


Abstract
We describe solid-phase cloning (SPC) for high-throughput assembly of expression plasmids. Our method allows PCR products to be put directly into a liquid handler for capture and purification using paramagnetic streptavidin beads and conversion into constructs by subsequent cloning reactions. We present a robust automated protocol for restriction enzyme based SPC and its performance for the cloning of >60 000 unique human gene fragments into expression vectors. In addition, we report on SPC-based single-strand assembly for applications where exact control of the sequence between fragments is needed or where multiple inserts are to be assembled. In this approach, the solid support allows for head-to-tail assembly of DNA fragments based on hybridization and polymerase fill-in. The usefulness of head-to-tail SPC was demonstrated by assembly of >150 constructs with up to four DNA parts at an average success rate above 80%. We report on several applications for SPC and we suggest it to be particularly suitable for high-throughput efforts using laboratory workstations.
INTRODUCTION
Within molecular biology and all of its associated sciences, the manipulation, design and transfer of genetic material is essential, in particular the assembly of DNA constructs for the expression of recombinant proteins. Once discovered, the ability to cut and paste DNA by restriction endonucleases (REases) and ligase (1) has transformed molecular biology and enabled new means of diagnostics and treatment of disease, as well as large-scale genomics projects. Cloning with restriction enzymes and ligase is still more than 40 years after its introduction considered a highly stable and validated method and a standard method in many labs all over the world. Today over 19 000 REases are listed in REBASE and reagents targeting over 200 different specificities are commercially available (2). Despite being among the most accurate enzymes, thanks to their preferential binding and catalysis, star activity or digestion of additional sites is a general phenomenon among REases when subjected to non-optimal reaction conditions such as: ionic strength, replacement of preferred cations, dimethyl sulfoxide (DMSO) or glycerol presence, or to high enzyme concentration (2) which might be the case during typical in vitro reactions. The wish to increase the utility and robustness of the enzymes to work under a broader span of reaction conditions has led to the development of engineered enzymes and buffer supplements such as spermidine reducing star-activity (3) as well as tailored gel and column-based purification kits allowing buffer exchange and removal of unwanted components between cloning steps. This has greatly enhanced the versatility of RE based cloning although some of these steps are less suitable for automation.
When several DNA parts are to be put together this requires identification of multiple unique restriction sites to avoid cutting at undesired locations. Since a complex one-pot reaction with several enzymes active at the same time becomes more difficult, multiple-insertion experiments are often done by cloning one insert at a time followed by validation and plasmid preparation after each insertion, making this a time consuming effort. Since REases require a specific recognition sequence (cloning site), this has to be introduced into vectors or open reading frames (ORFs), thereby causing nucleic or amino acid sequence scars between the insertion points. Several cloning methods have been developed with the goal of standardizing DNA parts so that they can be easily shared between laboratories. BioBrick and BglBrick techniques make use of ‘scars’ to allow for the re-usage of only a handful of restriction enzymes (4,5). However, the sequence scars between insertion points are undesirable for some applications. Golden Gate is one of several cloning methods that can eliminate scars through use of TypeIIS restriction enzymes that can remove their own recognition sites during cloning (6,7). However, for all RE based methods, limitations regarding the availability of restriction sites still remain.
Other cloning methods have taken a different route, excluding restriction enzymes completely, e.g. the exonuclease based Ligation Independent Cloning (8), TA-cloning (9,10), USER cloning (11), cloning by overlap extension polymerase chain reaction (PCR) (12,13) and cloning methods based on recombinases (14). A more recent and improved variant of exonuclease-aided cloning is Gibson assembly (15) providing sequence independent assembly of multiple DNA parts, while CPEC (16) is an overlap extension PCR option for the same purpose. Other alternatives for sequence independent cloning of many DNA parts are, SLiCE (17) and Ligase Cycling Reaction (18). These methods can be used in conjunction with counter-selection markers such as the toxin gene ccdB to reduce template background (14) and some, such as USER cloning, can be performed with crude PCR products. However, most of the protocols require insert purification, elimination of template vectors, concentration determination, followed by calculations and preparation of the right ratios of the DNA parts to be assembled. This has prompted us to seek alternative routes based on solid phase approaches, which allow DNA pieces and enzyme(s) to react in optimal buffers and at optimal temperature(s) in an automated series of reactions.
Immobilization of DNA has shown to be very useful for several applications including next generation DNA sequencing (19,20) genotype/phenotype coupling (21–23) microfluidics-based biosensors (24) and expression-profiling chips (25). Paramagnetic beads have also been used in cloning applications as a physical handle on the DNA construct, eliminating the need for gel or spin column purification steps in automated sub-cloning when transferring one insert from a vector to another using restriction enzymes (26). The great advantage of the solid phase in all of these applications is the ability to capture the desired product and allow for washing and buffer exchanges for a pure efficient reaction and product formation to take place.
Here we present new solid-phase cloning (SPC) schemes, where streptavidin-coated paramagnetic beads are used as solid support, allowing for automated assemblies of single and multiple DNA parts. First, we developed RE-based SPC to allow for large-scale cloning of single human genes from RNA into Escherichia coli expression vectors. This solid-phase method was used for the cloning of over 60 000 human gene fragments within the frame-work of the Human Protein Atlas (HPA) (27) program. The solid-phase allows optimal conditions for all reactions and generates pure inserts for ligation into expression vectors. Secondly, we developed head-to-tail SPC assembly for applications where a scar-free connection or insertion of multiple DNA parts is wanted. Utilizing the ability to selectively elute one DNA strand from a captured PCR-product, this approach allows for an exact assembly of constructs using hybridization and polymerase overlap extension of bead-bound DNA without introducing scars between the pieces. We believe both of these new SPC methods to be highly suitable for high-throughput applications using automated laboratory workstations.
MATERIALS AND METHODS
RE-based SPC
Primer design
Primers for amplification of target sequences were designed with in-house developed software using the following criteria: strict design: Tm 58–62°C, length 17–24 nt, primer ends with G or C, max 4 consecutive identical nucleotides, max 4 di-nucleotide repeats, no palindromes longer than three base pairs (with three or more nucleotides between palindrome pairs), not more than three consecutive base pairs (max 2 G|C) between primer pairs in the 3′ complementarities, not more than five consecutive base pairs (max 3 G|C) within primer pairs, no perfect matches to other human transcripts, max 4 G/C of last 6 nucleotides in 3′ end. Semi-strict: same as strict but without max 4 G|C of last 6 nucleotides in 3′ end. Semi-loose: Tm 58–62°C, length 17–24 nt, primer ends with G|C, max 3 consecutive identical nucleotides. Loose: Tm 58–62°C and length 17–24 nt. Melting temperatures were calculated using the ‘4 + 2’ formula, Tm = 4 * (nG + nC) + 2 * (nA + nT). Sequences are found in the supplementary information.
Amplification of insert
Each human gene fragment was amplified from total RNA by two PCR reactions, first using unique primers to capture the gene of interest, then by generic primers to provide biotin-attachment to the insert. First amplification: 3.25 μl water, 10 μl Reaction mix (2x), 0.5 μl pooled total RNA (four different pools of human total RNA (1 μg/μl) were used, Clontech, Mountain View, California or Agilent Technologies Santa Clara, CA, USA), 3 μl reverse primer (4 pmol/μl) and 0.25 μl of Superscript III RT/Platinum Taq (Life Technologies, Bleiswijk, The Netherlands) by incubation at 50°C for 30 min before cooled to 4°C. Three microliter of forward primer (4 pmol/μl) was then added and the subsequent PCR program was carried out: 94°C 1 min, 15 cycles of 94°C 20 s, 55°C 30 s, 72°C 2 min, final extension 72°C 10 min, cooled to 4°C. Second amplification: 32 μl water, 5 μl dNTPs (2 mM), 5 μl Pfx50 buffer (10x), 1 μl of each generic primer (10 pmol/μl), 1 μl Pfx50 (5 U/μl, Life Technologies, Bleiswijk, Netherlands) and 5 μl of the first amplification, with PCR temperature profile: 95°C 1 min, 35 cycles of 95°C 30 s, 50°C 30 s, 68°C 1 min before cooled to 4°C.
Restriction of insert
A Magnatrix 8000+(NorDiag AS, Oslo, Norway) liquid handler was used for the following steps. For DNA capture on beads, 15 μl of the PCR product was immobilized using 20 μl of washed Dynabeads® M-270 Streptavidin in 35 μl 2× binding buffer (2 M NaCl, 10 mM Tris–HCl, 1 mM Ethylenediaminetetraacetic acid (EDTA), 0.1% Tween 20, pH 7.6). Beads were washed one time with wash buffer (WB) (water w/ 0.1% Tween 20). Insert was first cleaved with 0.05 μl NotI-HF (20 U/ μl, New England Biolabs, Ipswich, MA, USA) in 15.3 μl water, 2.2 μl WB, 2.2 μl NEB4 Buffer (10×) and 0.2 μl BSA at 37°C for 1 h. The temperature was lowered to 20°C and the beads were washed 1× with wash buffer. The insert was cleaved a second time with 0.1 μl AscI (10 U/ μl, New England Biolabs, Ipswich, MA, USA) in 15.3 μl water, 2.2 μl wash buffer (water w/ 0.1 μl Tween 20), 2.2 μl NEB4 Buffer (10×) at 37°C for 1 h. Temperature was lowered to 20°C. Supernatant was separated from beads and incubated at 70°C for 5 min. Temperature was lowered to 20°C.
Ligation and transformation
Five microliters of the prepared insert was ligated with 1 μl of cleaved expression vector (∼100 ng/μl) in 11.8 μl water, 2 μl T4 DNA ligase buffer (10×) and 0.2 μl T4 DNA ligase (5 U/μl, Thermo Fischer Scientific Inc., Waltham, MA, USA) at room temperature for 2 h. Ligation reaction was transferred to 150 μl of ice cold E. coli Rosetta(DE3) and was incubated for 30 min on ice. Cells were heat shocked at 41°C for 5 min if using deep well plate or 40°C for 4.5 min if using microcentrifuge tube. After 5 min of incubation on ice, 700 μl of pre-warmed TSB + Y was added before incubation at 37°C for 30–60 min, gentle mixing. Samples were centrifuged for 2 min at 700 RCF, ∼620 μl of media was removed and cells were re-suspended before plating on pre-warmed agar plates.
Head-to-tail SPC
Amplification of DNA parts
Primers (Integrated DNA Technologies, Heverlee, Belgium) were designed by hand (aided by IDT OligoAnalyzer for Tm values) with complementary regions of 30 bases for hybridization. Delta G-values for these overhangs spanned from 35.2 to 51 Kcal/mol (28) (calculated using default settings). One primer per amplification reaction had a biotinylated 5′-end and 5 pmol of each primer was used in a 50 μl PCR reaction. DNA parts were amplified with Phusion® Hot-Start Flex (2 U/μl, New England Biolabs, Ipswich, MA, USA) by the following PCR program: 98°C for 30 s, 30 cycles of 98°C 8 s, 25 s annealing with temperature depending on primer melting temperature and 72°C for 20 s/kb, before ending with 72°C for 7 min followed by 4°C hold. Sequences are detailed in the supplementary information.
DNA immobilization on beads
All of the PCR reactions for the backbone and insert parts were separately immobilized using 20 μg of Dynabeads® M-270 Streptavidin beads (Life Technologies, Bleiswijk, The Netherlands) in 50 μl immobilization buffer (2 M NaCl, 10% w/v PEG 6000, 10 mM Tris–HCl, 1mM EDTA, 0.1% Tween 20, pH 7.6) during 30 min. After immobilization, beads were washed 3× with 45 μl wash buffer (WBss) (1× TE supplemented with 0.01% Tween 20).
Strand elution
Backbone and insert beads were re-suspended in 20 μl of NaOH (0.15 M) and incubated at room temperature for 10 min, to allow for strand elution. Backbones beads were captured and the backbone part supernatant was discarded before beads were washed with 45 μl of NaOH (0.15 M). Insert beads were captured and the supernatant of these was mixed with the backbone beads. When using synthetic oligos as inserts (PAGE Ultramer(R) DNA Oligo, Integrated DNA Technologies, Heverlee, Belgium), 5 pmol of oligo was added to 20 μl of NaOH (0.15 M). No prior bead immobilization or strand elution was needed for the oligos.
Hybridization
A mixture of 20 μl HCl (0.15 M), 10 μl Tris–HCl (1 M, pH 7.4), 10 μl PEG 6000 (50% w/v) and 20 μl water was preheated at 77°C. To this mixture, the backbone beads with insert strands and sodium hydroxide were added. The temperature was thereby decreased by 1°C every 20 s until it reached 50°C. Mixing was done at 70°C and 60°C. Beads were then captured and supernatant discarded.
Extension
An extension mix containing 17.25 μl water, 2.5 μl Phusion buffer (10×), 2.5 μl dNTPs (2 mM) and 0.25 μl Phusion (2 U/ μl New England Biolabs, Ipswich, MA, USA) was pre-heated at 65°C before used to resuspend the beads from the hybridization. Phusion HF was used as preferred buffer for extension unless the insert was PCR amplified using a different buffer; in those instances the matching buffer (e.g. Phusion GC buffer) was used also during the extension. The beads were incubated at 65°C for 3 min before temperature was raised to 72°C. Sample was incubated at 72°C for 5 min (9 min if it was the last extension). Beads were then captured and supernatant discarded. Beads were washed with 3 × 45 μl WBss before repeating the ‘strand elution’ step to incorporate the next part.
Bead release
After the final extension, beads were washed with sodium dodecyl sulphate (SDS) wash buffer (1× TE w/ Tween 20 0.01% and SDS 2%). The SDS wash was incubated for 10 min. This wash was followed by three washes with regular WBss. Beads were subsequently incubated for 15 min at 37°C with 8.67 μl water, 1 μl FD Buffer and 0.33 μl Fast Digest enzyme (Thermo Fischer Scientific Inc, Waltham, MA, USA). NotI, AscI, NheI, EcoRI, AatII, BamHI and BsiWI have been used for bead release and no variance in outcome has been observed between them. A detailed view of the sequences near the bead and terminal ends is presented in Supplementary Table S1. The enzyme was heat-inactivated for 5 min at 80°C before returned to room temperature. Supernatant from the bead release was mixed with 34 μl water, 0.67 μl ATP (10 mM), 4 μl T4 Ligase buffer (10×), 1.33 μl T4 Ligase (Thermo Fischer Scientific Inc, Waltham, MA, USA) and incubated for 45 min at room temperature.
Transformation
Eight microliters from the ligation mixture was mixed with 2 μl of 5× 500 mM KCl, 150 mM CaCl2, 250 mM MgCl2 (KCM) and incubated on ice for 3 min before 10 μl of TOP10 chemically competent cells were added (29). Cells were left for 20 min on ice before heat shocked at 42°C for 1 min, put on ice for 3 min and thereafter 200 μl of TSB + Y was added and cells were incubated at 37°C for 1 h before plating.
RESULTS
Solid-phase cloning
Magnetic solid-phase support enables easy DNA capture, washing and switching to preferred reaction conditions between assembly steps. The procedures of the two methods are outlined in Figure 1. The restriction based method (Figure 1A) uses the paramagnetic beads to purify and sequentially wash, buffer exchange and digest PCR products, thereby providing a very simple and robust workflow for generation of ligated constructs without need for manual gel or spin-column based purification steps. Briefly, RE-based SPC starts by capturing PCR amplified inserts on streptavidin-coated paramagnetic beads, which then are sequentially washed, buffer-exchanged and incubated at appropriate temperatures for serial solid-phase digestions using independent REases in a liquid handler.
Figure 1.
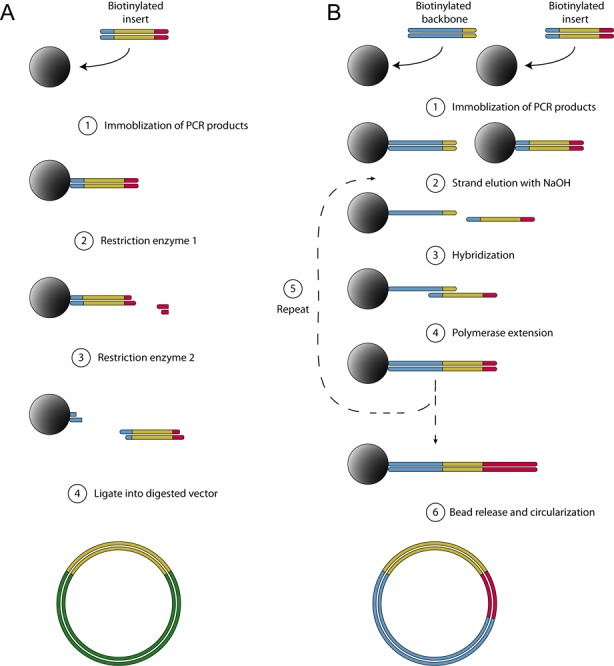
Schematic outline of solid-phase cloning. (A) RE-based: biotinylated PCR products are captured and purified on streptavidin-coated paramagnetic beads (1). Solid-phase digestion of immobilized DNA, followed by washing away of cut pieces and buffer exchange (2). A second restriction enzyme cleaves at a site close to the beads (3). The supernatant is separated from the beads and the restriction enzyme is heat-inactivated. This purified and cleaved PCR product is ligated into a vector (4). (B) Head-to-tail assembly: PCR amplified backbone and inserts are separately immobilized onto streptavidin-coated paramagnetic beads. (1) Non-biotinylated strands are eluted in NaOH. Eluted backbone strands are discarded while the eluted insert strands are kept. (2) The eluted insert strand hybridizes to complementary sequence on the bead-bound fragments. (3) A polymerase is added to extend the two strands. (4) To introduce more inserts, steps 1–3 are repeated. (5) The final construct is released from the beads by one restriction enzyme cutting at both ends, allowing it to be ligated and circularized.
The head-to-tail assembly (Figure 1B) uses the same type of capture of DNA for bead-based purification of PCR products on the solid support, followed by buffer exchange to NaOH to achieve DNA strand separation. By sequential addition and annealing of single-stranded DNA, carrying complementary end-regions, multiple DNA fragments are joined head-to-tail before release and circularization.
RE-based SPC
In total, 60 735 human gene fragments (Protein Epitope Signature Tags) (27) were amplified from RNA pools as cDNA and cloned into expression vectors by RE based SPC with an average success rate of 85% (Figure 2A). 10% were failed due to PCR-related issues, not generating a single band of expected size and 5% of the sequenced clones did not pass the sequencing validation. If no amplicon of correct size could be generated by any of the four RNA pools, the cloning outcome was annotated as PCR fail. If the correct amplicon length was detected but no correct sequence was found in any of the RNA pools, cloning outcome was annotated as sequencing fail. At the most, six colonies were sequenced for each RNA pool. Details can be found in the supplementary information. All cloning attempts were grouped into bins based on the length, guanine-cytosine (GC) or linguistic sequence complexity (LC) (30) content of the insert. Cloning performance did not show a straight correlation with insert length and the success rate was well above 80% for most bins although a slight decline was observed toward longer insert with a success rate of 70% for the longest (Figure 2B). A grouping by GC-content (Figure 2C) demonstrated that inserts with low GC-content performed substantially better than the more GC-rich inserts. The best performing bins, representing inserts with GC-content below 55%, had an average cloning success rate above 90%. A distinct decrease in success rate from 90 to 10% was unveiled when increasing GC content from 55 to 80%.
Figure 2.
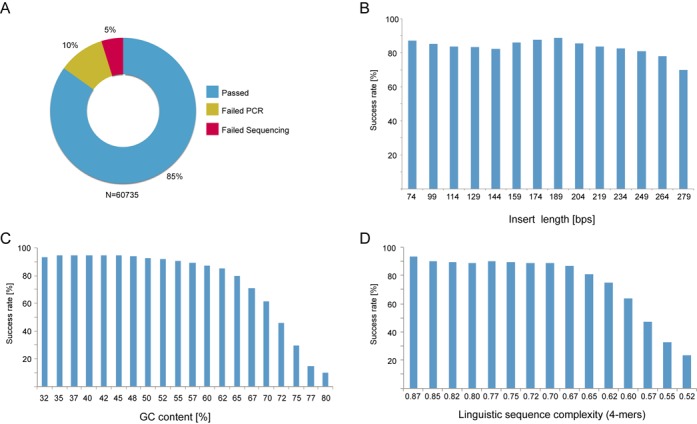
Results from restriction enzyme-based SPC. (A) Out of 60 735 cloning experiment, 51 556 were successful and 9173 unsuccessful. (B) The length of inserts did not have a clear impact on the success rate although a slight decline could be perceived for the longer inserts. (C) High GC-content of inserts was clearly affecting the overall cloning success rate negatively. The success rate dropped from above 90 down to 10% as the GC content increased from 55 to 80%. (D) Similar decline in success rate was also observed for sequences with LC-values below 0.67.
The effect of complexity on the cloning success rate was analyzed analogously by binning (Figure 2D). Here we found the most successful inserts having a high LC-value and a clear drop in success rate beginning with LC-values around 0.67. GC-content and complexity of a sequence is linked, hence these two characteristics were analyzed simultaneously in order to determine which one is of most importance (Figure 3). GC-content was found to be the most important with a distinct trend showing a decline in success rate with a higher percentage of GC while LC-values were more scattered. A logistic regression analysis test using GC content and linguistic complexity as independent variables and the cloning success status as a dichotomous outcome variable showed that both variables have a significant effect on the success rate (BGC = 49.0, P < 0.0001 and BLC = 25.7, P < 0.0001) and that there is significant interaction between them (BGC:CL = −57.6, P < 0.0001). Although both variables significantly affect the success rate, the coefficients indicate a greater effect from GC content than from linguistic complexity. Of the 6252 constructs failing the PCR-step, 4.5% had neither a successfully cloned construct amplified from the same transcript nor a template verified by deep sequencing of the RNA pools (31).
Figure 3.
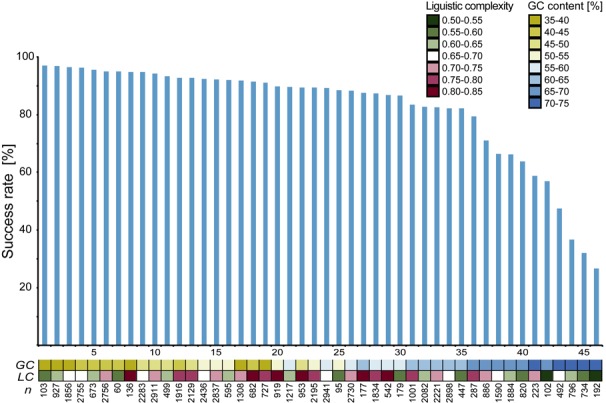
Success rate of RE-based experiments (blue bars, 1–46) grouped by insert characteristics (squares). The colored boxes indicate the magnitude of linguistic complexity content (green-red) or GC-content (yellow-blue) of inserts. Inserts with a low GC-content were unambiguously more successful compared to inserts with a high percentage of GC.
Head-to-tail SPC
In order to establish a robust head-to-tail SPC protocol working regardless of DNA parts being assembled, several sections of the protocol were subjected to improvements. Originally, extension was done at 37°C and although the results were passable when working with only two DNA parts, the efficiency rapidly subsided when attempting to assemble more parts, in particular due to miss-hybridizations. In order to reduce this risk, a set of thermostable polymerases with recommended extension temperature at 72°C were evaluated. DyNAzyme II and DyNAzyme EXT were found to be working well (Figure 5A). Unfortunately, these polymerases have limited proofreading capabilities and the disadvantageous addition of a non-templated adenine residue at the 3′ by these enzymes would cause problems when assembling multiple fragments. At first, the polymerases Deep Vent® and Phusion® with proof-reading capability, yielded few transformants (Figure 5A). Polymerases have varying binding affinity toward DNA, such as the high-fidelity enzyme Deep Vent which binds 200× stronger toward DNA compared to normal Taq polymerase (32). Such stronger affinity could make a polymerase more laborious to remove when used for extension of immobilized DNA. Incomplete removal of these polymerases could be troublesome since they potentially could blunt sticky ends before ligation or hinder bead release. Several different washing protocols were therefore investigated to improve the release of the enzyme from DNA, e.g. repeating washes (up to 9×) or supplementation of herring sperm DNA to the buffer, all giving none or marginal improvements except for addition with SDS. Phusion, Deep Vent and 9° North yielded correct colonies by addition of SDS to the final wash buffer (data not shown). Phusion was chosen based on its advantageous proofreading capability, and it was observed that 2% SDS wash buffer provided the best results (Figure 5B). Bead amounts, hybridization protocols and incubation times were also parameters subjected to improvements (data not shown).
Figure 5.
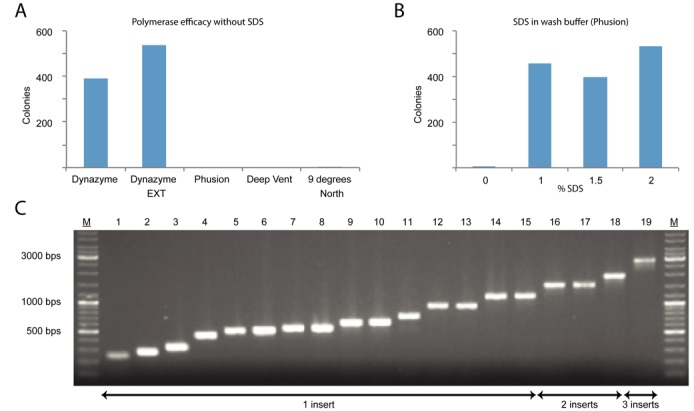
Results from head-to-tail SPC. (A) Comparison of compatibility of thermostable polymerases with the head-to-tail method. The polymerases with best proof-reading capabilities, Phusion and Deep Vent, all failed to generate transformants with the standard head-to-tail protocol. (B) Activity was retained for protocols using Phusion after supplementation of the washing buffer with SDS. (C) Colony screens of inserted region representing a selection of assemblies of various lengths and number of inserts assembled by head-to-tail SPC. Final construct sizes spanned 2.9 to 7.2 kbps.
Ten different two-fragment constructs of various lengths were assembled (Table 1. 1–10, in Figure 5C, bands 4 and 7–11 are examples of these). Supplementary Table S2 presents a more detailed view of Table 1, including insert lengths, number of picked colonies and success rate. All constructs, generated colonies with correct product. The average success rate per colony was 87% and the largest construct assembled was 7.2 kbp. The protocol was adapted for automated cloning using a liquid handling robot and several key features was shared with the RE-based SPC (Figure 4A and B). Both methods make use of liquid handlers with a temperature block and magnetic capture capabilities. RE-based SPC was set up on a laboratory workstation (see ‘Materials and Methods’ section for details) to prepare cloning inserts for 96 clones in parallel. With existing head-to-tail protocols, 12 constructs can be made in parallel assembling up to four DNA fragments on the Magnatrix 1200 workstation or up to 96 constructs of two fragments on Magnatrix 8000+ workstation. These can easily be reprogrammed to make more constructs or constructs with different number of inserts depending on application. The standard protocol for head-to-tail SPC takes 2 h and 30 min to go from PCR products to ligated plasmids and the 96-construct assembly takes 6 h. Detailed flow-charts for adaptation to other workstations can be found in the supplementary material (Supplementary Figure S1).
Table 1. Overview of assembled head-to-tail SPC constructs.
Figure 4.
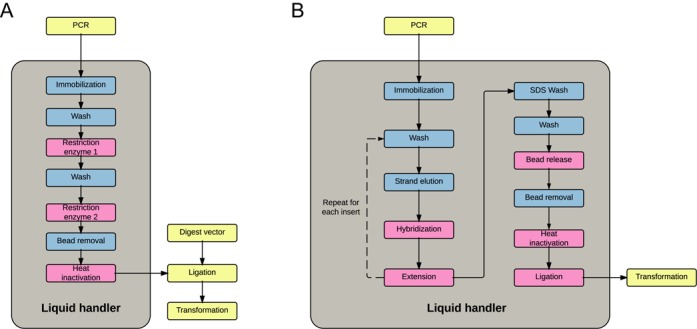
Unit-operations for RE-based and head-to-tail SPC. The light gray boxes indicate operations performed inside and yellow boxes indicate operations performed outside liquid handlers. Pink boxes are operations requiring temperature blocks while blue boxes indicate operations carried out in room temperature. (A) Restriction enzyme based SPC: PCR amplified inserts are automatically purified digested and subsequently ligated into a predigested vector. (B) Head-to-tail SPC: PCR amplified inserts and backbones are automatically purified, sequentially assembled by overlap extension and circularized into a plasmid. Transformation of Escherichia coli is done outside of the robot for both protocols.
Head-to-tail SPC was adapted for the high-throughput cloning application of reformatting antibody single-chain proteins into full-length heavy and light IgG1 proteins. The cloning involved assembly of the variable light and heavy regions for 48 single chain fragments into two corresponding full-length IgG vectors, yielding 96 plasmids (Table 1. 11). All variable fragments and backbone vectors including the Fc fragments were PCR amplified and thereafter directly loaded onto the liquid handler for reformatting with no prior PCR-purification. Of these assemblies, 95/96 were correct with an 87% success rate per colony. To affirm the potential of assembling multiple DNA parts, combinations of fusion proteins were cloned (Table 1. 12–15 and Figure 5C lanes 16–19 are four examples). Up to four DNA parts were assembled with over 80% of the colonies having all the inserts assembled. Equal results were obtained when assembling the same plasmid with Gibson Assembly® (Supplementary Figure S2). Another cloning application is the modular assembly of expression vectors by combination of standard components, such as promoters, selection marker or reporter protein. In order to see if it was possible to construct expression vectors using head-to-tail cloning, three vectors pHisZamp, pHisZKm and pAFF8c were assembled using four DNA fragments. One fragment carried the antibiotic resistance, one the origin of replication, one a promoter for recombinant production and one a gene to be expressed. Seventy-five percent of the assemblies were correct for these three vector assemblies (Table 1. 16–18). Transformation frequencies for three DNA parts were up to of 105 cfu/μg of backbone part using heat-shock transformation. If the inserts are short, it is possible to synthesize single-stranded oligos that are ready to be used without requiring PCR and strand elution. Synthetic oligos were used to specifically tailor 17 expression vectors for protein secretion in mammalian cells by introducing signal peptide, protease site and different purification tags (Table 1. 19–20, Figure 5C lane 2 is one example). Within antibody engineering precise construction of fusion proteins is essential to avoid unwanted amino acids which might introduce unwanted glycosylation sites, immunogenic residues or residues affecting protein stability or scale-up processes. For this purpose we evaluated the cloning method for generation of engineered antibodies and proteins carrying an additional GFP11 peptide (33) at precise positions of the c-termini using synthetic oligos as insert parts (Table 1. 19–25 and Figure 5C constructs 1, 3, 5 and 6). The success rate was on average 91% for these 26 constructs made with synthetic oligo inserts. Another antibody engineering application is the conversion of monoclonal antibodies into bi-specific moieties targeting multiple targets by genetic fusion of additional binding-domains to it. Head-to-tail SPC was evaluated for this purpose and allowed for generation of bi-specific antibody constructs by three piece cloning of affinity proteins to the C-terminal of the heavy or light chain of an antibody, using three different linker lengths (Table 1. 26 and Supplementary Figure S3). The linkers and small affinity proteins were all inserted as synthetic oligos on PCR amplified antibody-vectors.
DISCUSSION
There is a need for high-throughput automated cloning methods within several sectors of molecular biotechnology including, genomics, proteomics, protein engineering, metabolic engineering and structural genomics. RE-based SPC was developed to provide an efficient and reliable cloning of genes and gene fragments. It has so far performed over 60.000 cloning tasks and has become a reliable workflow with an overall success-rate of 85%. The head-to-tail SPC was developed to maintain the automated simplicity of RE-based SPC, but to reduce the dependency on restriction enzymes and addition of extra nucleotides due to the restriction sites. One of the benefits of head-to-tail SPC is its inherent compatibility with synthetic oligos, allowing for direct incorporation of shorter inserts by annealing to the immobilized ssDNA on the bead, thereby omitting the PCR step. Supplementary Table S3 lists examples of applications and how these benefit from assembly method properties including: need for multiple inserts, need for scar-free assembly or benefit from usage of synthetic oligos.
For the RE-based SPC, high GC-content affected the success rate of the cloning. At a GC-content above 55% a decline in success rate from 90 to 10% was observed. High GC-content is known to complicate polymerase extension due to secondary structures with high melting temperatures. This could explain failure at the PCR step. The observed effect of LC-content on success rate (Figure 2D) is probably explained by higher GC-content among inserts with low LC-values, hence the scattered distribution when comparing the two factors simultaneously (Figure 3). Given these results, we would recommend paying extra attention to inserts with high GC-content, either by changing PCR conditions (protocol parameters and DMSO supplementation) or if possible considering optimization of codons for these inserts. In the protocol described, all cloning steps until ligation are done in the workstation; future work could include automation of the remaining ligation and transformation steps for fully automated cloning process. In a previous report (26) a 10-fold improvement in ligation efficiency was observed when bead-bound vector and insert fragments were brought into proximity by a magnet during ligation, which might further improve the efficacy of this protocol as well. The method could also further benefit from utilizing typeIIS restriction enzymes, which would allow for a higher degree of freedom to choose sequences in the overhang regions.
For the head-to-tail SPC, the assembly of two DNA parts rely on polymerase extension, thus it seamlessly connects DNA-pieces without introducing additional bases or amino acids into the construct and expressed products. In our study, we show that the elution of high-fidelity polymerases from bead-bound DNA is possible by changing a basic wash buffer to an SDS-containing buffer. This is in agreement with previous studies where T4 Ligase have been washed off from DNA immobilized on a streptavidin chip by the addition of SDS (34). This change in the protocol remarkably improved the success rate and allowed for a more robust protocol. Hybridization and extension at elevated temperature reduces the risk of miss-hybridizations and problematic structures of DNA end-sequences, such as hairpin loops, preventing assembly. The transformation frequency around 104 – 105 cfu/μg backbone vector with chemically competent cells indicate a high degree of successful constructs and a possibility to use this method for construction of combinatorial libraries. Head-to-tail SPC requires only one restriction enzyme regardless of the number of DNA parts to be put together. This site is needed for the release of the construct from the beads once assembly has been completed, and it may be chosen freely at the point of design. The protocol could also be used completely without REases by dsDNA elusion upon streptavidin-biotin interruption, which might be beneficial for applications where linear products are preferred. Another possible route is to use bead attached DNA for direct transformation of mammalian cells as previously reported (35). Head-to-tail assemblies containing all the correct parts have been considered successful, however some of these contain errors at the nucleotide level. Such errors have almost exclusively been found in primer regions or as part of synthetic oligos making us believe that these are errors originating from oligo-synthesis (sequencing errors were observed three to four times outside such regions) rather than as a result of the PCR-reaction.
While our standardized head-to-tail protocol (same setup for all inserts) has provided reliable results for all constructs, the method allows for the possibility to incorporate unique conditions for the hybridization and extension of each insert, if needed for specific applications. Additional additives during hybridization and polymerase extension, such as DMSO, could improve the efficiency during these steps of the method. Future studies should focus on the detailed analysis of a larger set of head-to-tail SPC experiments to possibly improve and automate the oligo design based on parameters including overhang characteristics, DNA secondary structure and predictable off-target annealing. Many of the other cloning alternatives presented could benefit from a solid-phase support. In theory, any assembly being carried out in vitro could be turned into a solid-phase method. As such, a big multi-step assembly could be divided into a couple of smaller subassemblies in an automated fashion. By avoiding the ‘everything-in-one-pot’ situation one could reduce the risk of incorrect annealing and thereby envision the correct assembly of even larger constructs.
As DNA synthesis is improving in product-length and becoming more affordable, the ordering of complete constructs have become a real alternative to in house cloning for some applications. One clear example of where cloning still is more efficient is the high-throughput single-chain transfer using head-to-tail SPC. Here, using eight oligos, we setup a system for the routine assembly of 96 full-length heavy and light chains of antibodies in just a few hours, compared to the time and cost of synthesizing all of them. We believe that our method, allowing for direct incorporation of synthetic oligos also will benefit from the current development with reduction in oligo cost and increase in synthesis length.
In conclusion, we demonstrate the usefulness of immobilization of DNA to a solid support for cloning of vectors. Two alternative approaches have been described both adapted to automation in laboratory workstations, but with large differences in strategy allowing cloning with or without the use of restriction enzymes. To improve the cost-effectiveness of the method, streptavidin beads can be regenerated as previously described (36), by gentle disruption of the streptavidin-biotin bond by heating in deionized Milli-Q water to 70°C. We believe these two methods to be well suited for high-throughput cloning and useful for a wide variety of applications in nucleic acid based research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Novo Nordisk Foundation Center for Biosustainability and the entire staff of the HPA for their efforts.
FUNDING
Novo Nordisk Foundation; Knut and Alice Wallenberg Foundation; VINNOVA ProNova Center. Funding for open access charge: Novo Nordisk Foundation Center for Biosustainability.
Conflict of interest statement. None declared.
REFERENCES
- 1.Cohen S.N., Chang A.C.Y., Boyer H.W., Helling R.B. Construction of Biologically functional bacterial plasmids in vitro. Proc. Nat. Acad. Sci. U.S.A. 1973;70:3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loenen W.A.M., Dryden D.T.F., Raleigh E.A., Wilson G.G., Murray N.E. Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Res. 2014;42:3–19. doi: 10.1093/nar/gkt990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pingoud A. Spermidine increases the accuracy of type II restriction endonucleases. Eur. J. Biochem. 1985;147:105–109. doi: 10.1111/j.1432-1033.1985.tb08725.x. [DOI] [PubMed] [Google Scholar]
- 4.Shetty R.P., Endy D., Knight T.F. Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2008;2:1–12. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson J.C., Dueber J.E., Leguia M., Wu G.C., Goler J.A., Arkin A.P., Keasling J.D. BglBricks: a flexible standard for biological part assembly. J. Biol. Eng. 2010;4:1–12. doi: 10.1186/1754-1611-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engler C., Kandzia R., Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008;3:e3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engler C., Gruetzner R., Kandzia R., Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aslanidis C., de Jong P.J. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchuk D., Drumm M., Saulino A., Collins F.S. Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res. 1991;19:1154. doi: 10.1093/nar/19.5.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou M., Gomez-Sanchez C.E. Universal TA Cloning. Curr. Issues Mol. Biol. 2000;2:1–7. [PubMed] [Google Scholar]
- 11.Geu-Flores F., Nour-Eldin H.H., Nielsen M.T., Halkier B.A. USER fusion: a rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res. 2007;35:e55. doi: 10.1093/nar/gkm106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 13.Bryksin A.V., Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques. 2010;48:463–465. doi: 10.2144/000113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartley J., Temple G., Brasch M. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson D.G., Young L., Chuang R., Venter J.C., Iii C.A.H., Smith H.O., America N. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 16.Quan J., Tian J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 2011;6:242–251. doi: 10.1038/nprot.2010.181. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y., Werling U., Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012;40:e55. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kok S., Stanton L., Slaby T. Rapid and reliable DNA assembly via ligase cycling reaction. ACS Synth. Biol. 2014;3:97–106. doi: 10.1021/sb4001992. [DOI] [PubMed] [Google Scholar]
- 19.Uhlen M. Magnetic separation of DNA. Nature. 1989;340:733–374. doi: 10.1038/340733a0. [DOI] [PubMed] [Google Scholar]
- 20.Ronaghi M., Uhlén M., Nyrén P. A sequencing method based on real-time pyrophosphate. Science. 1998;281:363–365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 21.Kojima T., Takei Y., Ohtsuka M., Kawarasaki Y., Yamane T., Nakano H. PCR amplification from single DNA molecules on magnetic beads in emulsion: application for high-throughput screening of transcription factor targets. Nucleic Acids Res. 2005;33:e150. doi: 10.1093/nar/gni143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gan R., Yamanaka Y. Microbeads display of proteins using emulsion PCR and cell-free protein synthesis. Biotechnol. Prog. 2008;24:1107–1114. doi: 10.1002/btpr.43. [DOI] [PubMed] [Google Scholar]
- 23.Quan J., Saaem I., Tang N., Ma S., Negre N., Gong H., White K.P., Tian J. Parallel on-chip gene synthesis and application to optimization of protein expression. Nat. Biotechnol. 2011;29:449–452. doi: 10.1038/nbt.1847. [DOI] [PubMed] [Google Scholar]
- 24.Yeung S.-W., Lee T.M.-H., Cai H., Hsing I.-M. A DNA biochip for on-the-spot multiplexed pathogen identification. Nucleic Acids Res. 2006;34:e118. doi: 10.1093/nar/gkl702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth M.E., Feng L., McConnell K.J., Schaffer P.J., Guerra C.E., Affourtit J.P., Piper K.R., Guccione L., Hariharan J., Ford M.J., et al. Expression profiling using a hexamer-based universal microarray. Nat. Biotechnol. 2004;22:418–426. doi: 10.1038/nbt948. [DOI] [PubMed] [Google Scholar]
- 26.Hudson E.P., Nikoshkov A., Uhlen M., Rockberg J. Automated solid-phase subcloning based on beads brought into proximity by magnetic force. PLoS One. 2012;7:e37429. doi: 10.1371/journal.pone.0037429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhlén M., Björling E., Agaton C., Szigyarto C.A.-K., Amini B., Andersen E., Andersson A.-C., Angelidou P., Asplund A., Asplund C., et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics. 2005;4:1920–1932. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Kibbe W.A. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:W43–W46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung C., Miller R. A rapid and convenient method for the preparation and storage of competent bacterial cells. Nucleic Acids Res. 1988;16:3580. doi: 10.1093/nar/16.8.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trifonov E.N. Making Sense of the Human Genome. In: Sarma RH, Sarma, editors. Structure & Methods. Vol. 1. Albany: Adenine Press; 1990. pp. 69–77. [Google Scholar]
- 31.Fagerberg L., Hallström B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K., et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong H., Kucera B., Jack W.E. Characterization of a DNA Polymerase from the Hyperthermophile Archaea Thermococcus litoralis. J. Biol. Chem. 1993;268:1965–1975. [PubMed] [Google Scholar]
- 33.Cabantous S., Terwilliger T.C., Waldo G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 34.Nilsson P., Persson B., Uhlen M., Nygren P. Real-time monitoring of DNA manipulations using biosensor technology. Anal. Biochem. 1995;224:400–4098. doi: 10.1006/abio.1995.1057. [DOI] [PubMed] [Google Scholar]
- 35.Isalan M., Santori M., Gonzalez C., Serrano L. Localized transfection on arrays of magnetic beads coated with PCR products. Nat. Methods. 2005;2:113–118. doi: 10.1038/nmeth732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmberg A., Blomstergren A., Nord O., Lukacs M., Lundeberg J., Uhlén M. The biotin-streptavidin interaction can be reversibly broken using water at elevated temperatures. Electrophoresis. 2005;26:501–510. doi: 10.1002/elps.200410070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.