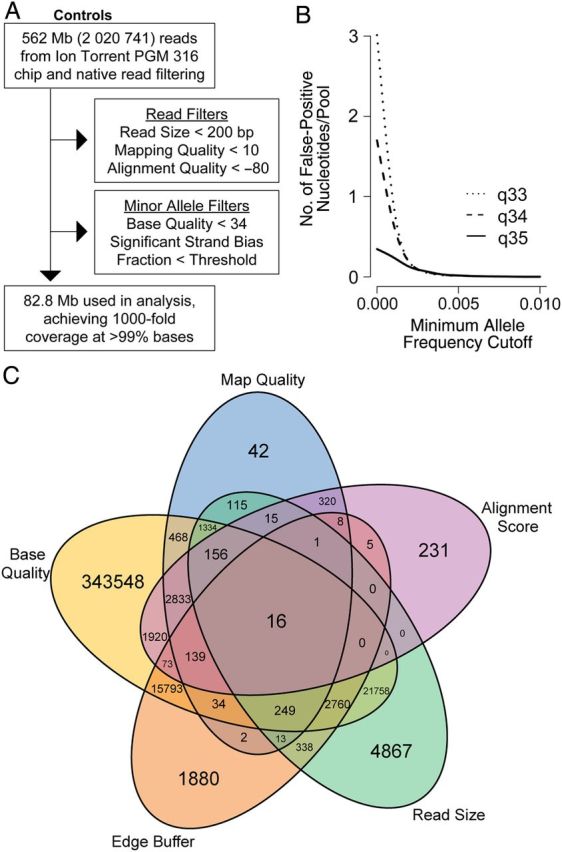
Sequencing results of a mixture of parasite strains 3D7 and Cam91. A, Schematic approach and output of quality-filtering program applied to sequencing reads. B, Kernel density plot of the number of false-positive nucleotides that would be detected across the length of our amplicon, using 3 different minimum Phred base quality cutoffs (q33, q34, and q35) at various minimum percentage minor allele frequency cutoffs. C, Venn diagram of the number of bases censored by the 5 main quality metrics (using the q34 filter) applied to the sequencing reads in panel A.
We examined multiple-base Phred score quality cutoffs, using sequences that were generated from mixtures of parasite isolates 3D7 and Cam91 (Figure 1B). On the basis of this analysis, we selected a high cutoff of 34 for the minimum base quality. Using these criteria, we maintained sufficient base depth to ensure accuracy of the frequency estimates and high specificity by setting MAF cutoffs of 0.5% (for known alleles, based on MalariaGen [19] and work by Ariey et al [6]) or 1.0% (for novel alleles). These minimum frequency cutoffs are similar to those reported for variant discovery in human immunodeficiency virus genomes [20, 21] and, on the basis of our 12 controls, predict an expected false-discovery rate of <0.6 and 0.1 false calls for known and novel alleles, respectively, across all 14 experimental pools.
Amplification and Sequencing of Field Parasites
We made 14 pools of P. falciparum gDNA from 14 different study sites in sub-Saharan Africa collected between 2002 and 2011; each pool comprised between 35 and 273 P. falciparum infections (Supplementary Table 1). To prepare a pool, we combined 3 µL of gDNA from each parasite isolate after extraction from a dried blood spot, using either Chelex-100 or a commercial kit. The K13-propeller fragments were processed as described above and sequenced on an Ion Torrent 318 chip. For Sanger sequencing of selected isolates, we used the PCR, sequencing, and read scoring protocols described above.
Analysis of Mutations
We scanned sequencing reads for the co-occurrence of mutations in a single read because mutant K13-propeller haplotypes in Cambodia uniformly harbored a single coding mutation [6]. We used the pysam module to interrogate our quality-filtered sequencing reads by collating all reads that contributed to minor allele calls at each base position and then identified the pairwise intersect of reads involved for all minor allele calls. To infer the relationships between populations, we used allele frequency estimates at each polymorphic site (n = 23) in each population (n = 14) to calculate pairwise Nei's chord distances (DA) [22], using PowerMarker (v3.25) [23]. The DA-derived distance matrix was used to construct 100 bootstrap replicates from which a consensus tree was computed using MEGA (v6.0) [24] and rendered using APE for R [25]. The analysis was repeated regionally between West Africa (for samples from Gambia, Ghana, Burkina Faso, and 2 populations in Mali), Central Africa (for samples from 4 populations in the Democratic Republic of the Congo), and East Africa (for samples from Uganda, Tanzania, Malawi, and 2 populations in Kenya).
Ethics
Original studies were approved by their respective governing ethics boards (Supplementary Table 1). Molecular testing of parasites was approved by the University of North Carolina.
RESULTS
Determination of False Allele Discovery Rate
In the analysis of known control mixtures of the 2 clonal lines, the initial 2 020 741 reads were filtered for quality and artifacts to maximize specificity while maintaining sensitivity and accuracy (Figure 1A and “Methods” section). Individual base quality had the greatest impact on coverage and specificity of minor allele detection (Figure 1C). Therefore, we assessed 3 levels of base quality filtering in conjunction with all other filtering. To quantify the false-positive rates for each base quality level, we determined the number of falsely detected alleles exceeding a given MAF cutoff across all 12 control reactions and determined the per base rate by dividing this number by the total number of nucleotides sequenced. This per base rate was multiplied by the length of the amplicon to determine the number of false alleles likely detected in a single experimental pool (Figure 1B). Specificity increased with increasing quality threshold, but there was minimal gain in specificity with q35, and base depth declined significantly, impacting allele frequency estimation and sensitivity and leading us to select q34 as our experimental filter threshold. Based on q34 error rates, the MAF cutoffs of 0.5% for known alleles and 1.0% for unknown alleles were chosen for maximum specificity so that <1 false positive would be expected across all 14 experimental pools. Also, in the controls, all observed false positives between the 0.5% and 1% cutoffs were at or near the 0.5% cutoff, suggesting an absence of high-frequency PCR errors. In the end, 1 996 507 aligned reads containing 82 787 771 bases successfully passed our quality filters, resulting in >1000-fold coverage at >99% of bases among our controls.
Quantification of Allele Frequencies in Laboratory Mixtures
From the control mixtures, the observed frequencies of the C580Y allele were closely correlated with expected frequencies, with correlation coefficients of >0.99 and a regression line with a R2 value of 0.98 (Figure 2). In the template mixture with an expected frequency of C580Y of 0.39%, the observed frequencies were 0.33% and 0.24%, indicating that the sequencing and read analysis quantified MAF accurately and did so at low allele frequencies.
Figure 2.

Expected and observed frequencies of the C580Y substitution in mixtures of parasite strains 3D7 (C580) and Cam91 (580Y). Gray diamonds are proportions of alleles in 2 independent replicates for each mixture; the solid line is a linear regression line fitted to these points, with the shaded area indicating the error estimate for the line (R2 = 0.98). The horizontal lines indicate the minimum percentage minor allele cutoffs that were ultimately used for known (dotted) and novel (dashed) loci.
P. falciparum K13-propeller Mutations in Field Parasites
We next applied the described sequencing approach to parasite isolates collected in 14 studies across sub-Saharan Africa (Supplementary Table 1). After applying the quality filters described above, we analyzed 115 463 298 bases and again achieved >1000-fold coverage at >99% of bases in the amplified region (Supplementary Figure 1); for these analyses, we used our empirically derived cutoffs (see “Methods and Discussion” sections) to censor MAFs below 0.5% at previously described polymorphic loci and below 1% in previously unknown polymorphic loci.
We observed 23 polymorphisms in 21 codons in the 14 parasite populations: 14 were nonsynonymous mutations, 8 were synonymous mutations, and 1 encoded a premature stop codon (Table 1). Six mutations have been described previously: 5 are in the MalariaGen database, and 1 was reported by Ariey et al [6]. By K13-propeller blade, blades 3 (n = 6) and 5 (n = 4) contained the greatest number of coding substitutions (Table 1). Parasite populations harbored 1–5 polymorphisms; this number did not correlate with the number of parasite isolates in a pool (correlation coefficient = 0.30).
Table 1.
P. falciparum K13-propeller Polymorphisms Observed in 14 Sub-Saharan African Sites
| K13-propeller Blade, Amino Acid Locus | Nucleotide Locus | Reference Allele | Mutant Allele | West Africa |
Central Africa |
East Africa |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Basse, Gambia | Kita, Mali | San, Mali | Navrongo, Ghana | Ziniaré, Burkina Faso | Bas-Congo, DRC | Kinshasa, DRC (2006) | Kinshasa, DRC (2007) | Bandundu, DRC | Tororo, Uganda | Kisumu, Kenya | Siaya, Kenya | Fukayosi, Tanzania | Machinga, Malawi | ||||
| Blade 1 | |||||||||||||||||
| G449D | 1346 | G | A | … | … | 5.32 | … | … | … | … | … | … | … | … | … | … | … |
| C469Ca | 1407 | C | T | … | … | … | … | … | … | … | … | … | … | 1.24 | … | … | 0.87 |
| W470X | 1410 | G | A | … | … | 5.03 | … | … | … | … | … | … | … | … | … | … | … |
| R471R | 1413 | T | C | … | … | … | … | … | … | … | 9.79 | … | … | … | … | … | … |
| Blade 2 | |||||||||||||||||
| G496Ga | 1488 | T | C | … | 5.77 | 0.78 | … | … | … | … | 1.64 | … | … | … | … | … | … |
| R513R | 1539 | T | C | … | … | … | … | … | … | … | 1.15 | … | … | … | … | … | … |
| V520A | 1559 | T | C | 2.14 | 1.65 | 1.31 | 1.74 | 2.05 | 2.11 | 1.83 | 2.19 | 3.32 | … | 2.18 | 1.93 | 2.07 | 2.03 |
| S522Ca | 1564 | A | T | … | … | … | … | … | … | … | … | … | 2.57 | … | … | … | … |
| Blade 3 | |||||||||||||||||
| C542Y | 1625 | G | A | … | … | … | … | 1.01 | … | … | … | … | … | … | … | … | … |
| G544R | 1630 | G | A | … | … | … | 1.31 | … | … | … | … | … | … | … | … | … | … |
| G545E | 1634 | G | A | … | 3.17 | … | … | … | … | … | … | … | … | … | … | … | … |
| P553La | 1658 | C | T | … | … | … | … | … | … | … | … | … | … | 0.53 | … | … | 0.59 |
| P553P | 1659 | G | A | … | 4.78 | … | … | … | … | … | … | … | … | … | … | … | … |
| A557S | 1669 | G | T | … | … | … | … | … | 36.31 | … | … | … | … | … | … | … | … |
| R561C | 1681 | C | T | … | … | 1.14 | … | … | … | … | … | … | … | … | … | … | … |
| Blade 4 | |||||||||||||||||
| A578Sb | 1732 | G | T | … | … | … | … | … | … | … | … | … | 1.03 | … | … | … | … |
| V581V | 1743 | T | C | … | … | … | … | … | 7.09 | … | … | … | … | … | … | … | … |
| V589V | 1767 | C | T | … | … | … | … | … | … | … | … | 1.21 | … | … | … | … | … |
| Blade 5 | |||||||||||||||||
| A617T | 1849 | G | A | … | … | … | … | … | 1.75 | … | … | … | … | … | … | … | … |
| A617V | 1850 | C | T | … | 1.31 | … | … | … | … | … | … | … | … | … | … | … | … |
| V637A | 1910 | T | C | … | … | … | … | … | 3.22 | … | … | … | … | … | … | … | … |
| G638R | 1912 | G | A | 1.72 | … | … | … | 2.21 | … | … | … | … | … | … | … | … | … |
| Q654Qa | 1962 | A | G | … | … | … | … | … | … | … | … | … | … | … | … | … | 0.52 |
Data are the proportion of reads within each population that harbored the indicated mutation. Minor alleles were censored below 1% for novel loci and below 0.5% for known polymorphic loci. Empty cells indicate mutations that were not observed.
Abbreviation: DRC, Democratic Republic of the Congo.
a Polymorphisms previously reported in the MalariaGen beta release of the Plasmodium falciparum Community Project [19].
b Substitutions previously observed by Ariey et al [6] in polymerase chain reaction–amplified Cambodian parasites.
Notably, the C580Y, R539T, or Y493H substitutions that were associated with delayed parasite clearance in Southeast Asia were not observed, nor was the M476I mutation that was selected in vitro in a Tanzanian parasite [6]. The single polymorphism we observed that was reported by Ariey et al [6] was P553L, present in low frequencies in our pools from Kisumu, Kenya, and Machinga, Malawi. In San, Mali, we also observed 2 alternate substitutions at residues that were mutated in Cambodian parasites: G449D (A in Cambodia [6]) and R561C (H in Cambodia [6]).
Population Genetics of K13-propeller Mutations
Eighteen of the 23 mutations that we observed were private alleles restricted to single parasite populations. These 18 alleles included the 3 mutations that achieved the highest site frequencies: A557S at 36.3% in Bas-Congo, Democratic Republic of the Congo; R471R at 9.8% in Kinshasa, Democratic Republic of the Congo; and V581V at 7.1% in Bas-Congo. Only 5 alleles were shared between sites: the V520A substitution was most prevalent at 13 sites at frequencies of 1.3%–3.3%, and other shared alleles were present at a maximum of 3 sites.
We confirmed the presence of the A557S allele in Bas-Congo, using bidirectional Sanger sequencing. The A557S mutation was present in 1 of 13 isolates selected randomly from the original pool of 50.
In Cambodian parasites, K13-propeller genes did not contain >1 coding mutation [6]. Nine of our parasite populations harbored >1 coding substitution (Table 1). We interrogated the quality-filtered sequencing reads from these sites, and, similar to results from Cambodia, coding substitutions did not co-occur in reads spanning 2 polymorphic loci in 8 of the parasite populations; only in Bas-Congo did a small fraction of reads that spanned residues 557 and 617 harbor coding substitutions at both loci. However, we cannot exclude the possibility of chimeric sequencing reads, owing to the limited number of reads that spanned this 180-bp segment (n = 101) and the frequencies of the potential parental haplotypes. Therefore, our data largely support the notion that mutant K13-propeller haplotypes harbor only single coding mutations in the sequenced segment.
The geographic prevalences of coding mutations, silent mutations, and wild-type allele proportions suggested that polymorphisms were less frequent in East than in Central or West Africa (Figure 3). We explored the genetic relationships between K13-propeller alleles within the parasite populations, using a population genetic allele-based clustering algorithm. In this analysis, there was no significant differentiation of mutant alleles between parasite populations. Between 3 populations—Fukayosi, Tanzania, Siaya, Kenya, and Kinshasa (2006)—the pairwise DA values were 0 (indicating no genetic differentiation), and the greatest distance (between Bas-Congo and Kita, Mali) was only 0.0146. Furthermore, a neighbor-joining tree computed with these distances demonstrated no geographic clustering of alleles between populations, suggesting no significant spread of K13 polymorphisms (Supplementary Figure 2). Similar low genetic distances were seen when analyzed on a regional scale.
Figure 3.

Epidemiology of Plasmodium falciparum K13-propeller mutations in 14 sub-Saharan African sites. Proportions within each geographic site of wild-type alleles, coding mutations, and silent mutations. Proportions calculated with the assumption that each K13-propeller haplotype contained only a single mutation, as observed here and in Cambodia [6]. Abbreviation: DRC, Democratic Republic of the Congo.
DISCUSSION
Our survey of K13-propeller polymorphisms in P. falciparum captured a diversity of mutations across and within sub-Saharan African parasite populations. Notably, we did not detect any polymorphisms that are clearly associated with either artemisinin resistance in Cambodian parasites or with artemisinin tolerance in vitro, and we observed only 1 polymorphism that was shared with those previously reported from Cambodia. Overall, most polymorphisms encoded nonsynonymous substitutions but were present at low frequencies and restricted to single geographic sites; this allelic heterogeneity does not suggest directional selection resulting from drug pressure. In the short term, the absence of known molecular markers of artemisinin resistance augurs favorably for the antimalarial efficacy of ACTs in Africa. However, it is difficult to predict how soon resistance mutations may appear in Africa, and molecular surveillance can provide a framework to rapidly monitor for the emergence or importation of resistance alleles.
Ariey et al [6] reported that (1) prolonged parasite survival ex vivo was associated with the Y493H, I543T, R539T, and C580Y mutations; (2) in vivo delayed parasite clearance was associated with the Y493H, R539T, and C580Y mutations; and (3) artemisinin tolerance in vitro was associated with the M476I mutation, which also indicated that K13-propeller mutations can produce an artemisinin-resistance phenotype in vitro with an African parasite genetic background. A subsequent report supports the causal role of K13-propeller mutations in conferring resistance, because the introduction to a susceptible parasite of the C580Y substitution prolonged the survival of the parasite in vitro in the presence of artemisinin [7]. We observed none of these 5 mutations in our survey of >1000 African parasite samples. Furthermore, we observed only 1 K13-propeller mutation (P553L) that was previously reported from Cambodia, and this mutation was rare both in Cambodia and in Africa. The paucity of shared mutant alleles between African and Cambodian parasites suggests that there exists a large reservoir of K13-propeller polymorphisms globally.
Despite the allelic diversity of the K13-propeller gene, our data do not suggest widespread selection for an artemisinin resistance polymorphisms in Africa at the time these samples were collected. Such a process would be expected to increase frequencies of mutations that confer resistance and, concomitantly, to reduce allelic heterogeneity through linkage disequilibrium. For other antimalarials, the loss of allelic diversity at the locus mediating resistance has accompanied the geographic spread of P. falciparum resistance to chloroquine [26], pyrimethamine [27], and sulfadoxine [28]. In contrast, in our data, mutations did not appear to cluster geographically, likely owing to the paucity of allele sharing between parasite populations. Only 5 of 23 polymorphisms were shared between any 2 sites, and although 1 polymorphism (V520A) was detected in 13 of 14 populations, the other 4 were present in a maximum of 3 sites. Additionally, in the parasite populations harboring the highest frequencies of individual coding mutations (from Bas-Congo province and San), we observed at least 4 other polymorphisms, suggesting that the high frequency of coding mutations was not coincident with a loss of other polymorphisms. These observations appear analogous to data from Cambodia, where it appears that minor alleles were abundant but regionally circumscribed prior to recent replacement by artemisinin resistance mutations. Because these mutant alleles have unknown phenotype consequences, we cannot exclude the possibility that the observed allele frequencies represent incipient sweeps, and further studies are needed to better characterize this variation.
The function of the K13-propeller of P. falciparum is largely unknown, and direct predictions of function are precluded by the diverse functions of kelch-containing proteins in other species. Within the K13-propeller gene, mutations clustered in domains similar to those reported from Cambodia, including those associated with resistance in domains 2, 3, and 4 (Table 1). Two coding substitutions are particularly notable. First, the A557S substitution achieved a high frequency (36.3%) in Bas-Congo province but was absent in other contemporary parasite populations, including those from neighboring sites in Kinshasa and Bandundu province. This mutation was confirmed using Sanger sequencing of individual isolates. The phenotype of this mutation is unknown, but given its high frequency and position in a K13-propeller domain that harbored 2 artemisinin resistance substitutions (R539T and I543T) in Cambodia parasites, we suggest it merits further characterization. Second, in San, we observed a nonsense mutation at codon 470 in 5% of reads. Presumably, a K13-propeller protein truncated in the first propeller domain would manifest a substantial loss of function in the parasite; such mutations have been observed in the kelch-containing human gene KEAP1 and have been associated with lung carcinoma [29], suggesting biological plausibility. This unusual polymorphism also merits further characterization.
The World Health Organization's Global Plan for Artemisinin Resistance Containment prioritizes the monitoring of ACT efficacy to detect resistance [30], and this is most credibly quantified using clinical efficacy data. This approach may be limited by resource constraints [31]. Resource-intensive studies might be prefaced by molecular surveillance to detect foci of parasites bearing resistance genotypes that would facilitate targeting of in vivo and ex vivo investigations. Such molecular surveillance would ideally include efficient and scalable methods to quantify parasite alleles.
Our protocol using pooled second-generation sequencing is sensitive, inexpensive, and scalable. In just over 2 months, we designed the protocol, piloted it by evaluating laboratory parasites, and applied it to field parasites to generate K13-propeller allele frequencies from >1100 parasites. Traditional genotyping approaches may not be suitable to detect K13-propeller polymorphisms [8]. The polymorphisms associated with an artemisinin-resistant phenotype are numerous, unlinked, and occur on a background of allelic diversity. These facts may technically undermine traditional approaches that only interrogate specific alleles. Because several independent polymorphisms can produce the artemisinin-resistant phenotype, ongoing surveillance will need to detect the appearance of new mutations, shifts in frequencies of existing African variants, and the importation of known Asian resistance mutations. Although our pooled approach does not allow direct investigations of individual clinical or molecular phenotypes, it can easily be followed by K13-propeller gene resequencing to enable more-comprehensive surveillance of the emergence or importation of resistance genotypes.
Our study had several limitations. Although studies of defined parasite mixtures accurately quantified allele frequencies, frequency estimates in the field isolates may have been biased by the varied densities of the input parasites. Nevertheless, in a prior study using a similar protocol, allele frequencies were closely correlated with those obtained by Sanger sequencing of individual isolates [13]. Because both PCR enzymes and second-generation sequencing introduce nucleotide errors, many reads contained errors that could have falsely identified minor alleles. To mitigate this risk, we (1) used high-fidelity polymerase, (2) empirically designed a quality-assurance algorithm by using our validation sequencing reactions of clonal parasite lines to minimize false discovery of alleles, and (3) used this algorithm for reads obtained from field parasites.
In this first large-scale survey of P. falciparum molecular markers of artemisinin resistance, we found no previously identified artemisinin resistance mutations in contemporary African parasite populations. Although the overall frequency of wild-type K13-propeller genes was high, we identified numerous novel coding substitutions that are of unclear phenotype. A better understanding of the biological and clinical impact of these genotypes will require coordinated clinical and molecular genetic investigations. Complementing these studies with ongoing, large-scale molecular epidemiologic surveillance will enhance our ability to monitor artemisinin resistance. Together, these integrated efforts may help forestall the spread of resistance and enhance the global durability of artemisinin therapies.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Kyaw L. Thwai and Oksana Kharabora (University of North Carolina, Chapel Hill), for their assistance in the laboratory; Dr Augustin Okenge (Programme National de Lutte Contre de SIDA, Kinshasa, Democratic Republic of the Congo) and Dr Jeremie Mwonga (Laboratoire National de Reference SIDA et IST, Kinshasa), for help in obtaining access to samples from the Democratic Republic of the Congo; MR4, for providing us with the 3D7 strain of P. falciparum that was originally contributed by D. J. Carucci; Drs David Saunders and Chanthap Lon (Armed Forces Research Institute of Medical Sciences, Bangkok, Thailand), for the isolate Cam91; Abraham Hodgson and Stephen Quaye (Navrongo Health Research Center, Ghana), for their efforts in the coordination of the malaria in pregnancy trial in Ghana and the collection of samples, respectively; Dr Brenda Temple (R. L. Juliano Structure Bioinformatics Core, University of North Carolina), for assistance with the protein model; Alfredo Mayor (Barcelona Center for International Health Research, Spain), for helpful review of the manuscript; and the hundreds of participants in the original clinical studies.
Disclaimer. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. The funders had no role in the analysis of data, preparation of the manuscript, or the decision to seek publication.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH; grants K08AI100924 [to S. M. T.], R01AI089819 [to J. J. J.], R56AI097909 [to S. R. M.], and U19AI089674), the NIH (KL2RR031981 to J. A. B. and training grant T32GM0088719 to C. M. P.), and the Malaria in Pregnancy Consortium, which is funded through a grant from the Bill and Melinda Gates Foundation to the Liverpool School of Tropical Medicine (grant 46099 to F. O. t. K.). Additional sources of funding for the original clinical studies are specified in Supplementary Table 1.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol. 2009;7:864–74. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. Geneva: World Health Organization; 2012. World malaria report 2011. [Google Scholar]
- 3.Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phyo AP, Nkhoma S, Stepniewska K, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–6. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lon C, Manning JE, Vanachayangkul P, et al. Efficacy of two versus three-day regimens of dihydroartemisinin-piperaquine for uncomplicated malaria in military personnel in northern Cambodia: an open-label randomized trial. PLoS One. 2014;9:e93138. doi: 10.1371/journal.pone.0093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ariey F, Witkowski B, Amaratunga C, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol. 2014;32:819–21. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- 8.White NJ. Malaria: a molecular marker of artemisinin resistance. Lancet. 2014;383:1439–40. doi: 10.1016/S0140-6736(14)60656-5. [DOI] [PubMed] [Google Scholar]
- 9.Dorsey G, Staedke S, Clark TD, et al. Combination therapy for uncomplicated falciparum malaria in Ugandan children: a randomized trial. JAMA. 2007;297:2210–9. doi: 10.1001/jama.297.20.2210. [DOI] [PubMed] [Google Scholar]
- 10.A head-to-head comparison of four artemisinin-based combinations for treating uncomplicated malaria in African children: a randomized trial. PLoS Med. 2011;8:e1001119. doi: 10.1371/journal.pmed.1001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopera-Mesa TM, Doumbia S, Chiang S, et al. Plasmodium falciparum clearance rates in response to artesunate in Malian children with malaria: effect of acquired immunity. J Infect Dis. 2013;207:1655–63. doi: 10.1093/infdis/jit082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maiga AW, Fofana B, Sagara I, et al. No evidence of delayed parasite clearance after oral artesunate treatment of uncomplicated falciparum malaria in Mali. Am J Trop Med Hyg. 2012;87:23–8. doi: 10.4269/ajtmh.2012.12-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor SM, Parobek CM, Aragam N, et al. Pooled deep sequencing of Plasmodium falciparum isolates: an efficient and scalable tool to quantify prevailing malaria drug-resistance genotypes. J Infect Dis. 2013;208:1998–2006. doi: 10.1093/infdis/jit392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vinayak S, Alam MT, Mixson-Hayden T, et al. Origin and evolution of sulfadoxine resistant Plasmodium falciparum. PLoS Pathog. 2010;6:e1000830. doi: 10.1371/journal.ppat.1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey JA, Mvalo T, Aragam N, et al. Use of massively parallel pyrosequencing to evaluate the diversity of and selection on Plasmodium falciparum csp T-Cell epitopes in Lilongwe, Malawi. J Infect Dis. 2012;206:580–7. doi: 10.1093/infdis/jis329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rantala AM, Taylor SM, Trottman PA, et al. Comparison of real-time PCR and microscopy for malaria parasite detection in Malawian pregnant women. Malar J. 2010;9:269. doi: 10.1186/1475-2875-9-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium M. P. falciparum Community Project. 2014. http://www.malariagen.net/apps/pf/2.0/ Accessed 10 March 2014.
- 20.Vandenhende MA, Bellecave P, Recordon-Pinson P, et al. Prevalence and evolution of low frequency HIV drug resistance mutations detected by ultra deep sequencing in patients experiencing first line antiretroviral therapy failure. PLoS One. 2014;9:e86771. doi: 10.1371/journal.pone.0086771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibson RM, Meyer AM, Winner D, et al. Sensitive deep sequencing-based HIV-1 genotyping assay to simultaneously determine susceptibility to protease, reverse transcriptase, integrase, and maturation inhibitors, as well as HIV-1 coreceptor tropism. Antimicrob Agents Chemother. 2014;58:2167–85. doi: 10.1128/AAC.02710-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol. 1983;19:153–70. doi: 10.1007/BF02300753. [DOI] [PubMed] [Google Scholar]
- 23.Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21:2128–9. doi: 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 24.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 26.Wootton JC, Feng X, Ferdig MT, et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–3. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- 27.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- 28.Pearce RJ, Pota H, Evehe MS, et al. Multiple origins and regional dispersal of resistant dhps in African Plasmodium falciparum malaria. PLoS Med. 2009;6:e1000055. doi: 10.1371/journal.pmed.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.WHO. Geneva: World Health Organization; 2011. Global plan for artemisinin resistance containment. [Google Scholar]
- 31.Vestergaard LS, Ringwald P. Responding to the challenge of antimalarial drug resistance by routine monitoring to update national malaria treatment policies. Am J Trop Med Hyg. 2007;77:153–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.