

Abstract
Since the first reports in early 2013, >440 human cases of infection with avian influenza A(H7N9) have been reported including 122 fatalities. After the isolation of the first A(H7N9) viruses, the nucleotide sequences became publically available. Based on the coding sequence of the influenza virus A/Shanghai/2/2013 hemagglutinin gene, a codon-optimized gene was synthesized and cloned into a recombinant modified vaccinia virus Ankara (MVA). This MVA-H7-Sh2 viral vector was used to immunize ferrets and proved to be immunogenic, even after a single immunization. Subsequently, ferrets were challenged with influenza virus A/Anhui/1/2013 via the intratracheal route. Unprotected animals that were mock vaccinated or received empty vector developed interstitial pneumonia characterized by a marked alveolitis, accompanied by loss of appetite, weight loss, and heavy breathing. In contrast, animals vaccinated with MVA-H7-Sh2 were protected from severe disease.
Keywords: Influenza A(H7N9) virus, vaccine, Modified Vaccinia virus Ankara
In spring 2013, avian influenza A(H7N9) caused an outbreak of severe respiratory illness among humans in China. These viruses reemerged during winter 2013–2014 to cause a second outbreak [1–3]. So far, >440 human cases have been reported, of which 122 had a fatal outcome [4]. Based on the absence of a multibasic cleavage site in the viral hemagglutinin (HA) protein and the phenotype of the virus in the intravenous pathogenicity index test, the A(H7N9) virus is categorized as having low pathogenicity [5]. Despite the low-pathogenicity phenotype, the virus is able to attach to the lower respiratory tract in humans, and A(H7N9)-infected patients present at the hospital with severe respiratory illness [1, 6]. Furthermore, it was demonstrated that intratracheal inoculation of ferrets with an influenza A(H7N9) virus causes severe bronchointerstitial pneumonia [7]. More widespread circulation of these viruses is feared, and therefore there is interest in the development of vaccines that could prevent infection or mitigate disease severity.
The conventional production of such (pre)pandemic vaccines fell short during the last pandemic, involving influenza A(H1N1) [8, 9]. The evaluation of (pre) pandemic vaccines against avian influenza virus A(H5N1) and A(H7N9) indicated that these vaccines are poorly immunogenic and that adjuvants are required to improve their immunogenicity [10, 11]. However, the use of adjuvants may have some limitations and adverse side effects [12].
These issues underscore the need for new production platforms with the capacity to produce large quantities of efficacious vaccines in a short period [13]. So far, various H7 vaccine candidates have been evaluated for their immunogenicity and protective capacity, with variable success [14–22]. Recombinant DNA technology may facilitate the production of purified recombinant viral proteins, DNA vaccines, but also viral vectors, such as modified vaccinia virus Ankara (MVA) [18–21, 23].
MVA is a highly attenuated and replication-deficient orthopoxvirus that is biologically and genetically well characterized, has successfully been developed as a next-generation smallpox vaccine, and has been evaluated as a viral vector for numerous prophylactic and therapeutic vaccines [24, 25]. Recombinant MVA has also been tested extensively as a vaccine candidate against various influenza viruses [26]. Recently, it was shown that influenza A(H5N1) vaccines based on the MVA technology were safe and immunogenic in mice, chickens, and macaques. Currently, a prototype MVA-H5 vaccine is under clinical development in a phase 1/2a clinical trial [27–31]. In addition, MVA-based universal influenza vaccines that elicit heterosubtypic immunity are in various stages of development [32–35].
Here we evaluate the first A(H7N9) viral vector vaccine, based on MVA, in a ferret model for influenza A(H7N9) virus–induced pneumonia. The immunogenicity and protective capacity of the vaccine was investigated after 1 and 2 immunizations. To assess the possibility for dose sparing, a 100-fold lower vaccine dose was also investigated.
MATERIAL AND METHODS
Vaccine Construction
The HA gene sequence of influenza virus A/Shanghai/2/2013 (GISAID accession number: EPI439502; H7-Sh2) underwent codon optimization for stable insertion and expression in the context of MVA. Silent mutations were introduced in H7-Sh2 to remove runs of ≥4 G/C and stop signal sequences (TTTTTNT) for early MVA-specific transcription. The tailor-made H7-Sh2 sequence with HpaI and NotI restriction sites added to the 5′ and 3′ termini was obtained as a synthetic gene (Baseclear, Leiden, the Netherlands) and cloned into the MVA vector plasmid pMKIIIred [36] under control of the synthetic promoter psynII and containing the mCherry sequence as a marker gene, resulting in pMKIIIred-H7 (Figure 1) [37]. Subsequently, chicken embryo fibroblasts (CEFs) cultured under serum-free conditions in VP-SFM medium (Life Technologies, Breda, the Netherlands) were infected with MVA and transfected with pMKIIIred-H7 DNA, using Fugene HD (Promega, Leiden, the Netherlands) to generate recombinant MVA containing the H7-Sh2 sequence and mCherry as a fluorescent marker. Recombinant viruses were clonally isolated in plaque passages on CEF screening for foci of red fluorescent cells. Finally, the mCherry marker gene was removed from the viral genomes by means of a second step of intragenomic homologous recombination, resulting in the final marker-free recombinant MVA-H7-Sh2. To generate vaccine preparations, MVA-H7-Sh2 was purified by ultracentrifugation through sucrose, resuspended in physiological saline, and stored at −80°C. Virus amplifications, titrations, and quality control experiments were performed as described previously [36]. Expression of the H7 protein was confirmed using immunocytochemical and Western blot analyses as described previously, using hyperimmune rabbit serum against influenza virus A/Seal/Massachussets/1/80 (Figure 1F) [36].
Figure 1.

Construction of the modified vaccinia virus Ankara (MVA) H7-Sh2 vector (A–E) and H7 protein expression (F). The hemagglutinin (HA) gene sequence of influenza virus A/Shanghai/2/2013 (H7-Sh2) underwent codon optimization, obtained as synthetic gene (A), and cloned between the flank 2 and flank 1 regions of the MVA vector plasmid pMKIIIred under control of the PsynII promoter and containing the mCherry sequence as a marker gene, resulting in pMKIIIred-H7 (B). Subsequently, chicken embryo fibroblasts (CEFs) were infected with MVA (C) and transfected with pMKIIIred-H7 DNA, using Fugene HD (Promega, Leiden, the Netherlands) to generate recombinant MVA containing the H7-Sh2 sequence and mCherry as a fluorescent marker, inserted in the MVA genome through homologous recombination (D). Finally, the mCherry marker gene was removed from the viral genome by means of a second step of intragenomic homologous recombination, resulting in the final marker-free recombinant MVA-H7-Sh2 (E). H7 protein expression was confirmed by Western blot analysis of cell lysates from MVA-H7-Sh2–infected baby hamster kidney cells (BHK-21), using a hyperimmune rabbit serum against influenza virus A/Seal/Massachussets/1/80 and a goat anti-rabbit IRDye Infrared antibody (Westburg, Leusden, the Netherlands. F, Lane c, negative control; lane 1, MVA-H7-Sh2 1:50; lane 2, MVA-H7-Sh2 1:10; lane 3, MVA-H7-Sh2 undiluted).
Animals
Twenty-eight healthy female ferrets (Mustela putorius furo) approximately 12 months old were used and assigned to experimental groups as indicated below. Before use, the absence of antibodies against Aleutian disease virus and seasonal influenza viruses was confirmed. The animals were housed under standard conditions, provided with commercial food pellets and water ad libitum, and placed in biosafety level 3 isolator units just before challenge inoculation with A(H7N9) virus. An independent animal ethics committee (DEC consult) approved the experimental protocol before the start of the experiment.
Immunization and Challenge Infection
Animals were immunized once (group A [n = 6]) or twice (group B [n = 6]) intramuscularly with MVA-H7-Sh2 at 108 plaque-forming units (PFU) per dose and an interval of 3 weeks. Empty vector control (wild-type MVA [wtMVA] at a dose of 107 PFU; group C, 1 immunization [n = 3]; group D, 2 immunizations [n = 3]) and phosphate-buffered saline [PBS; group E [n = 4]) were used as negative control immunizations. To assess the possibility of dose sparing, a group of ferrets was immunized twice with 106 PFU of MVA-H7-Sh2 (group F [n = 6]). Before each immunization and before the challenge, blood specimens were collected from the animals to test for the induction of influenza virus–specific antibodies. Four weeks after the last immunization, the animals were inoculated under anesthesia (ketamine/medetomidine [reversed with atipamezole]) with 3 mL of 105.5 median tissue culture infective doses (TCID50) of influenza virus A/Anhui/1/2013(H7N9) by the intratracheal route. This virus was isolated from a fatal human case in China and kindly provided under conditions of the World Health Organization Pandemic Influenza Preparedness Framework. The virus was passaged 3 times in embryonated chicken eggs and passaged once in Madin-Darby canine kidney (MDCK) cells. The infectious titer was determined as described previously and expressed as TCID50 [38]. The challenge dose was based on the outcome of a dose-finding study showing substantial levels of virus replication in the upper respiratory tract and lower respiratory tract, as well as lung damage, but no mortality during a 4-day follow-up period. Nasal and pharyngeal swab specimens were collected before influenza virus inoculation (day 0) and on days 2 and 4 after inoculation and stored in transport medium (Hanks’ minimum essential medium with lactalbumin, glycerol, penicillin, streptomycin, polymyxin B, nystatin, and gentamicin) at −70°C until use. Four days after inoculation, all animals were euthanized. From the time of influenza virus inoculation onward, the animals were housed in biosafety level 3 containment facilities and monitored for clinical signs.
Serologic Analysis
The serum samples collected before immunization and on days 21 and 49 after immunization were tested for the presence of influenza A(H7N9) virus–specific antibodies by the hemagglutination inhibition (HI) assay with a 6 + 2 reassortant strain of influenza virus A/Anhui/1/2013 (containing the HA and neuraminidase [NA] of the A[H7N9] virus and the remaining 6 gene segments of influenza virus A/Puerto Rico/8/34[A/PR/8/34]) [39]. Sera were also tested for the presence of virus-neutralizing antibodies, using a virus microneutralization (VN) assay, performed as described previously and by using the reverse genetics virus described above [40]. Rabbit serum raised against influenza virus A/Seal/Massachussets/1/80(H7N7) was used as positive control in both assays. For the purpose of calculation, serum samples with an antibody titer of <10 were arbitrarily assigned a titer of 5. Seroconversion is defined as a postvaccination titer of ≥40 or a 4-fold rise in the antibody titer when the baseline titer was >10. Antibody titers of ≥40 were considered to be seroprotective.
Virus Replication in the Respiratory Tract and the Central Nervous System (CNS)
Upon necropsy of the animals on day 4 after inoculation, tissue samples were collected from their right lung lobes and accessory lobe, nasal turbinates, trachea, bronchi, and tracheobronchial lymph nodes and stored at –70°C until further processing. In addition to the respiratory tract, tissue specimens from the cerebrum and olfactory bulb were also collected. Tissue samples were homogenized and processed as described previously [41]. Ten-fold serial dilutions of nasal and pharyngeal swab supernatants (quadruplicate) and the homogenate supernatants of lungs (quintuplicate), other respiratory tract samples (triplicate; quadruplicate), and the CNS (triplicate; quadruplicate) were used to determine the presence of infectious virus titers in confluent layers of MDCK cells as described previously [38].
Pathological Examination and Immunohistochemical Analysis
Necropsies were performed according to standard protocols. The trachea was clamped before opening the thorax, and inflated lungs were assessed visually to determine the percentage of lung grossly affected. Lungs were weighed, and the relative lung weight was calculated as a percentage of the total body weight. Tissue specimens from the nasal turbinates, nasal septum, trachea, tracheobronchial lymph node, bronchus, lung, cerebrum (including the olfactory bulb), and cerebellum were collected and fixed in 10% neutral buffered formalin. To optimize histological assessment, the right lung was inflated with formalin before fixation, and after fixation right cranial and caudal lung lobes were sectioned in a longitudinal and cross-sectional plane in a standardized manner. Formalin-fixed tissue specimens were embedded in paraffin, cut to 4 µm, and either stained with hematoxylin-eosin (HE), for histopathological evaluation or with an immunoperoxidase-based method, using a primary monoclonal antibody (clone HB65 immunoglobulin G2a [IgG2a]; American Type Culture Collection) against the nucleoprotein of influenza A virus and a secondary goat–anti-mouse IG2a horseradish peroxidase antibody (Southern Biotech, Birmingham, AL), for immunohistochemical (IHC) evaluation. A positive control and IgG2a isotype controls of each slide were included. All slides were evaluated without prior knowledge of the identity of the animals, and HE slides were assessed semiquantitatively by using previously described criteria (Table 3) [42]. IHC findings were evaluated qualitatively to confirm the association of histological lesions with the presence of viral antigen.
Table 3.
Histopathological and Immunohistochemical (IHC) Scores
| Variable | Group, Mean ± SD |
|||||
|---|---|---|---|---|---|---|
| A (108 PFU MVA-H7-Sh2; n = 6) | B (108 PFU MVA-H7-Sh2; 2 doses; n = 6) | C (107 PFU wtMVA; n = 3) | D (107 PFU wtMVA; 2 doses; n = 3) | E (PBS; 2 doses; n = 3) | F (106 PFU MVA-H7-Sh2; 2 doses; n = 6) | |
| Proportion of lung affected, %a | 17.5 ± 22.3 | 5.2 ± 2.9 | 30.0 ± 15.0 | 41.7 ± 17.6 | 33.3 ± 27.5 | 24.2 ± 15.9 |
| Relative lung weight, % | 1.1 ± 0.2 | 1.0 ± 0.2 | 1.5 ± 0.1 | 1.7 ± 0.1 | 1.5 ± 0.1 | 1.5 ± 0.4 |
| Extent of alveolitisb | 1.5 ± 0.5 | 1.2 ± 0.4 | 2.3 ± 0.6 | 3.0 ± 0.0 | 2.0 ± 1.0 | 1.5 ± 0.8 |
| Severity of alveolitisc | 1.3 ± 0.5 | 1.3 ± 0.5 | 2.3 ± 0.6 | 2.3 ± 0.6 | 2.0 ± 0.0 | 1.2 ± 0.4 |
| Severity of bronchiolitis | 2.5 ± 0.5 | 2.2 ± 0.4 | 2.7 ± 0.6 | 2.3 ± 0.6 | 2.3 ± 0.6 | 2.5 ± 0.5 |
| Severity of bronchitis | 1.2 ± 0.4 | 1.2 ± 0.4 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.7 ± 0.6 | 0.7 ± 0.5 |
| Severity of tracheitis | 1.5 ± 0.5 | 1.3 ± 0.5 | 1.3 ± 0.6 | 0.7 ± 0.6 | 0.7 ± 0.6 | 0.8 ± 0.8 |
| Alveolar edemad | 0.2 ± 0.4 | 0.5 ± 0.5 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.7 ± 0.6 | 0.7 ± 0.5 |
| Alveolar hemorrhage | 0.5 ± 0.5 | 0.5 ± 0.5 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.7 ± 0.6 | 0.7 ± 0.5 |
| Type II hyperplasia | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 |
| Peribronchiolar infiltratese | 2.5 ± 0.5 | 2.5 ± 0.5 | 2.0 ± 0.0 | 2.3 ± 0.6 | 2.3 ± 0.6 | 2.2 ± 0.4 |
| Peribronchial infiltrates | 2.0 ± 0.9 | 1.5 ± 1.0 | 1.3 ± 0.6 | 1.3 ± 0.6 | 1.3 ± 0.6 | 1.0 ± 0.0 |
| Perivascular infiltrates | 1.7 ± 0.5 | 1.3 ± 0.5 | 1.3 ± 0.6 | 1.3 ± 0.6 | 1.7 ± 0.6 | 1.2 ± 1.0 |
| BALT hyperplasia | 1.5 ± 0.5 | 1.7 ± 0.5 | 1.0 ± 0.0 | 1.0 ± 1.0 | 1.3 ± 0.6 | 1.7 ± 0.5 |
| IHC analysis of alveolif | 1.0 ± 0.0 | 1.2 ± 0.4 | 2.0 ± 0.0 | 3.0 ± 0.0 | 2.7 ± 0.6 | 1.8 ± 0.8 |
| IHC analysis of bronchi(oli) | 1.8 ± 0.8 | 1.3 ± 0.5 | 2.0 ± 0.0 | 2.0 ± 0.0 | 2.7 ± 0.6 | 2.0 ± 0.0 |
a The percentage of lung affected was determined by examination of the intact lungs to determine consolidation of pulmonary parenchyma, characterized by depressed areas of increased consistency and (dark) red discoloration.
b The extent of alveolitis and alveolar damage was scored as follows: 0, 0%; 1, 1%–25%; 2, 25%–50%; 3, >50%.
c The severity of alveolitis, bronchiolitis, bronchitis, and tracheitis was scored as follows: 0, no inflammatory cells; 1, few inflammatory cells; 2, moderate numbers of inflammatory cells; 3, many inflammatory cells.
d The presence of alveolar edema, alveolar hemorrhage, and type II pneumocyte hyperplasia was scored as follows: 0, no; 1, yes.
e The extent of peribronchial, peribronchiolar, and perivascular infiltrates was scored as follows: 0, none; 1, 1–2 cells thick; 2, 3–10 cells thick; 3, >10 cells thick.
f The IHC score was defined as follows: 0, 0%; 1, <1%; 2, 5%–10%; 3, 10%–25%; 4, 25%–50%; 5, <50% cell showing positive nuclear staining. Bold values indicate a marked difference in the respective score for the indicated group(s) compared to score of the others groups.
Abbreviations: BALT, bronchial-associated tissue; MVA, modified vaccinia virus Ankara; PBS, phosphate-buffered saline; PFU, plaque-forming units; wtMVA, wild-type modified vaccinia virus Ankara.
Statistics
Differences in antibody titers and virus titers were tested for statistical significance, using the unpaired Student t test. Differences were considered statistically significant at P values of < .05.
RESULTS
Influenza Virus–Specific Immune Responses
After 1 immunization with 108 PFU MVA-H7-Sh2, 92% of the animals (groups A and B) seroconverted upon vaccination, as measured by the HI assay (Table 1). In these groups, 75% of the animals reached titers of ≥40, which are considered to be protective.
Table 1.
Serological Findings After Modified Vaccinia Virus (MVA) H7-Sh2 Immunization
| Immunization Group,a Finding | HI Titer |
VN Titer |
||
|---|---|---|---|---|
| Dose 1 | Dose 2 | Dose 1 | Dose 2 | |
| A (108 PFU MVA-H7-Sh2) | ||||
| Seroconversion, proportionb | 6/6 | NA | 6/6 | NA |
| Seroprotection, proportionc | 5/6 | NA | 3/6 | NA |
| GMT ± SD | 50.4 ± 1.8d | … | 28.3 ± 1.5 | … |
| B (108 PFU MVA-H7-Sh2; 2 doses) | ||||
| Seroconversion, proportionb | 5/6 | 6/6 | 6/6 | 6/6 |
| Seroprotection, proportionc | 4/6 | 6/6 | 4/6 | 5/6 |
| GMT ± SD | 31.7 ± 2.8d | 127.0 ± 2.0d | 44.9 ± 2.0d | 160.0 ± 3.5d |
| F (106 PFU MVA-H7-Sh2; 2 doses) | ||||
| Seroconversion, proportionb | 1/6 | 3/6 | 2/6 | 5/6 |
| Seroprotection, proportionc | 0/6 | 2/6 | 0/6 | 1/6 |
| GMT ± SD | 6.3 ± 1.8 | 12.6 ± 2.8 | 7.9 ± 2.0 | 17.8 ± 2.0 |
No HI or VN antibody titers were detected against influenza A/Anhui/1/2013(H7N9) in sera from the animals in the phosphate-buffered saline and wild-type MVA groups.
Abbreviations: GMT, geometric mean titer; HI, hemagglutination inhibition; NA, not applicable; PFU, plaque-forming units; VN, virus microneutralization.
a Postimmunization sera were obtained 4 weeks after immunization. Animals were immunized at the moment of the second immunization in group B and F. The sera after the first immunization in groups B and F were obtained 3 weeks later (ie, on the day of the second immunization).
b Defined as a ≥4-fold increase in antibody titer after immunization. The numbers for seroconversion are cumulative: an animal that seroconverted after the first immunization is also accounted for as having seroconverted after the 2nd immunization.
c Defined as an antibody titer of ≥40.
d Significantly higher than that of group F.
A second vaccination boosted the serum antibody titers, as seen in animals from group B. The HI geometric mean titer (GMT; ±SD) increased from 31.7 ± 2.8 to 127 ± 2.0. All animals in this group reached antibody titers of ≥40. In general, the results obtained with the HI assay were in agreement with those obtained by the VN assay. After 2 immunizations, a GMT (±SD) of 160 ± 3.5 was achieved.
To assess whether dose sparing was possible with MVA-H7-sh2, ferrets were also immunized twice with a 100-fold lower dose. However, only 1 of 6 animals (group F) seroconverted after the first immunization, and the GMT (±SD) of the group was 6.3 ± 1.8. The second immunization, with a vaccine dose diluted 1:100, increased the GMT (±SD) to 12.6 ± 2.8, but only 3 of 6 ferrets seroconverted, and 2 reached HI serum antibody titers of ≥40. Again, similar results were obtained with the VN assay, in which a GMT (±SD) of 17.8 ± 2.0 was reached. Antibodies directed to influenza virus A/Anhui/1/2013 were not detectable in any of the control animals that received wtMVA (groups C and D) or PBS (group E) by the HI or VN assay.
Clinical Signs
Two days after inoculation with influenza virus A/Anhui/1/13, all animals but those immunized once or twice with 108 PFU MVA-H7-Sh2 had loss of appetite. No other signs of disease were noticeable. On day 4 after challenge inoculation, 3 different clinical phenotypes were observed: group B animals (which received 2 doses of MVA-H7-Sh2 108 PFU) did not display any signs of disease (ie, they ate well, had normal posture, and had normal breathing); group A animals (which received 1 dose of MVA-H7-Sh2 108 PFU) and group F animals (which received 2 doses of MVA-H7-Sh2 106 PFU) ate slightly less, were a bit lethargic, but displayed normal posture and normal breathing (normal respiratory rate and depth); and group C animals (which received 1 dose of, wtMVA 107 PFU), group D animals (which received 2 doses of wtMVA 107 PFU), and group E animals (which received PBS) were lethargic, showed heavy breathing (hyperpnea), and alternately showed a hunched or flat-stretched posture. Most animals that experienced clinical signs also had weight loss, with mean decreases (±SD) from the prevaccination weight of 9.7% ± 5.1% in group C, 14.1% ± 3.5% in group D, 8.3% ± 3.4% in group E, and 8.6% ± 3.7% in group F. In contrast, animals in groups A and B were protected from weight loss (mean weight gain [±SD] from the prevaccination period, 2.8% ± 8.0% and 4.3% ± 5.5%, respectively).
Virus Replication in the Upper and Lower Respiratory Tract
Swab specimens were collected from the nose and pharynx on days 0, 2, and 4 after inoculation to asses viral shedding from the upper respiratory tract. All samples obtained on day 0 were negative. Apart from 1 animal in group E, which had a virus titer in its nose on day 2 after inoculation (104.8), no virus was present in any of the other nasal swab specimens on days 2 and 4. The pharyngeal swab specimens collected on day 2 after inoculation were virus positive for animals from all 6 groups. In groups A, B, and F, all of which were immunized with MVA-H7-Sh2, virus titers were lower on day 4 after inoculation, and in groups C, D, and E, pharyngeal virus titers increased between days 2 and 4 after inoculation (Table 2).
Table 2.
Virus Replication Inside and Outside of the Respiratory Tract, by Specimen
| Group, Variable | Pharyngeal Swab, by Collection Time |
Nasal Turbinates | Bronchi | Tracheobronchial Lymph Nodes | Cerebrum | Olfactory Bulb | |
|---|---|---|---|---|---|---|---|
| Day 2 | Day 4 | ||||||
| A (108 PFU MVA-H7-Sh2) | |||||||
| Log10 TCID50/g tissue, mean ± SD | 3.9 ± 1.2 | 2.2 ± 1.8 | 2.1 ± 2.1 | 5.0 ± 1.9 | … | 1.6 ± 1.0 | 1.8 ± 1.0 |
| Detectable virus, ferrets, % | 100 | 50 | 17 | 100 | Not detected | 33 | 17 |
| B (108 PFU MVA-H7-Sh2; 2 doses) | |||||||
| Log10 TCID50/g tissue, mean ± SD | 2.2 ± 1.8a | 0.7 ± 0.3b | … | 3.1 ± 2.2c | 1.7 ± 0.3 | … | … |
| Detectable virus, ferrets, % | 50 | 33 | Not detected | 67 | 33 | Not detected | Not detected |
| C (107 PFU wtMVA) | |||||||
| Log10 TCID50/g tissue, mean ± SD | 1.1 ± 1.0a | 3.2 ± 1.7 | 1.3 ± 0.1 | 6.8 ± 1.0 | 2.5 ± 1.2 | … | … |
| Detectable virus, ferrets, % | 33 | 100 | 33 | 100 | 67 | Not detected | Not detected |
| D (107 PFU wtMVA; 2 doses) | |||||||
| Log10 TCID50/g tissue, mean ± SD | 2.8 ± 2.0 | 3.3 ± 2.0 | 1.7 ± 0.4 | 6.4 ± 0.2 | 3.2 ± 1.6 | 1.2 ± 0.0 | 1.4 ± 0.2 |
| Detectable virus, ferrets, % | 67 | 100 | 67 | 100 | 67 | 33 | 33 |
| E (PBS; 2 doses) | |||||||
| Log10 TCID50/g tissue, mean ± SD | 2.8 ± 1.6 | 3.1 ± 2.4 | 2.5 ± 1.6 | 6.6 ± 1.1 | … | 1.3 ± 0.1 | … |
| Detectable virus, ferrets, % | 100 | 66 | 100 | 100 | Not detected | 33 | Not detected |
| F (106 PFU MVA-H7-Sh2; 2 doses) | |||||||
| Log10 TCID50/g tissue, mean ± SD | 2.2 ± 1.7 | 1.1 ± 1.3 | 1.2 ± 0.2 | 6.6 ± 0.6 | 1.8 ± 1.1 | … | … |
| Detectable virus, ferrets, % | 83 | 33 | 17 | 100 | 17 | Not detected | Not detected |
Virus titers were determined in tissue samples obtained on day 4 after inoculation.
Abbreviations: MVA, modified vaccine virus Ankara; PBS, phosphate-buffered saline; PFU, plaque-forming units; TCID50, median tissue culture infective dose; wtMVA, wild-type modified vaccinia virus Ankara.
a Significantly lower than that for group A.
b Significantly lower than those for groups C–E.
c Significantly lower than those for groups C–F.
The highest virus titers were observed in the trachea and lung samples (Figure 2). Mean virus titers in groups A and B were significantly lower for both organs, and 50% and 67% of the animals in these respective groups tested negative for the presence of infectious virus in their trachea. Also, in lung tissue specimens from 2 of 6 animals from group B, virus was undetectable. In addition, no animals from this group had virus detectable in nasal turbinate specimens, compared with 17%–100% of the animals in the other groups (Table 2). Virus was detectable in bronchi specimens from animals in all groups, but the lowest mean titers (±SD) were observed in animals from groups A and B (105.0 ± 101.9 and 103.1 ± 102.2, respectively). Sporadically, low virus titers were detected in tracheobronchial lymph node specimens and CNS specimens (from the cerebrum and olfactory bulb) from some animals, but there was no clear pattern regarding the distribution of animals that tested positive over the experimental groups (Table 2).
Figure 2.

Virus replication in the trachea (A) and lung (B). A single asterisk indicates that the virus titers in tracheal specimens from the animals in the 2 indicated groups are significantly lower than those of the other 4 groups. A double asterisk indicates that the virus titers in lung specimens from the animals in the 2 indicated groups are significantly lower than those of all other groups except the group that received a single dose of wild-type modified vaccinia virus Ankara (wtMVA). Abbreviations: PBS, phosphate-buffered saline; PFU, plaque-forming units; TCID50, median tissue culture infective dose.
Histopathological Changes and IHC Findings
Macroscopically observed lesions consisted of multifocal to coalescing consolidation of pulmonary parenchyma, characterized by depressed areas of increased consistency and (dark) red discoloration. The total percentage of lung tissue affected varied between groups, with group B showing only minimal lesions and group D most severely affected (Table 3). All infections resulted in a similar expression of pulmonary pathology, characterized by alveolitis, bronchiolitis, and, to a lesser extent, bronchitis. Alveolar lesions consisted of thickening of septae and filling of alveolar spaces with variable quantities of (alveolar) macrophages and neutrophils, in severe cases accompanied by karyolytic and karyorrhectic (necrotic) cellular debris, red blood cells (hemorrhage), proteinaceous fluid (edema), and eosinophilic fibrillar material (fibrin; Figure 3). In addition, multifocally alveolar epithelium showed hyperplasia of type II pneumocytes. Bronchiolar lesions consisted of necrosis of epithelial cells, luminal accumulations of aforementioned inflammatory cells, and cellular debris, frequently accompanied by peribronchiolar accumulations of lymphocytes and fewer plasma cells, macrophages, and neutrophils. Bronchi were similarly affected but to a far lesser extent and variably showed hyperplasia of bronchial-associated tissue (BALT). Infrequently, perivascular spaces were infiltrated, predominantly with lymphocytes (perivascular cuffing).
Figure 3.
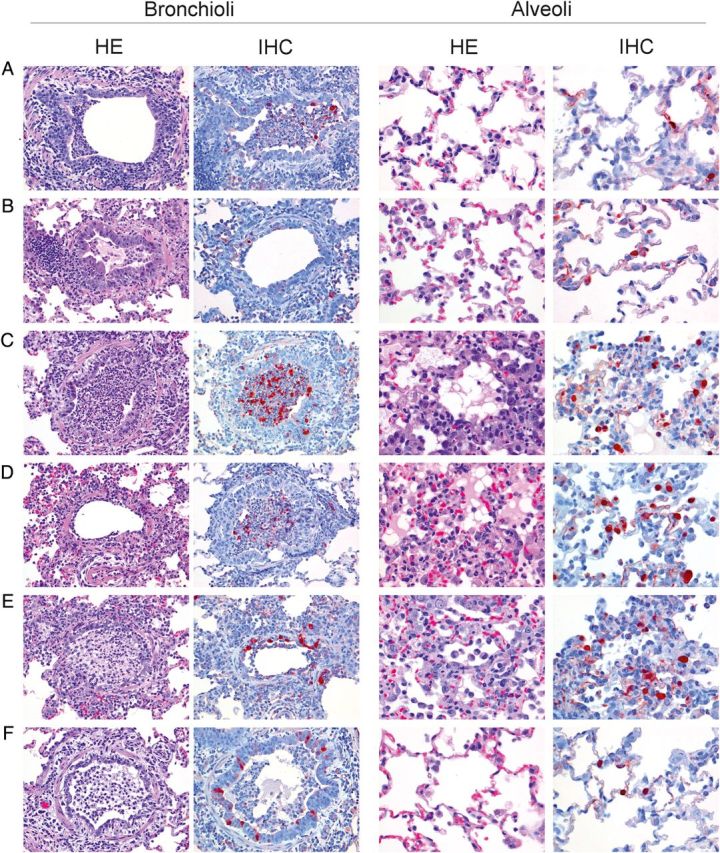
Histopathological findings (by hematoxylin-eosin staining) and antigen expression (by immunohistochemical analysis) in bronchioli (original magnification, ×20) and alveoli (original magnification, ×40) of groups (A–F). Histopathological findings for bronchioli show peribronchiolar inflammatory infiltrates, epithelial necrosis, and luminal accumulations of predominantly neutrophils and necrotic debris in all groups. Antigen expression in bronchioli is of similar extent for groups A and C–F and mild for group B. Histopathological analysis of the alveoli shows a small increase in alveolar macrophages in groups A (108 plaque-forming units [PFU] modified vaccinia virus Ankara [MVA] H7-Sh2), B (108 PFU MVA-H7-Sh2; 2 doses), and F (106 PFU MVA-H7-Sh2; 2 doses). Groups C (107 PFU wild-type MVA [wtMVA]), D (107 PFU wtMVA; 2 doses), and E (phosphate-buffered saline; 2 doses) show marked interstitial and luminal accumulations of predominantly neutrophils and macrophages, (epithelial) necrosis, and edema. Antigen expression in alveoli is minimal to mild for groups A, B, and F and marked for groups C, D, and E.
Remarkable differences between groups predominated in the extent and severity of alveolar lesions, with groups C, D, and E showing significantly more extensive and more severely affected alveolar tissue. The severity of lesions in bronchioli was remarkably similar between groups. Groups A, B, and F showed slightly more BALT hyperplasia (consistent with an active immune response; Figure 3). Semiquantitative assessment of IHC staining showed little positive nuclear staining in alveolar tissue specimens, especially from groups C and F and to a lesser extent from group E, compared with remaining groups. Bronchioli showed fewer differences in IHC scores among groups, which was consistent with the histological findings.
DISCUSSION
Here we describe the construction and preclinical evaluation of the first A(H7N9) influenza vaccine based on the MVA viral vector technology. Ferrets immunized with the MVA-H7-Sh2 vaccine developed protective antibody titers against the A(H7N9) influenza virus strain A/Anhui/1/2013 that was used for challenge infection. Animals that received 1 or 2 doses of 108 PFU seroconverted, and after 2 immunizations all animals developed HI antibody titers of ≥40, which are considered to be protective. Indeed, animals with postvaccination HI titers of ≥40 displayed reduced virus replication in the respiratory tract, especially in the lungs. The histopathological changes caused by challenge infection were less severe in these animals, particularly in the alveoli. Thus, MVA-H7-Sh2 immunization protected animals against weight loss and respiratory symptoms and the development of interstitial pneumonia and alveolitis but to a lesser extent protected them against the development of bronch(iol)itis. A few animals in the control groups and the group that received a single immunization with MVA-H7-Sh2 had low virus titers in CNS specimens (ie, olfactory bulb and brain tissues). Spread to the CNS may, although to a minimal extent, be the result of infection of cells in the olfactory epithelium, as was described previously [43, 44] after intranasal inoculation of ferrets with A(H5N1) viruses. In the present study, the ferrets were inoculated via the intratracheal route to model A(H7N9)-induced pneumonia. Since the nasal turbinates also tested positive for the presence of infectious virus 4 days after inoculation, the olfactory bulb may have become infected indirectly.
Administration of a 100-fold lower dose of MVA-H7-Sh2 proved to be less immunogenic than the normal dose. Clearly, the MVA-H7-Sh2–induced antibody response is strongly dose dependent, which may be explained in part by the replication deficiency of the vectors. With a lower dose, fewer cells become infected and express the transgene. However, in mice it was demonstrated that, with an MVA-based A(H5N1) vaccine candidate, substantial dose sparing could be achieved [28].
Recently, we demonstrated that, with an MVA-based vaccine, partial protection of ferrets against infection with a 2009 pandemic influenza A(H1N1) virus could be achieved. Immunization with 108 PFU of MVA-H7-Sh2 seems to afford stronger protection against virus replication in the respiratory tract than that afford by the MVA-H1 vaccine against infection with 2009 pandemic influenza A(H1N1) virus [41]. However, in both models the MVA-based vaccines did not induce sterile immunity, as was observed against challenge infection with A(H5N1) influenza viruses in other species, including mice and cynomolgus macaques [27–29, 45].
Reduction of A(H7N9) virus titers, especially in the upper respiratory tract, after challenge infection by MVA-H7-Sh2 vaccination may reduce the risk of airborne transmission of these viruses [46, 47]. In contrast to the partial protection from virus replication in the upper and lower respiratory tracts shown here, Chen et al have shown that a live-attenuated A(H7N9) vaccine was capable of providing sterile immunity in ferrets against homologous and heterologous challenge infection [48]. Although it was not tested, mucosal immunoglobulin A antibodies most likely contributed to the protective capacity of the vaccine in their model, in which vaccine and challenge virus were both inoculated intranasally. It would be of interest to test MVA-H7-Sh2 in a similar model to determine whether it can be used for the induction of mucosal immunity.
Apart from vaccines based on the actual novel A(H7N9) strain, vaccines based on historic H7Nx viruses have also been shown to induce cross-reactive antibodies that are able to neutralize the novel A(H7N9) virus [15]. Goff et al demonstrated that a Newcastle disease virus expressing the HA gene of a historical A(H7N3) virus induced cross-reactive antibodies in mice that afforded protection against infection with A(H7N1) influenza virus isolated in 1993 [18]. Chu et al demonstrated that a historic A(H7N9) virus–derived vaccine partially protected mice against infection with influenza virus A/Anhui/1/2013. Furthermore, live-attenuated A(H7N3) and A(H7N7) vaccines provided partial protection against the novel A(H7N9) strain in ferrets [49].
These examples illustrate the benefits of virus repositories containing prototypic strains of the respective HA and NA subtypes found in nature. Such a repository should be updated regularly on the basis of influenza surveillance data in poultry, swine, and humans. The HA nucleotide sequence of a prototypic strain is already enough to produce a viral-vector vaccine for viruses of that particular subtype, and a logical next step would be to not only have an influenza virus repository [50], but to also establish a vaccine repository based, for example, on a versatile vaccine production platform such as MVA. To accelerate the further clinical development of MVA-based influenza vaccines, further expansion of the safety and efficacy track record of MVA as a viral vector is required. Finally, once advanced regulatory frameworks are in place that reach beyond national guidelines (eg, pandemic influenza vaccine registration by the European Medicines Agency), MVA-based vaccine production technology could facilitate rapid availability of vaccines for emerging novel influenza viruses, including A(H7N9).
Notes
Acknowledgments. We thank C. Van de Sandt and P. van Run for providing excellent technical assistance.
Financial support. This work was supported by the European Union (for project FLUNIVAC [project ID 103972; to H. L. M. d. G., G. F. R., and G. S.] and project FLUPLAN [project ID 101920; to J. H. C. M. K. and A. D. M. E. O.), the German Center for Infection Research (TTU01.802), and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (contract HHSN272201400008C to R. A. M. F.).
Potential conflict of interest. R. A. M. F. and A. D. M. E. O. are holders of certificates of shares in ViroClinics Biosciences. To avoid any possible conflict of interests, Erasmus Medical Center policy dictates that the shares as such are held by the Stichting Administratiekantoor Erasmus Personeelsparticipaties. The board of this foundation is appointed by the Board of Governors of the Erasmus Medical Center and exercises all voting rights with regard to these shares. G. F. R. and A. D. M. E. O. are employed part time by Viroclinics Biosciences as a consultant and chief scientific officer, respectively. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Gao R, Cao B, Hu Y, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–97. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 2.Li Q, Zhou L, Zhou M, et al. Preliminary report: epidemiology of the avian influenza A (H7N9) outbreak in China. N Engl J Med. 2013;370:520–32. doi: 10.1056/NEJMoa1304617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen E, Chen Y, Fu L, et al. Human infection with avian influenza A(H7N9) virus re-emerges in China in winter 2013. Euro Surveill. 2013;18:1–10. doi: 10.2807/1560-7917.es2013.18.43.20616. [DOI] [PubMed] [Google Scholar]
- 4.Center of Excellence for Influenza Research and Surveillance (CEIRS) Resources. Cases of H7N9 influenza in China by week of onset (May 16, 2014) CIDRAP Newsletter.
- 5.Liu J, Xiao H, Wu Y, et al. H7N9: a low pathogenic avian influenza A virus infecting humans. Curr Opin Virol. 2014;5c:91–7. doi: 10.1016/j.coviro.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Riel D, Leijten LM, de Graaf M, et al. Novel avian-origin influenza A (H7N9) virus attaches to epithelium in both upper and lower respiratory tract of humans. Am J Pathol. 2013;183:1137–43. doi: 10.1016/j.ajpath.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreijtz JH, Kroeze EJ, Stittelaar KJ, et al. Low pathogenic avian influenza A(H7N9) virus causes high mortality in ferrets upon intratracheal challenge: a model to study intervention strategies. Vaccine. 2013;31:4995–9. doi: 10.1016/j.vaccine.2013.06.071. [DOI] [PubMed] [Google Scholar]
- 8.Collin N, de Radigues X World Health Organization H1N1 Vaccine Task Force. Vaccine production capacity for seasonal and pandemic (H1N1) 2009 influenza. Vaccine. 2009;27:5184–6. doi: 10.1016/j.vaccine.2009.06.034. [DOI] [PubMed] [Google Scholar]
- 9.International Federation of Pharmaceutical Manufacturers and Associations. Authoritative new study reveals global pandemic influenza vaccine capacity [news release] 23 February 2009. http://www.oliverwyman.com/content/dam/oliver-wyman/global/en/files/archive/pr/OW_HLS_Press_2009_IVC.pdf. Accessed 20 May 2010.
- 10.Couch RB, Decker WK, Utama B, et al. Evaluations for in vitro correlates of immunogenicity of inactivated influenza a H5, H7 and H9 vaccines in humans. PLoS One. 2012;7:e50830. doi: 10.1371/journal.pone.0050830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Groot AS, Ardito M, Terry F, et al. Low immunogenicity predicted for emerging avian-origin H7N9: implication for influenza vaccine design. Hum Vaccin Immunother. 2013;9:950–6. doi: 10.4161/hv.24939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller E, Andrews N, Stellitano L, et al. Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009 influenza vaccine: retrospective analysis. BMJ. 2013;346:f794. doi: 10.1136/bmj.f794. [DOI] [PubMed] [Google Scholar]
- 13.Kreijtz JH, Osterhaus AD, Rimmelzwaan GF. Vaccination strategies and vaccine formulations for epidemic and pandemic influenza control. Hum Vaccin. 2009;5:126–35. doi: 10.4161/hv.5.3.6986. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Jia N, Li J, et al. Optimal designs of an HA-based DNA vaccine against H7 subtype influenza viruses. Hum Vaccin Immunother. 2014;10:1949–58. doi: 10.4161/hv.28795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu DH, Sakoda Y, Nishi T, et al. Potency of an inactivated influenza vaccine prepared from A/duck/Mongolia/119/2008 (H7N9) against the challenge with A/Anhui/1/2013 (H7N9) Vaccine. 2014;32:3473–9. doi: 10.1016/j.vaccine.2014.04.060. [DOI] [PubMed] [Google Scholar]
- 16.Yan J, Villarreal DO, Racine T, et al. Protective immunity to H7N9 influenza viruses elicited by synthetic DNA vaccine. Vaccine. 2014;32:2833–42. doi: 10.1016/j.vaccine.2014.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bart SA, Hohenboken M, Della Cioppa G, Narasimhan V, Dormitzer PR, Kanesa-Thasan N. A cell culture-derived MF59-adjuvanted pandemic A/H7N9 vaccine is immunogenic in adults. Sci Transl Med. 2014;6:1–10. doi: 10.1126/scitranslmed.3008761. 234ra55. [DOI] [PubMed] [Google Scholar]
- 18.Goff PH, Krammer F, Hai R, et al. Induction of cross-reactive antibodies to novel H7N9 influenza virus by recombinant Newcastle disease virus expressing a North American lineage H7 subtype hemagglutinin. J Virol. 2013;87:8235–40. doi: 10.1128/JVI.01085-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krammer F, Albrecht RA, Tan GS, et al. Divergent H7 immunogens offer protection from H7N9 virus challenge. J Virol. 2014;88:3976–85. doi: 10.1128/JVI.03095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klausberger M, Wilde M, Palmberger D, et al. One-shot vaccination with an insect cell-derived low-dose influenza A H7 virus-like particle preparation protects mice against H7N9 challenge. Vaccine. 2014;32:355–62. doi: 10.1016/j.vaccine.2013.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Margine I, Palese P, Krammer F. Expression of functional recombinant hemagglutinin and neuraminidase proteins from the novel H7N9 influenza virus using the baculovirus expression system. J Vis Exp. 2013;81:e51112. doi: 10.3791/51112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fries LF, Smith GE, Glenn GM. A recombinant viruslike particle influenza A (H7N9) vaccine. N Engl J Med. 2013;369:2564–6. doi: 10.1056/NEJMc1313186. [DOI] [PubMed] [Google Scholar]
- 23.Hahn T, Courbron D, Hamer M, et al. Rapid manufacture and release of a GMP batch of avian influenza A(H7N9) virus-like particle vaccine made using recombinant baculovirus-Sf9 insect cell culture technology. Bioprocessing J. 2013;12:4–17. [Google Scholar]
- 24.Kreijtz JH, Gilbert SC, Sutter G. Poxvirus vectors. Vaccine. 2013;31:4217–9. doi: 10.1016/j.vaccine.2013.06.073. [DOI] [PubMed] [Google Scholar]
- 25.Moss B. Reflections on the early development of poxvirus vectors. Vaccine. 2013;31:4220–2. doi: 10.1016/j.vaccine.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rimmelzwaan GF, Sutter G. Candidate influenza vaccines based on recombinant modified vaccinia virus Ankara. Expert Rev Vaccines. 2009;8:447–54. doi: 10.1586/erv.09.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreijtz JH, Suezer Y, de Mutsert G, et al. Recombinant modified vaccinia virus Ankara expressing the hemagglutinin gene confers protection against homologous and heterologous H5N1 influenza virus infections in macaques. J Infect Dis. 2009;199:405–13. doi: 10.1086/595984. [DOI] [PubMed] [Google Scholar]
- 28.Kreijtz JH, Suezer Y, de Mutsert G, et al. MVA-based H5N1 vaccine affords cross-clade protection in mice against influenza A/H5N1 viruses at low doses and after single immunization. PLoS One. 2009;4:e7790. doi: 10.1371/journal.pone.0007790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreijtz JH, Suezer Y, van Amerongen G, et al. Recombinant modified vaccinia virus Ankara-based vaccine induces protective immunity in mice against infection with influenza virus H5N1. J Infect Dis. 2007;195:1598–606. doi: 10.1086/517614. [DOI] [PubMed] [Google Scholar]
- 30.Veits J, Romer-Oberdorfer A, Helferich D, et al. Protective efficacy of several vaccines against highly pathogenic H5N1 avian influenza virus under experimental conditions. Vaccine. 2008;26:1688–96. doi: 10.1016/j.vaccine.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 31.Hessel A, Schwendinger M, Holzer GW, et al. Vectors based on modified vaccinia Ankara expressing influenza H5N1 hemagglutinin induce substantial cross-clade protective immunity. PLoS One. 2011;6:e16247. doi: 10.1371/journal.pone.0016247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sutter G, Wyatt LS, Foley PL, Bennink JR, Moss B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine. 1994;12:1032–40. doi: 10.1016/0264-410x(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 33.Berthoud TK, Hamill M, Lillie PJ, et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin Infect Dis. 2011;52:1–7. doi: 10.1093/cid/ciq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hessel A, Savidis-Dacho H, Coulibaly S, et al. MVA vectors expressing conserved influenza proteins protect mice against lethal challenge with H5N1, H9N2 and H7N1 viruses. PLoS One. 2014;9:e88340. doi: 10.1371/journal.pone.0088340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brewoo JN, Powell TD, Jones JC, et al. Cross-protective immunity against multiple influenza virus subtypes by a novel modified vaccinia Ankara (MVA) vectored vaccine in mice. Vaccine. 2013;31:1848–55. doi: 10.1016/j.vaccine.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kremer M, Volz A, Kreijtz JH, Fux R, Lehmann MH, Sutter G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol Biol. 2012;890:59–92. doi: 10.1007/978-1-61779-876-4_4. [DOI] [PubMed] [Google Scholar]
- 37.Wyatt LS, Earl PL, Liu JY, et al. Multiprotein HIV type 1 clade B DNA and MVA vaccines: construction, expression, and immunogenicity in rodents of the MVA component. AIDS Res Hum Retroviruses. 2004;20:645–53. doi: 10.1089/0889222041217428. [DOI] [PubMed] [Google Scholar]
- 38.Rimmelzwaan GF, Baars M, Claas EC, Osterhaus AD. Comparison of RNA hybridization, hemagglutination assay, titration of infectious virus and immunofluorescence as methods for monitoring influenza virus replication in vitro. J Virol Methods. 1998;74:57–66. doi: 10.1016/s0166-0934(98)00071-8. [DOI] [PubMed] [Google Scholar]
- 39.Palmer D, Dowle W, Coleman M, Schild G. Atlanta: US Department of Health, Education, and Welfare; 1975. Haemagglutination inhibition test. Advanced laboratory techniques for influenza diagnosis. Procedural Guide; pp. 25–62. [Google Scholar]
- 40.McCullers JA, Van De Velde LA, Allison KJ, Branum KC, Webby RJ, Flynn PM. Recipients of vaccine against the 1976 “swine flu” have enhanced neutralization responses to the 2009 novel H1N1 influenza virus. Clin Infect Dis. 2010;50:1487–92. doi: 10.1086/652441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kreijtz JH, Suzer Y, Bodewes R, et al. Evaluation of a modified vaccinia virus Ankara (MVA)-based candidate pandemic influenza A/H1N1 vaccine in the ferret model. J Gen Virol. 2010;91:2745–52. doi: 10.1099/vir.0.024885-0. [DOI] [PubMed] [Google Scholar]
- 42.van den Brand JM, Kreijtz JH, Bodewes R, et al. Efficacy of vaccination with different combinations of MF59-adjuvanted and nonadjuvanted seasonal and pandemic influenza vaccines against pandemic H1N1 (2009) influenza virus infection in ferrets. J Virol. 2011;85:2851–8. doi: 10.1128/JVI.01939-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodewes R, Kreijtz JH, van Amerongen G, et al. Pathogenesis of influenza A/H5N1 virus infection in ferrets differs between intranasal and intratracheal routes of inoculation. Am J Pathol. 2011;179:30–6. doi: 10.1016/j.ajpath.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schrauwen EJ, Herfst S, Leijten LM, et al. The multibasic cleavage site in H5N1 virus is critical for systemic spread along the olfactory and hematogenous routes in ferrets. J Virol. 2012;86:3975–84. doi: 10.1128/JVI.06828-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreijtz JH, Suezer Y, de Mutsert G, et al. Preclinical evaluation of a modified vaccinia virus Ankara (MVA)-based vaccine against influenza A/H5N1 viruses. Vaccine. 2009;27:6296–9. doi: 10.1016/j.vaccine.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Richard M, Schrauwen EJ, de Graaf M, et al. Limited airborne transmission of H7N9 influenza A virus between ferrets. Nature. 2013;501:560–3. doi: 10.1038/nature12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu L, Bao L, Deng W, et al. Novel avian-origin human influenza A(H7N9) can be transmitted between ferrets via respiratory droplets. J Infect Dis. 2014;209:551–6. doi: 10.1093/infdis/jit474. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Baz M, Lu J, et al. Development of a high-yield live attenuated H7N9 influenza virus vaccine that provides protection against homologous and heterologous H7 wild-type viruses in ferrets. J Virol. 2014;88:7016–23. doi: 10.1128/JVI.00100-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Q, Chen Z, Cheng X, Xu L, Jin H. Evaluation of live attenuated H7N3 and H7N7 vaccine viruses for their receptor binding preferences, immunogenicity in ferrets and cross reactivity to the novel H7N9 virus. PLoS One. 2013;8:e76884. doi: 10.1371/journal.pone.0076884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keawcharoen J, Spronken MI, Vuong O, et al. Repository of Eurasian influenza A virus hemagglutinin and neuraminidase reverse genetics vectors and recombinant viruses. Vaccine. 2010;28:5803–9. doi: 10.1016/j.vaccine.2010.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]