

ABSTRACT
A high-throughput phenotypic screen based on a Citrobacter freundii AmpC reporter expressed in Escherichia coli was executed to discover novel inhibitors of bacterial cell wall synthesis, an attractive, well-validated target for antibiotic intervention. Here we describe the discovery and characterization of sulfonyl piperazine and pyrazole compounds, each with novel mechanisms of action. E. coli mutants resistant to these compounds display no cross-resistance to antibiotics of other classes. Resistance to the sulfonyl piperazine maps to LpxH, which catalyzes the fourth step in the synthesis of lipid A, the outer membrane anchor of lipopolysaccharide (LPS). To our knowledge, this compound is the first reported inhibitor of LpxH. Resistance to the pyrazole compound mapped to mutations in either LolC or LolE, components of the essential LolCDE transporter complex, which is required for trafficking of lipoproteins to the outer membrane. Biochemical experiments with E. coli spheroplasts showed that the pyrazole compound is capable of inhibiting the release of lipoproteins from the inner membrane. Both of these compounds have significant promise as chemical probes to further interrogate the potential of these novel cell wall components for antimicrobial therapy.
IMPORTANCE The prevalence of antibacterial resistance, particularly among Gram-negative organisms, signals a need for novel antibacterial agents. A phenotypic screen using AmpC as a sensor for compounds that inhibit processes involved in Gram-negative envelope biogenesis led to the identification of two novel inhibitors with unique mechanisms of action targeting Escherichia coli outer membrane biogenesis. One compound inhibits the transport system for lipoprotein transport to the outer membrane, while the other compound inhibits synthesis of lipopolysaccharide. These results indicate that it is still possible to uncover new compounds with intrinsic antibacterial activity that inhibit novel targets related to the cell envelope, suggesting that the Gram-negative cell envelope still has untapped potential for therapeutic intervention.
INTRODUCTION
The biosynthesis of the bacterial cell wall is a well-established target for antibacterial agents such as β-lactams, one of the oldest and clinically most prevalent classes of antibiotics. However, with the rise of infections caused by multidrug-resistant bacteria, there is a widely recognized need for new antibacterial compounds (1–3). The bacterial cell wall is an excellent target for development of antibiotics, because its synthesis is conserved across bacterial pathogens and absent from mammalian cells. This cell wall consists of peptidoglycan, a mesh-like structure that plays an essential role in maintaining cell integrity, which is composed of repeating β-(1,4)-N-acetylglucosamine-β-(1,4)-N-acetylmuramic acid disaccharide strands cross-linked through peptide stems. Whereas several advances have been made in reconstituting the biosynthetic steps for peptidoglycan synthesis in vitro for chemical interrogation, the later steps in this pathway that utilize membrane-bound enzymes make biochemical high-throughput screening challenging (4). Thus, phenotypic or cell-based screening to find novel inhibitors of cell wall biogenesis is an attractive alternative to target-based enzymatic screens. Phenotypic screening not only overcomes the biochemical hurdles associated with assays needing membrane proteins but also permits interrogation of the entire pathway at once and selects for compounds that penetrate into the cell, which has been recognized as a significant hurdle in target-based antibacterial drug discovery (5, 6).
In some Gram-negative bacteria, exposure to β-lactam antibiotics induces expression of the chromosomally encoded AmpC β-lactamase (7, 8). In these organisms, AmpC expression is repressed by a divergently transcribed repressor, AmpR, under normal growth conditions. Treatment with β-lactam antibiotics disrupts the balance of peptidoglycan synthesis due to the inhibition of the transpeptidase activity of penicillin-binding proteins (PBPs), which are involved in the final stage of peptidoglycan synthesis (reviewed in reference 9). Continued peptidoglycan turnover reactions in the absence of synthesis lead to the accumulation of anhydromuramyl peptides, which bind to the AmpR regulator, causing a derepression of the expression of the AmpC β-lactamase. Sun et al. showed that inhibitors of other steps of cell wall biogenesis are also capable of inducing AmpC β-lactamase production and that AmpC can be used as a reporter to detect cell wall-active compounds (10, 11).
In a previous report using a hypersensitive Escherichia coli strain, we identified a novel inhibitor of lipoprotein transport to the outer membrane (12). Here we describe the adaptation of the AmpC β-lactamase reporter system (11) for phenotypic screening in a high-throughput, 384-well format, which identified another novel inhibitor of lipoprotein trafficking as well as a novel inhibitor of lipopolysaccharide (LPS) synthesis. Compound 1 targets LpxH, which is involved in the biosynthesis of lipid A, the outer membrane anchor of LPS (13) (Fig. 1). Compound 2 was found to inhibit the function of the LolCDE complex, which is required for transport of lipoproteins to the outer membrane (14, 15) (Fig. 1). Identification of these inhibitors indicates that this screening assay is not restricted to identifying compounds that directly affect peptidoglycan synthesis but also has a broader scope in finding inhibitors of indirectly related cell envelope biosynthesis.
FIG 1.

Chemical structures of compounds 1 and 2.
MATERIALS AND METHODS
Bacterial strain construction.
To engineer the reporter construct, the ampR-ampC locus from a clinical isolate of Citrobacter freundii was amplified by PCR using primers CfrAmpRAmpCFEcoRI and CfrAmpRAmpCRBsaI (see Table S1 in the supplemental material) and Roche High Fidelity master mix, according to the manufacturer's instructions. The resulting PCR product was cloned into the pCR4 Blunt TOPO vector (Life Technologies, WI) to obtain pAN118. The sequence of the ampR-ampC insertion was verified by DNA Sanger sequencing using a Life Sciences 3100 series genetic analyzer. The ampR-ampC region of pAN118 was subcloned into the EcoRI site of the low-copy-number vector pWSK129 (16) to create the reporter construct used for screening, i.e., pAN116. The reporter plasmid pAN116 was then transformed into E. coli W3110 ΔampC ΔacrB to create the screening strain ARC4150. The ampC and acrB gene deletions were constructed using λ Red-mediated recombination in E. coli strain BW25113 containing plasmid pKD46, as previously described (17). Primers used are shown in Table S1 in the supplemental material. Recombinants were selected on Luria-Bertani (LB) agar medium containing 25 μg/ml kanamycin, and deletions were verified by PCR. The ampC deletion was then moved by P1 phage transduction into E. coli W3110 (18). The kanamycin resistance gene was excised from the chromosome by using the FLP recombinase expressed from pCP20, as previously described (17). The acrB deletion was subsequently moved by P1 phage transduction into the W3110 ΔampC strain, and the kanamycin resistance gene was removed as described above. The artifact control strain ARC4151 was created by transforming the empty vector pWSK129 into W3110 ΔampC ΔacrB.
To create the LpxH overexpression strain, lpxH from E. coli MG1655 was amplified by PCR as described above, with an upstream primer encoding an EcoRI site and a downstream primer encoding a HindIII site (see Table S1 in the supplemental material). The resulting PCR product was digested and cloned into the EcoRI and HindIII sites of pPSV35 (19). This plasmid and the empty vector (pPSV35) were then transformed by electroporation into E. coli MG1655 ΔtolC, with selection on LB agar containing 12 μg/ml of gentamicin.
High-throughput screening.
The screening strain ARC4150 and the artifact strain ARC4151 were grown in LB with 25 μg/ml kanamycin at 37°C to an optical density at 600 nm (OD600) of 0.8. The cells were mixed with an equal volume of 20% glycerol, divided into aliquots, flash frozen in a dry ice and ethanol bath, and stored at −80°C. On each day of screening, cells were thawed at room temperature and diluted 1:20 in LB broth supplemented with 25 μg/ml kanamycin to obtain a final OD600 of 0.02. The cells were grown at 37°C with shaking at 200 rpm to an OD600 of 0.08. Thirty-microliter aliquots of cells were then dispensed using a multidrop dispenser (Thermo Scientific, Waltham, MA) into each well of 384-well plates containing test compounds. For details on how the screening plates were prepared, please see the supplemental material. The final compound concentration was 50 μM, with a dimethyl sulfoxide (DMSO) concentration of 1.25% (vol/vol). After the plates were incubated at room temperature for 2 h, 10 μl of reaction buffer (20 mM Tris-Cl, pH 8.0, 20 μg/ml lysozyme, and 0.1 mM nitrocefin) was added to each well and incubated at room temperature for another hour. The reaction was stopped with cloxacillin at a final concentration of 10 μg/ml, and the A490 and OD600 of the plates were read using an EnVision multilabel plate reader (PerkinElmer, Waltham, MA). The active compounds were rescreened for a 7-point concentration-response curve with a final compound concentration range of 200 μM to 3 μM.
Susceptibility testing.
MICs were determined according to the guidelines of the Clinical and Laboratory Standards Institute (20). A preliminary toxicity assessment was made by measuring the antiproliferation activity of compounds of interest against Candida albicans and the human lung carcinoma cell line A549 and their induction of lysis of sheep red blood cells as previously described (21, 22).
Inhibition of cellular biosynthetic processes.
Inhibition of cell wall, fatty acid, DNA, RNA, and protein biosynthesis processes was measured as previously described, using an E. coli W3110 ΔtolC ΔlysA strain (23).
Microscopy.
Linnaeus Bioscience performed bacterial cytological profiling as described previously (24). E. coli ATCC 25922 ΔtolC was grown at 30°C with shaking until early log phase (OD600 of 0.15 to 0.2). Cells were then mixed with a compound at 2× to 10× the MIC and rolled in test tubes at 30°C for 120 min. Cells were subsequently stained, concentrated by centrifugation, and observed by fluorescence microscopy.
Resistant mutant selection.
Mutants resistant to compounds 1 and 2 were raised against E. coli MG1655 ΔtolC and E. coli ATCC 25922 ΔtolC, respectively. One-hundred-microliter aliquots of cells (9.65 × 108 CFU/ml for E. coli MG1655 ΔtolC and 5.7 × 109 CFU/ml for E. coli ATCC 25922 ΔtolC) were plated on LB agar containing 8×, 16×, 32×, and 64× the MIC of the test compound. In addition, 10-fold serial dilutions of the culture were spread on plates without antibiotic selection to determine the total number of CFU per milliliter of sample. Plates were incubated at 37°C for 24 to 48 h. The resistance frequency was calculated as the number of CFU per milliliter on the compound-containing plates divided by the total number of CFU per milliliter of bacterial culture. Resistant colonies were confirmed by plating on LB agar containing the compound at 8× to 64× the MIC. Resistant isolates were passaged on LB agar without selection three times prior to determining the MICs for compounds 1 and 2 to ensure that the isolates were stably resistant mutants. Resistant mutants were then subjected to whole-genome sequencing using an Illumina MiSeq V2 instrument to identify mutations as previously described (25).
Spheroplast release assays.
E. coli MG1655 ΔtolC cells were grown in LB medium at 37°C to an OD600 of 1.0. The cells were converted into spheroplasts as described previously (26). The Lpp release assay was performed as described previously (12). Briefly, suspensions containing 2 × 108 spheroplasts were incubated with or without 3.5 μg His-tagged LolA in the presence of DMSO, 1.4 μg globomycin, or compound 2 at 30°C for 1 min. Two hundred fifty microliters of LB containing 0.3 M sucrose and 10 μg/ml DNase I was added and incubated at 30°C for 30 min. The spheroplasts were pelleted by centrifugation at 16,000 × g for 2 min. The supernatant was then diluted 3-fold with 7.15 mM MgCl2 and ultracentrifuged at 100,000 × g for 30 min to remove the membranes. The supernatants were then analyzed by SDS-PAGE and immunoblotting with anti-Lpp and anti-OmpA antibodies.
RESULTS
An AmpC reporter strain specifically detects inhibitors of cell wall biogenesis.
It was previously shown that the inducible AmpC protein from C. freundii can be used as a sensor for inhibitors of cell wall biosynthesis (11). Here we used a similar inducible AmpC protein from C. freundii, introduced into E. coli, to develop a high-throughput assay with a 384-well format. The ampR-ampC region from C. freundii was cloned into a low-copy-number plasmid and introduced into an E. coli screening strain. A strain of E. coli lacking its chromosomal copy of ampC was used to reduce the signal background, as the endogenous E. coli AmpC system produces a low level of noninducible expression of AmpC (27). Initially, we tested a waaP deletion screening strain, which affects permeability via an LPS defect, but we found a higher level of background expression of the AmpC reporter. This result was similar to that previously reported by Sun et al., who also noted an increase in background for their envA (lpxC) screening strain (11). Consequently, we decided instead to employ an RND efflux pump mutant (ΔacrB) in combination with the ampC chromosomal deletion to increase the screen sensitivity, without affecting the expression level of the AmpC reporter system (28). Several methods of preparing cells and various reaction buffers were evaluated and compared to those employed by Sun et al. to select the optimal assay conditions that gave a robust signal and a format amenable to our high-throughput robotic screening system. A Tris-Cl, pH 8, buffer with 20 μg/ml lysozyme was found to yield the highest level of ampC induction compared to the previously used Z salts (sodium phosphate-based buffer system) (11; data not shown). Several detergents, such as cetyltrimethylammonium bromide (CTAB) and sodium deoxycholate, were also tested in the reaction buffer; however, under these conditions, some nitrocefin precipitation was observed and no improvement in signal was seen, so these reagents were abandoned. The optimal conditions for our robotic system were found to be growth of the reporter strain to an OD600 of 0.08 and subsequent incubation with the test compounds for 2 h at room temperature, after which a detection reagent consisting of Tris-Cl, pH 8, lysozyme, and the colorimetric β-lactam nitrocefin was added (data not shown). Cleavage of nitrocefin by the AmpC β-lactamase resulted in a colorimetric change, which was detected by measuring the A490. Because the amount of nitrocefin cleavage changes over time, a β-lactamase inhibitor (cloxacillin) was added after incubation at 25°C for 1 h to stabilize the signal. The A490 values were normalized to the cell density (OD600) to account for compounds that caused slower cell growth or cell lysis.
Antibiotics that inhibit a variety of classes of cellular targets were profiled to characterize induction of the AmpC reporter strain (Fig. 2). The assay was able to detect inhibitors of multiple steps of cell wall biogenesis, such as phosphomycin, which inhibits the first committed step of peptidoglycan synthesis performed by MurA, as well as cefoxitin, which inhibits one of the last steps in peptidoglycan synthesis, the transpeptidation reaction carried out by PBPs. The maximal expression level of the AmpC reporter (relative to baseline) increased 1.8- and 1.4-fold for phosphomycin and cefoxitin, respectively. In addition to inhibition of peptidoglycan synthesis, inhibitors of other factors required for outer membrane synthesis were also found to induce the AmpC reporter strain. For example, CHIR-090, an inhibitor of LpxC (29), which performs the first committed step in the biosynthesis of the lipid A component of outer membrane LPS, was also detected by the AmpC reporter strain, showing an approximately 1.5-fold induction. Some of the compounds displayed concentration curves that had a bell shape due to cell death which occurred at concentrations higher than the MIC. Conversely, antibiotics that inhibit cellular targets that are not involved in cell wall or outer membrane biogenesis, such as the translation inhibitors tetracycline and chloramphenicol or the gyrase inhibitor ciprofloxacin, did not show induction of the reporter (data not shown). These data demonstrate that this reporter strain is specific and suitable for detecting molecules that disrupt components of the E. coli envelope.
FIG 2.
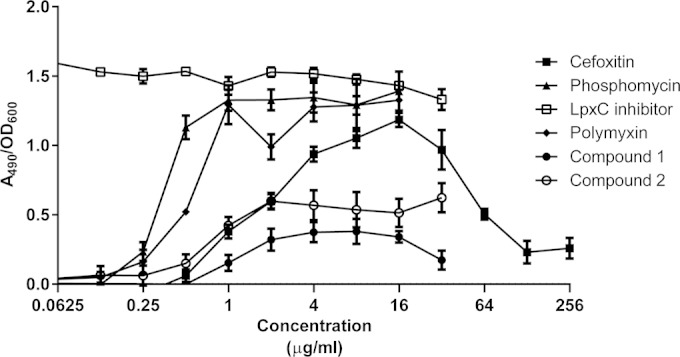
AmpC induction by cell wall inhibitors. The graph shows the induction of the C. freundii ampC reporter plasmid in the presence of cell wall-active compounds. Expression of ampC was detected upon cleavage of the colorimetric β-lactam analog nitrocefin, as measured by the A490. Induction was calculated as the A490/OD600 ratio to correct for cell density. The LpxC inhibitor was CHIR-90 (29). The effects of compounds 1 and 2 on the reporter are also shown.
High-throughput screening by AmpC reporter assay.
A high-throughput screen to identify compounds that induce the AmpC reporter strain was conducted in a 384-well format. Approximately 1.2 million compounds from the AstraZeneca collection were screened at a 50 μM test concentration. This phenotypic screen resulted in a robust assay with a Z′ value (mean ± standard deviation) of 0.79 ± 0.04 over the 31 screening runs of the entire campaign (30). A chemical triage process was applied to remove compounds with undesirable physical-chemical properties, commercial antibiotics, and known inhibitors of cell wall biosynthesis. The remaining compounds were tested in a 7-point concentration-response assay for determination of potency. As some compounds are colored and absorb light at 490 nm (the wavelength used to detect the cleaved nitrocefin product), an orthogonal artifact assay was used in cases where compounds were tested against the E. coli ΔampC ΔacrB strain lacking the C. freundii ampR-ampC reporter construct. To correct for artifactual A490 signals, the concentration-response data were compared to the artifact assay data and normalized by subtraction if an increase in signal was detected in the artifact assay, as previously described (31). The resulting hits were screened against the yeast strain Candida albicans and the A549 mammalian cell line and for lysis of sheep red blood cells to remove promiscuous or broadly toxic compounds.
Profiles of two compounds from the AmpC reporter screen.
Two compounds were selected for further characterization and to confirm their mode of action as inhibitors of cell envelope biogenesis. Compound 1 contains both indoline and piperazine scaffolds and has a molecular weight of 453.5 and a measured logD of 2.7 (Fig. 1). Compound 2 has a pyrazole core, a molecular weight of 345.4, and a measured logD of 4.3 (Fig. 1). Both compounds have high human serum protein binding levels (>99% bound). Compounds 1 and 2 were resynthesized (see the supplemental material) and retested in the AmpC reporter assay to confirm their activity. Compound 1 induced the reporter 0.4-fold, and compound 2 induced the reporter 0.6-fold over the baseline signal (Fig. 2). Both compounds were also profiled for antibacterial activity against Gram-negative and Gram-positive species. Strong inhibition of growth was observed in an E. coli efflux mutant (ATCC 25922 ΔtolC), with a MIC of 0.25 μg/ml for compound 1 and 0.125 μg/ml for compound 2 (Table 1). Compound 2 also had moderate activity against the wild-type E. coli strain ATCC 25922, with a MIC of 8 μg/ml, and weak activity (MIC of 32 μg/ml) against Haemophilus influenzae. Neither compound 1 nor compound 2 was active against Staphylococcus aureus or the yeast strain C. albicans, and these compounds did not cause lysis of sheep red blood cells; however, both compounds inhibited the proliferation of the human cell line A549 when cells were exposed for 72 h (90% cytotoxic concentration [CC90] of 23 μM for compound 1 and 84 μM for compound 2) (Table 1 and data not shown).
TABLE 1.
Antibacterial activities of compounds 1 and 2
| Species | Strain | Description | MIC (μg/ml) |
|
|---|---|---|---|---|
| Compound 1 | Compound 2 | |||
| Escherichia coli | ATCC 25922 | Wild type | >64 | 8 |
| ATCC 25922 ΔtolC | Efflux mutant | 0.25 | 0.125 | |
| Haemophilus influenzae | ATCC 49247 | Wild type | >64 | 32 |
| Pseudomonas aeruginosa | PAO1 | Wild type | >64 | >64 |
| Staphylococcus aureus | ATCC 29213 | Wild type | >64 | >64 |
| Candida albicans | ATCC 90028 | Counterscreen target | >64 | >64 |
In order to confirm the results of the AmpC reporter assay, compounds 1 and 2 were assayed for inhibition of the incorporation of cellular pathway-specific radioactive precursors (23). Both were found to inhibit the incorporation of [3H]diaminopimelic acid, which is a component of the E. coli peptidoglycan, indicating that the activity of these compounds is related to the inhibition of cell wall biogenesis (Table 2). Although a decline in [14C]acetic acid incorporation was also observed in the presence of compound 1, suggesting a potential inhibition of fatty acid biosynthesis, among the 15 independent resistant mutants that were analyzed by whole-genome sequencing, mutations were found exclusively in the lpxH locus (see below).
TABLE 2.
Inhibition of E. coli macromolecular synthesis pathways
| Compound | Target | Incorporation IC50 (μg/ml)a |
||||
|---|---|---|---|---|---|---|
| Protein | Cell wall | Fatty acid | RNA | DNA | ||
| [14C]leucine | [3H]DAPb | [14C]acetic acid | [3H]uridine | [3H]thymidine | ||
| Erythromycin | Protein synthesis | 30 | >256 | >256 | >256 | >256 |
| Ampicillin | Cell wall synthesis | >256 | 16 | >256 | >256 | >256 |
| Triclosan | Fatty acid synthesis | >256 | 1.5 | 0.0156 | 2 | 2 |
| Rifampin | Transcription | 64 | >256 | >256 | 64 | >256 |
| Ciprofloxacin | DNA replication | 30 | >256 | >256 | 1 | 0.02 |
| Carbonyl cyanide m-chlorophenylhydrazone (CCCP) | Membrane potential | 50 | 0.15 | 0.07 | 0.25 | 0.3 |
| Compound 1 | Cell wall synthesis | >256 | 20 | 0.4c | >256 | >256 |
| Compound 2 | Cell wall synthesis | >64 | 16d | >64 | >64 | >64 |
Incorporation of radiolabeled precursors was measured in E. coli ΔtolC ΔlysA. IC50, 50% inhibitory concentration.
DAP, diaminopimelic acid.
The IC50 is based on a maximum inhibition of <40%.
The IC50 is based on a maximum inhibition of <42% due to the compound solubility.
Nonejuie et al. have shown that antibacterial compounds cause distinct changes in cellular morphology depending on their mode of action (24). The effects of compounds 1 and 2 on the morphology of E. coli ATCC 25922 ΔtolC were examined to investigate which cellular pathways they inhibit. The cells were exposed to either 2% DMSO (vehicle control), compound 1, compound 2, or meropenem at 2× to 10× the MIC for 120 min at 30°C. The cells were then subjected to fluorescence microscopy, with FM4-64 staining of membranes, DAPI (4′,6-diamidino-2-phenylindole) staining to visualize the nucleoid, and Sytox green staining to detect membrane permeabilization. All three compounds gave distinctly different morphologies, despite the fact that each inhibits a component related to cell wall biogenesis (Fig. 3). Compound 1 caused the cells to elongate relative to the DMSO control-treated cells, as well as giving faint staining of the interior of the cell with Sytox green, which indicates a loss of membrane integrity. Compound 2 also caused cell elongation, but in contrast to the cells treated with compound 1, the cells treated with compound 2 were quite swollen, and the nucleoids appear to be less condensed than those of the vehicle control cells. Some of the cells were also very brightly stained with Sytox green, which indicates a loss of membrane integrity. Both these morphologies are quite different from that with meropenem, which targets PBPs (reviewed in reference 32). In the presence of meropenem, the majority of cells were elongated, with distinct bulges in the middle, and the nucleoids were decondensed compared to those of the vehicle control cells, as previously described (24).
FIG 3.

Morphology and staining of E. coli ΔtolC strain in the presence of inhibitor compounds. E. coli ATCC 25922 ΔtolC was treated with the indicated compounds for 120 min and then stained with FM4-64 (red), DAPI (blue), and Sytox green (green) as previously described (24). An overlay of FM4-64 and DAPI is also shown for each compound. Bars, 1 μm.
Mode of action of compound 1.
To further define the cellular target of compound 1, resistant mutants were generated using an E. coli MG1655 ΔtolC strain. The frequencies of resistance ranged from 2.1 × 10−8 at 8× the MIC to 4.7 × 10−8 at 64× the MIC for compound 1. Fifteen stably resistant mutants were subjected to whole-genome sequencing using an Illumina platform. All 15 mutants were found to have single amino acid changes in LpxH. LpxH is essential for cell viability and catalyzes the fourth step in the biosynthesis of lipid A, the outer membrane anchor of LPS (13, 33, 34). The mutations mapped to four different residues in LpxH: G48, L84, F141, and R149. The MIC of compound 1 for each of these four mutants increased >512-fold compared to that for the parent strain (Table 3). The susceptibilities of these LpxH mutants did not change for control antibiotics, such as levofloxacin, meropenem, and tetracycline, indicating that the mechanism of resistance is specific to compound 1. Interestingly, these LpxH mutants also showed no change in susceptibility to the LpxC inhibitor PF1090, which targets an earlier step in the same pathway as that of LpxH (35). These results strongly suggest that this compound inhibits bacterial cell growth through inhibition of LpxH activity.
TABLE 3.
Susceptibility of compound 1 to resistant isolates (LpxH mutants) and LpxH-overexpressing strains
| Compound | MIC (μg/ml) for strain |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Parenta | LpxH (G48D) | LpxH (L84R) | LpxH (F141L) | LpxH (R149H) | pPSV35b | pPSV35 plus 50 μM IPTG | pPSV35-LpxHc | pPSV35-LpxH plus 50 μM IPTG | |
| Compound 1 | 0.25 | >128 | >128 | >128 | >128 | 0.125 | 0.125 | 0.125 | >128 |
| Levofloxacin | 0.008 | 0.008 | 0.008 | 0.008 | 0.004 | 0.004 | 0.004 | 0.004 | 0.004 |
| Meropenem | 0.016 | 0.031 | 0.016 | 0.016 | 0.008 | 0.031 | 0.031 | 0.031 | 0.031 |
| PF1090 | 0.008 | 0.008 | 0.008 | 0.008 | 0.004 | 0.004 | 0.004 | 0.004 | 0.004 |
| Tetracycline | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.125 | 0.25 | 0.0625 | 0.125 |
E. coli MG1655 ΔtolC.
E. coli MG1655 ΔtolC plus empty vector (pPSV35) control.
E. coli MG1655 ΔtolC/pPSV35 expressing LpxH under the control of an IPTG-inducible promoter.
To expand on these results, high-copy-number suppression by use of LpxH was used to confirm it as the cellular target of compound 1. lpxH from E. coli was cloned into a plasmid under the control of an IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible promoter (pPSV35) (19) and transformed into E. coli MG1655 ΔtolC. In the presence of 50 μM IPTG, the MIC of compound 1 against a strain overexpressing LpxH increased to >128 μg/ml, while other antibacterial compounds that do not inhibit LpxH showed no change in MIC (Table 3). These data are consistent with compound 1 having a mode of action through inhibition of LpxH, as the susceptibility to this inhibitor decreased upon an increased copy number of LpxH in the cell.
Mode of action of compound 2.
Mutants resistant to the pyrazole compound 2 were raised in E. coli ATCC 25922 ΔtolC (a pathogenic strain of E. coli with an engineered deletion of the efflux pump TolC) to define its mode of action. The frequency of resistance was determined to be 7.3 × 10−7 at 32× the MIC. At concentrations lower than 32× the MIC, confluent growth was observed. Whole-genome sequencing was performed on 10 stably resistant mutants by using an Illumina platform. Each isolate carried a mutation in a single gene locus. Mutations were mapped to either lolC, lolE, the predicted promoter region of lpp, or a locus predicted to carry two overlapping, divergent genes, annotated as the Z2510 and Z2511 genes, as further defined below.
The Lol mutations corresponded to either a single amino acid change, G254V, in LolC, or three different amino acid changes in LolE: G195S, P365C, and D367Y. LolC and LolE are both essential for cell viability and are members of an ABC transporter complex, LolCDE, which is responsible for releasing lipoproteins from the inner membrane for transport to the outer membrane (14, 36). All of these mutations in either LolC or LolE caused very large increases in the MIC of compound 2 (>1,024-fold) but not in those of other classes of antibiotics (Table 4). Interestingly, a mutation in the predicted promoter region of lpp was also isolated. This mutation caused a 32-fold increase in the MIC of compound 2. Lpp is one of the most abundant E. coli outer membrane lipoproteins and interacts both covalently and noncovalently with peptidoglycan to stabilize the cell surface structure (37, 38). When lipoprotein trafficking to the outer membrane is disrupted, Lpp accumulates in the inner membrane, where it binds covalently to peptidoglycan and is lethal for the cell (39, 40). It has been shown that mutations in Lpp can cause resistance to globomycin or myxovirescin, antibiotics that inhibit the signal peptidase II protein (LspA), which cleaves the signal peptide from lipoproteins (41–43). This is a step in lipoprotein maturation that occurs prior to LolCDE releasing lipoproteins for transport to the outer membrane. In order to confirm that this lpp promoter mutation (presumably due to decreased lpp transcription) is responsible for resistance to compound 2, an E. coli strain carrying a deletion in lpp was tested for susceptibility to compound 2. The E. coli MG1655 ΔtolC Δlpp strain (12) showed a 64-fold increase in the MIC of compound 2 relative to that for the E. coli MG1655 ΔtolC parental strain (data not shown), indicating that a loss of Lpp in the cell led to compound 2 resistance. There was no change in susceptibility of the lpp mutant to compounds that do not inhibit lipoprotein transport to the outer membrane, as expected (Table 4). We previously identified a structurally distinct inhibitor of the LolCDE complex (12), with mutations conferring resistance mapped to LolC (N265K) and LolE (L371P). These mutants were found to be resistant to compound 2 as well. The MIC of compound 2 increased 64-fold against each of these mutants (data not shown).
TABLE 4.
Relative sensitivities of compound 2-resistant isolates
| Compound | MIC (μg/ml) for strain |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parenta | LolC (G254V) | LolE (G195S) | LolE (P365C) | LolE (D367Y) | Plppb (A49G) | Z2510c (G28R) | Z2510 (A32P) | Z2510 (G46D) | Z2510 (G119fs)d | |
| Compound 2 | 0.125 | >128 | >128 | >128 | >128 | 4 | 4 | 8 | 8 | 4 |
| Levofloxacin | 0.004 | 0.004 | 0.004 | 0.004 | 0.004 | 0.004 | 0.016 | 0.016 | 0.016 | 0.008 |
| Meropenem | 0.016 | 0.016 | 0.016 | 0.016 | 0.016 | 0.016 | 0.016 | 0.016 | 0.016 | 0.016 |
| Tetracycline | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 1 | 1 | 1 |
E. coli ATCC 25922 ΔtolC.
Strain with a mutation in the promoter of lpp.
Z2510 is a putative transcriptional repressor from the AcrR family.
fs, frameshift mutation.
In addition to mutations in loci related to outer membrane lipoproteins, five different mutations (four single amino acid changes and one frameshift mutation) in Z2510 were also isolated. Z2510 is a putative AcrR family transcriptional repressor of unknown function that is encoded by some pathogenic strains of E. coli. Additionally, there is a putative open reading frame, annotated Z2511, that is divergent from and partially overlaps the Z2510 open reading frame. Therefore, these mutations would also affect Z2511. The Z2510 and Z2511 genes are positioned near an operon predicted to encode an efflux system, suggesting that they may be involved in regulating this process. These mutations caused 32- to 64-fold increases in the MIC of compound 2 (Table 4). The MIC of levofloxacin was also increased 2- to 4-fold in these strains, which suggests that these mutations cause a non-target-related efflux-mediated change in susceptibility to some antibiotics. Also, E. coli strains that lack Z2510 and Z2511, such as some E. coli K-12 strains, remained susceptible to compound 2 (data not shown), suggesting that these resistant mutants are not indicative of the mode of inhibition by compound 2.
Taken together, these data suggest that compound 2 inhibits bacterial growth by targeting LolCDE to block lipoprotein transport to the outer membrane. To determine whether compound 2 prevents trafficking of lipoproteins from the inner membrane to the outer membrane, we measured Lpp release from spheroplasts to purified LolA in the presence of compound 2. As a control, globomycin was also tested, as it inhibits a previous step in lipoprotein processing. Spheroplasts were prepared from E. coli and incubated with purified His-tagged LolA. The spheroplasts were then removed by centrifugation, and the supernatant was analyzed by SDS-PAGE and Western blotting with anti-Lpp antibody. The amount of Lpp released from the spheroplasts indicates the lipoprotein release activity. The blot was also probed for OmpA, an outer membrane protein not dependent on LolCDE for transport to the outer membrane. As shown in Fig. 4, the appearance of Lpp in the supernatant was dependent on the presence of both spheroplasts and purified LolA. Both globomycin and compound 2 decreased the amount of Lpp that was released from the spheroplasts. There was a slight decrease in the amount of OmpA released from the spheroplasts in the presence of compound 2 and globomycin, indicating that these compounds may also have some effects on protein synthesis or translocation across the inner membrane by the Sec machinery. To examine the effects of the LolC and LolE mutations that confer resistance to compound 2, spheroplasts were prepared from the E. coli ΔtolC LolE L371P and LolC N265K resistant mutants. Compound 2 was not able to inhibit release of Lpp from these mutant spheroplasts at the same concentration as that used to inhibit release from spheroplasts in the susceptible parent strain (Fig. 4). These data are consistent with compound 2 inhibiting E. coli growth through the LolCDE complex. Globomycin, which does not target LolCDE, was still capable of inhibiting Lpp release from the mutant E. coli spheroplasts, as expected (41).
FIG 4.

Compound 2 inhibits Lpp release from spheroplasts. Spheroplasts were prepared from E. coli MG1655 ΔtolC (parent) and the LolE (L371P) and LolC (N265K) mutant strains. Spheroplasts were incubated with purified LolA in the presence of DMSO (vehicle control), globomycin, or compound 2. The amount of Lpp released to LolA was detected by SDS-PAGE and subsequent immunoblotting with anti-Lpp antibodies (upper panel). OmpA, whose release from spheroplasts is independent of LolA, was also detected with an anti-OmpA antibody (lower panel).
DISCUSSION
An assay using an inducible AmpC reporter system as a sensor for molecules that inhibit processes related to E. coli cell wall biogenesis was first developed and validated for high-throughput screening. In conjunction with the cell wall phenotypic reporter, the strain also had a deletion of an RND efflux pump to improve detection of weak inducers of the reporter. The system exhibited a surprisingly broad response to a range of disturbances in the cell envelope assembly process, identifying novel inhibitors of both outer membrane lipoprotein transport and LPS biosynthesis. Therefore, this assay is capable of identifying inhibitors of a number of different targets in a single screening campaign, including many targets, such as the Lol transport system, that are not amenable to screening by an enzyme assay with a cell-free system. The results of this AmpC reporter screen also illustrate that disruption of systems involved in envelope assembly affects peptidoglycan biosynthesis and turnover processes, as demonstrated recently by a role for lipoproteins in the functioning of key cell wall (PBP) synthetic enzymes (44).
One of the inhibitors identified in this reporter screen was the sulfonyl piperazine compound 1, with antibacterial activity against an efflux mutant of E. coli. Mutations in mutants resistant to this compound mapped to LpxH, which suggests that the mode of action of this compound is to inhibit the production of LPS. Overexpression of LpxH in an efflux mutant of E. coli abolished sensitivity to compound 1, further implicating LpxH as the target of this compound. Numerous inhibitors of LpxC, which performs the first committed step in this pathway, have been reported (29, 35, 45); however, none of these compounds have advanced beyond phase I clinical testing. To our knowledge, compound 1 is the first reported inhibitor of LpxH. LpxH is essential for growth in E. coli and is present in most gammaproteobacteria; however, it has never been exploited as a target for therapeutic intervention (13, 33, 34). Due to its lipophilicity and high protein binding level in serum, compound 1 lacks the physical properties necessary for dosing in mammalian systems. The narrow antibacterial spectrum of this compound also limits its clinical use. It is possible that compound 1 does not permeate the outer membrane of some Gram-negative organisms of high unmet medical need, such as Pseudomonas aeruginosa, which is well known for its impermeable outer membrane and active efflux systems. Additionally, the high rate of spontaneous resistance to compound 1 indicates that targeting this protein alone may not lead to a single-agent therapy, but an LpxH inhibitor may need to be combined with another agent targeting a different step. In that regard, compound 1 may have promise as a chemical probe of LpxH function and for further assessment of the potential of LpxH as a target for antibacterial therapy.
Another inhibitor identified in the AmpC reporter screen was the pyrazole compound 2, with antibacterial activity primarily against E. coli. Mutations occurring at relatively high frequencies in mutants resistant to compound 2 mapped primarily to single amino acid changes in the LolC and LolE proteins, which form a complex with two copies of the LolD ATPase protein to form an inner membrane ABC transporter. This LolCDE complex is essential for cell viability and is responsible for releasing lipoproteins from the inner membrane to LolA, a periplasmic molecular chaperone, for transport to the outer membrane component, LolB (14, 36). Compound 2 was found to directly inhibit release of lipoproteins from spheroplasts to purified LolA, which confirms the genetic evidence found by mapping resistance mutations. These data suggest that the mode of action of compound 2 is to inhibit lipoprotein transport to the outer membrane. In addition, mutations in the lpp locus, which encodes one of the major outer membrane lipoproteins in the cell, also conferred resistance to compound 2, which further suggests that compound 2 inhibits lipoprotein trafficking to the outer membrane, as mislocalization of Lpp to the inner membrane is lethal to E. coli (39, 40). Thus, mutations that decrease the amount of Lpp in the cell should alleviate some of the toxicity that occurs in the presence of the inhibitor. Resistance mutations were also found in an uncharacterized putative transcriptional repressor; however, how these mutations lead to resistance to compound 2 is currently not known. These mutations do not cause as large a shift in the MIC for compound 2 as those in LolC or LolE, and this gene is not found in many susceptible strains of E. coli. It is therefore likely that these mutations correspond to a modest resistance mechanism for compound 2.
Inhibitors of outer membrane lipoprotein trafficking have been reported previously (12); however, to date, none of these molecules have been used therapeutically. Compound 2 lacks the physical properties and antibacterial spectrum required for clinical use, but it can be employed as a tool to aid in exploration of lipoprotein transport to the outer membrane, such as the physiological impact of disruption on outer membrane integrity. Compound 2 also displayed relatively high resistance rates and large shifts in MIC with single-step mutations. These properties are similar to what was reported for a structurally distinct pyridine imidazole series that targets the LolCDE complex (12). Whether this observed emergence of high resistance is a property of the target or these particular compounds still remains to be determined. Unfortunately, crystal structures are not available for either of the targets, so the binding modes of both compounds to their targets are unknown at present. Thus, determining the possibility of modifying the compounds to pick up additional binding interactions and strengthen compound targeting must await structural information.
One challenge in antibiotic discovery is that it has become evident that the physicochemical properties of the chemical libraries used for screening are not favorable for finding new antibiotics (46, 47). In addition, it has been argued that because of preexisting mutations in bacterial populations, single-gene targets are susceptible to single-step, high-level resistance and will need to be paired with agents that target other functions (6, 48). The results for the two targets presented here support these ideas, as do the recently published results on clinical resistance to the Anacor/GlaxoSmithKline oxaborole compound, which targets the leucyl tRNA synthetase editing site (49). Multiple-therapeutic-compound strategies for single-gene targets are employed as a matter of course in tuberculosis therapy (50). Inasmuch as inhibition of either the LpxH or LolCDE target would affect the cell envelope integrity, these compounds may act to potentiate a partner antibiotic's efficacy by improving influx through the outer membrane.
The rise of antibacterial resistance, particularly among Gram-negative pathogens, signals a growing need for novel antibacterial compounds. The results described above demonstrate that the AmpC reporter screen, combined with improved sensitivity due to efflux pump inactivation, is capable of uncovering a broad range of new molecules with intrinsic antibacterial activity that inhibit novel, cell envelope-related targets. The present screening results also suggest that interfering with the biogenesis of the Gram-negative cell envelope still has significant, untapped potential for therapeutic intervention, possibly by exploiting these novel targets to resensitize cells to current antibiotics by disruption of outer membrane assembly.
Supplementary Material
ACKNOWLEDGMENTS
We thank the AstraZeneca Infection Bioscience Department for MIC testing, James Whiteaker and Robert McLaughlin for whole-genome sequencing, Amy Kutschke for helping with assays to measure inhibition of macromolecular synthesis pathways, and Helen Plant for helping with high-throughput screening. We also thank Boudewijn de Jonge and Kirsty Rich for input and editing of the manuscript. We thank Joe Pogliano and employees of Linnaeus Bioscience for their bacterial cytological profiling services and helpful discussions.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02552-14.
REFERENCES
- 1.Rex JH, Eisenstein BI, Alder J, Goldberger M, Meyer R, Dane A, Friedland I, Knirsch C, Sanhai WR, Tomayko J, Lancaster C, Jackson J. 2013. A comprehensive regulatory framework to address the unmet need for new antibacterial treatments. Lancet Infect Dis 13:269–275. doi: 10.1016/S1473-3099(12)70293-1. [DOI] [PubMed] [Google Scholar]
- 2.Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 3.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J Jr. 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 4.Bugg TD, Braddick D, Dowson CG, Roper DI. 2011. Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol 29:167–173. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 6.Silver LL. 2011. Challenges of antibacterial discovery. Clin Microbiol Rev 24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson JW, Fisher JF, Mobashery S. 2013. Bacterial cell-wall recycling. Ann N Y Acad Sci 1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng X, Lin J. 2013. Beta-lactamase induction and cell wall metabolism in Gram-negative bacteria. Front Microbiol 4:128. doi: 10.3389/fmicb.2013.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zervosen A, Sauvage E, Frere JM, Charlier P, Luxen A. 2012. Development of new drugs for an old target: the penicillin binding proteins. Molecules 17:12478–12505. doi: 10.3390/molecules171112478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeCenzo M, Kuranda M, Cohen S, Babiak J, Jiang ZD, Su D, Hickey M, Sancheti P, Bradford PA, Youngman P, Projan S, Rothstein DM. 2002. Identification of compounds that inhibit late steps of peptidoglycan synthesis in bacteria. J Antibiot (Tokyo) 55:288–295. doi: 10.7164/antibiotics.55.288. [DOI] [PubMed] [Google Scholar]
- 11.Sun D, Cohen S, Mani N, Murphy C, Rothstein DM. 2002. A pathway-specific cell based screening system to detect bacterial cell wall inhibitors. J Antibiot (Tokyo) 55:279–287. doi: 10.7164/antibiotics.55.279. [DOI] [PubMed] [Google Scholar]
- 12.McLeod SM, Fleming PR, MacCormack K, McLaughlin RE, Whiteaker JD, Narita S, Mori M, Tokuda H, Miller AA. 2015. Small-molecule inhibitors of Gram-negative lipoprotein trafficking discovered by phenotypic screening. J Bacteriol 197:1075–1082. doi: 10.1128/JB.02352-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babinski KJ, Ribeiro AA, Raetz CR. 2002. The Escherichia coli gene encoding the UDP-2,3-diacylglucosamine pyrophosphatase of lipid A biosynthesis. J Biol Chem 277:25937–25946. doi: 10.1074/jbc.M204067200. [DOI] [PubMed] [Google Scholar]
- 14.Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Annu Rev Microbiol 65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 15.Yakushi T, Masuda K, Narita S, Matsuyama S, Tokuda H. 2000. A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat Cell Biol 2:212–218. doi: 10.1038/35008635. [DOI] [PubMed] [Google Scholar]
- 16.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 17.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomason LC, Costantino N, Court DL. 2007. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol Chapter 1:Unit 1.17. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- 19.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard, 9th ed Document M07-A8, vol 29, no. 2 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 21.de Jonge BL, Walkup GK, Lahiri SD, Huynh H, Neckermann G, Utley L, Nash TJ, Brock J, San Martin M, Kutschke A, Johnstone M, Laganas V, Hajec L, Gu RF, Ni H, Chen B, Hutchings K, Holt E, McKinney D, Gao N, Livchak S, Thresher J. 2013. Discovery of inhibitors of 4′-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy. Antimicrob Agents Chemother 57:6005–6015. doi: 10.1128/AAC.01661-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keating TA, Newman JV, Olivier NB, Otterson LG, Andrews B, Boriack-Sjodin PA, Breen JN, Doig P, Dumas J, Gangl E, Green OM, Guler SY, Hentemann MF, Joseph-McCarthy D, Kawatkar S, Kutschke A, Loch JT, McKenzie AR, Pradeepan S, Prasad S, Martinez-Botella G. 2012. In vivo validation of thymidylate kinase (TMK) with a rationally designed, selective antibacterial compound. ACS Chem Biol 7:1866–1872. doi: 10.1021/cb300316n. [DOI] [PubMed] [Google Scholar]
- 23.Hameed PS, Manjrekar P, Chinnapattu M, Humnabadkar V, Shanbhag G, Kedari C, Mudugal NV, Ambady A, de Jonge BL, Sadler C, Paul B, Sriram S, Kaur P, Guptha S, Raichurkar A, Fleming P, Eyermann CJ, McKinney DC, Sambandamurthy VK, Panda M, Ravishankar S. 2014. Pyrazolopyrimidines establish MurC as a vulnerable target in Pseudomonas aeruginosa and Escherichia coli. ACS Chem Biol 9:2274–2282. doi: 10.1021/cb500360c. [DOI] [PubMed] [Google Scholar]
- 24.Nonejuie P, Burkart M, Pogliano K, Pogliano J. 2013. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci U S A 110:16169–16174. doi: 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan J, de Jonge BL, MacCormack K, Sriram S, McLaughlin RE, Plant H, Preston M, Fleming PR, Albert R, Foulk M, Mills SD. 2014. A novel high-throughput cell-based assay aimed at identifying inhibitors of DNA metabolism in bacteria. Antimicrob Agents Chemother 58:7264–7272. doi: 10.1128/AAC.03475-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuyama S, Tajima T, Tokuda H. 1995. A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J 14:3365–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Honore N, Nicolas MH, Cole ST. 1986. Inducible cephalosporinase production in clinical isolates of Enterobacter cloacae is controlled by a regulatory gene that has been deleted from Escherichia coli. EMBO J 5:3709–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikaido H. 1998. Antibiotic resistance caused by gram-negative multidrug efflux pumps. Clin Infect Dis 27(Suppl 1):S32–S41. doi: 10.1086/514920. [DOI] [PubMed] [Google Scholar]
- 29.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CR. 2005. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry 44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 31.Shapiro AB, Walkup GK, Keating TA. 2009. Correction for interference by test samples in high-throughput assays. J Biomol Screen 14:1008–1016. doi: 10.1177/1087057109341768. [DOI] [PubMed] [Google Scholar]
- 32.Lister PD. 2007. Carbapenems in the USA: focus on doripenem. Expert Rev Anti Infect Ther 5:793–809. doi: 10.1586/14787210.5.5.793. [DOI] [PubMed] [Google Scholar]
- 33.Babinski KJ, Kanjilal SJ, Raetz CR. 2002. Accumulation of the lipid A precursor UDP-2,3-diacylglucosamine in an Escherichia coli mutant lacking the lpxH gene. J Biol Chem 277:25947–25956. doi: 10.1074/jbc.M204068200. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Quinn PJ. 2010. Lipopolysaccharide: biosynthetic pathway and structure modification. Prog Lipid Res 49:97–107. doi: 10.1016/j.plipres.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Montgomery JI, Brown MF, Reilly U, Price LM, Abramite JA, Arcari J, Barham R, Che Y, Chen JM, Chung SW, Collantes EM, Desbonnet C, Doroski M, Doty J, Engtrakul JJ, Harris TM, Huband M, Knafels JD, Leach KL, Liu S, Marfat A, McAllister L, McElroy E, Menard CA, Mitton-Fry M, Mullins L, Noe MC, O'Donnell J, Oliver R, Penzien J, Plummer M, Shanmugasundaram V, Thoma C, Tomaras AP, Uccello DP, Vaz A, Wishka DG. 2012. Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections. J Med Chem 55:1662–1670. doi: 10.1021/jm2014875. [DOI] [PubMed] [Google Scholar]
- 36.Yasuda M, Iguchi-Yokoyama A, Matsuyama S, Tokuda H, Narita S. 2009. Membrane topology and functional importance of the periplasmic region of ABC transporter LolCDE. Biosci Biotechnol Biochem 73:2310–2316. doi: 10.1271/bbb.90451. [DOI] [PubMed] [Google Scholar]
- 37.Choi DS, Yamada H, Mizuno T, Mizushima S. 1986. Trimeric structure and localization of the major lipoprotein in the cell surface of Escherichia coli. J Biol Chem 261:8953–8957. [PubMed] [Google Scholar]
- 38.Inouye M, Shaw J, Shen C. 1972. The assembly of a structural lipoprotein in the envelope of Escherichia coli. J Biol Chem 247:8154–8159. [PubMed] [Google Scholar]
- 39.Inukai M, Takeuchi M, Shimizu K, Arai M. 1979. Existence of the bound form of prolipoprotein in Escherichia coli B cells treated with globomycin. J Bacteriol 140:1098–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yakushi T, Tajima T, Matsuyama S, Tokuda H. 1997. Lethality of the covalent linkage between mislocalized major outer membrane lipoprotein and the peptidoglycan of Escherichia coli. J Bacteriol 179:2857–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inukai M, Takeuchi M, Shimizu K, Arai M. 1978. Mechanism of action of globomycin. J Antibiot (Tokyo) 31:1203–1205. doi: 10.7164/antibiotics.31.1203. [DOI] [PubMed] [Google Scholar]
- 42.Xiao Y, Gerth K, Muller R, Wall D. 2012. Myxobacterium-produced antibiotic TA (myxovirescin) inhibits type II signal peptidase. Antimicrob Agents Chemother 56:2014–2021. doi: 10.1128/AAC.06148-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zwiebel LJ, Inukai M, Nakamura K, Inouye M. 1981. Preferential selection of deletion mutations of the outer membrane lipoprotein gene of Escherichia coli by globomycin. J Bacteriol 145:654–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lupoli TJ, Lebar MD, Markovski M, Bernhardt T, Kahne D, Walker S. 2014. Lipoprotein activators stimulate Escherichia coli penicillin-binding proteins by different mechanisms. J Am Chem Soc 136:52–55. doi: 10.1021/ja410813j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hale MR, Hill P, Lahiri S, Miller MD, Ross P, Alm R, Gao N, Kutschke A, Johnstone M, Prince B, Thresher J, Yang W. 2013. Exploring the UDP pocket of LpxC through amino acid analogs. Bioorg Med Chem Lett 23:2362–2367. doi: 10.1016/j.bmcl.2013.02.055. [DOI] [PubMed] [Google Scholar]
- 46.Gwynn MN, Portnoy A, Rittenhouse SF, Payne DJ. 2010. Challenges of antibacterial discovery revisited. Ann N Y Acad Sci 1213:5–19. doi: 10.1111/j.1749-6632.2010.05828.x. [DOI] [PubMed] [Google Scholar]
- 47.O'Shea R, Moser HE. 2008. Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem 51:2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 48.Silver LL. 2007. Multi-targeting by monotherapeutic antibacterials. Nat Rev Drug Discov 6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
- 49.O'Dwyer K, Spivak AT, Ingraham K, Min S, Holmes DJ, Jakielaszek C, Rittenhouse S, Kwan AL, Livi GP, Sathe G, Thomas E, Van Horn S, Miller LA, Twynholm M, Tomayko J, Dalessandro M, Caltabiano M, Scangarella-Oman NE, Brown JR. 2015. Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrob Agents Chemother 59:289–298. doi: 10.1128/AAC.03774-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rattan A, Kalia A, Ahmad N. 1998. Multidrug-resistant Mycobacterium tuberculosis: molecular perspectives. Emerg Infect Dis 4:195–209. doi: 10.3201/eid0402.980207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.