


Abstract
RNA research and therapy relies primarily on synthetic RNAs. We employed recombinant RNA technology toward large-scale production of pre-miRNA agents in bacteria, but found the majority of target RNAs were not or negligibly expressed. We thus developed a novel strategy to achieve consistent high-yield biosynthesis of chimeric RNAs carrying various small RNAs (e.g. miRNAs, siRNAs and RNA aptamers), which was based upon an optimal noncoding RNA scaffold (OnRS) derived from tRNA fusion pre-miR-34a (tRNA/mir-34a). Multi-milligrams of chimeric RNAs (e.g. OnRS/miR-124, OnRS/GFP-siRNA, OnRS/Neg (scrambled RNA) and OnRS/MGA (malachite green aptamer)) were readily obtained from 1 l bacterial culture. Deep sequencing analyses revealed that mature miR-124 and target GFP-siRNA were selectively released from chimeric RNAs in human cells. Consequently, OnRS/miR-124 was active in suppressing miR-124 target gene expression and controlling cellular processes, and OnRS/GFP-siRNA was effective in knocking down GFP mRNA levels and fluorescent intensity in ES-2/GFP cells and GFP-transgenic mice. Furthermore, the OnRS/MGA sensor offered a specific strong fluorescence upon binding MG, which was utilized as label-free substrate to accurately determine serum RNase activities in pancreatic cancer patients. These results demonstrate that OnRS-based bioengineering is a common, robust and versatile strategy to assemble various types of small RNAs for broad applications.
INTRODUCTION
RNA interference (RNAi) technologies have been widely utilized for genome function studies. There are also a number of RNAi-based therapies under clinical trials (1–3) in addition to an RNA aptamer (Pegaptanib) being approved by the U.S. Food and Drug Administration for the treatment of age-related macular degeneration (4). Currently RNAi agents and noncoding RNA (ncRNA) materials used for basic, translational and clinical research such as small interfering RNAs (siRNAs), short hairpin RNAs (shRNAs), RNA aptamers and microRNAs (miRs or miRNAs) are mainly produced through chemical synthesis (5–9), while other virus and non-virus-vector based strategies literally utilize DNA agents. Although organic synthesis of oligonucleotides may be automated, a multi-milligram dose of 22-nt double-stranded siRNA or miRNA agents for in vivo testing or projected therapy is very costly. It is also unclear to what extent chemical modifications would alter the structures, biological activities and safety profiles of these ncRNAs, despite that synthetic ncRNAs exhibit some favorable pharmacokinetic properties such as a longer half-life. In vitro transcription (10,11) is another way to produce RNA agents in variable lengths. However, in vitro transcription generally produces RNA molecules in a test tube on micrograms scale, thus the production of larger quantities of RNAs requires considerably more of the costly RNA polymerases.
With a great interest in developing new strategies to bioengineer ready-to-use RNAi agents on a large scale, a successful example has been reported very recently for the generation of fully-processed siRNAs from p19-expressing bacteria (12). On the other hand, tRNA (13–15) and rRNA (16) have been employed as scaffolds to produce a number of chimeric RNAs in common strains of bacteria, given the fact that tRNAs and rRNAs are present as stable RNA molecules in the cells. The recombinant RNA chimeras are thus isolated, and the target RNAs may be released in demand by corresponding RNase (13,14), ribozyme (15) or DNAzyme (16) for structural and biophysical analyses. These recombinant RNA technologies provide a novel way for a cost-effective and fast production of large quantities of recombinant RNAs (e.g. milligrams of RNA chimeras from 1 l bacteria culture).
We had taken the initiative to produce pre-miRNA chimeras (Figure 1a) in common strains of Escherichia coli using tRNA scaffold (17). We hypothesized that fusion tRNA/pre-miRNA isolated from bacteria might act as a ‘prodrug’ where pre-miRNA could be selectively processed to mature miRNA in human cells, and the tRNA scaffold would be degraded to tRNA fragments (tRFs). In the present study, we demonstrated that the majority of tRNA/pre-miRNA chimeras did not accumulate in bacteria or only at a negligible level, thus we developed a novel optimal ncRNA scaffold (OnRS)-based strategy to achieve a consistent high-yield production of chimeric RNAs in E. coli that offers the versatility to carry various types of functional small RNAs of interests such as miRNAs, siRNAs and RNA aptamers (Figure 1b). This approach is proven robust and shall have broad applications to engineering of target chimeric RNAi agents and RNA sensors that may be utilized as research tools and further developed as therapeutic agents and/or diagnostic tools.
Figure 1.
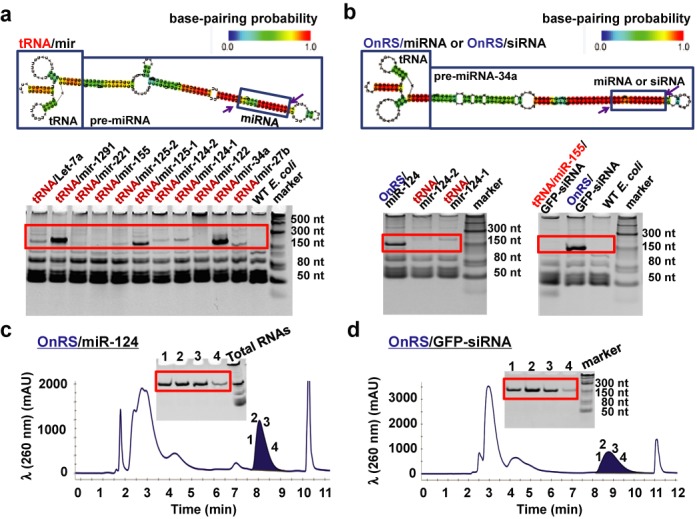
High-yield production of recombinant miRNA/siRNA agents in E. coli using OnRS-based technology. (a) Urea–PAGE analysis of total RNAs showed that there was large variability in the expression of chimeric pre-miRNAs in E. coli using the same tRNA scaffold. Total RNAs isolated from untransformed HST08 (WT) E. coli were used as a control. The heat color gradation indicates the base-pairing probability from 0 to 1. (b) The chimeric tRNA/mir-34a robustly expressed in E. coli was developed as an OnRS that offered a consistent high-level expression of chimeric miRNAs (e.g. OnRS/miR-124) and siRNAs (e.g. OnRS/GFP-siRNA) in E. coli (e.g. ∼15–20% of total RNAs). In contrast, there was no or minimal expression of miR-124 and GFP siRNA using the tRNA and tRNA/mir-155 scaffold, respectively. (c and d) Representative FPLC traces during the purification of OnRS/miR-124 and OnRS/GFP-siRNA, respectively. Inserts are corresponding urea–PAGE analyses of collected fractions (1, 2, 3 and 4) eluted at 8.3 and 8.7 min, respectively, which confirmed the purity of isolated recombinant ncRNAs.
MATERIALS AND METHODS
Bacterial culture
All E. coli stains were cultured at 37°C in LB broth supplemented with 100 μg/ml ampicillin. DH5α (Life Technologies, Grand Island, NY, USA) was used for cloning and HST08 (Clontech Laboratories, Mountain View, CA, USA) was employed for the production of multi-milligrams of chimeric RNAs. Other strains such as DH5α, Top 10 (Life Technologies, Grand Island, NY, USA) and BL21 (Sigma–Aldrich, St. Louis, MO, USA) were also used to evaluate ncRNA expression/accumulation.
Human cell culture
The human carcinoma cell line A549 was purchased from American Type Culture Collection (Manassas, VA, USA), and ES-2/GFP was from Cell Biolabs (San Diego, CA, USA). Both cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 Medium with 10% fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2 and 95% air.
Prediction of RNA secondary structures
The secondary structures of chimeric ncRNAs were predicted using the CentroidFold (http://www.ncrna.org/centroidfold) (18) and CentroidHomfold (http://www.ncrna.org/centroidhomfold) (19).
Construction of plasmids
Individual tRNA/pre-miRNA expression plasmids were cloned as we reported (17), following PCR amplification of target sequences from human genomic DNA using gene specific primers (IDT, San Diego, CA, USA) (Supplementary Table S1). To create OnRS/miR-124, OnRS/Neg, OnRS/GFP-siRNA and tRNA-miR-155/GFP-siRNA expression plasmids, the oligonucleotides (Supplementary Table S1) were annealed and amplified, then the amplicons were cloned into the vector pBSMrnaSeph (14) (kindly provided by Dr Luc Ponchon, France) linearized by endonucleases SalI-HF® and AatII (New England Biolabs, Ipswich, MA). To construct OnRS/MGA5 and OnRS/MGA3 expression plasmids, tRNA/mir-34a was used as a template for the amplification of target sequences using the oligonucleotides (Supplementary Table S1), and then the amplicons were inserted into pBSMrnaSeph vector linearized by SacII and EagI (New England Biolabs) which removed the sephadex aptamer from tRNA scaffold at the same time. All inserts were confirmed by Sanger sequencing analyses at UC Davis Genome Center.
Expression of chimeric RNAs in E. coli
Chimeric RNAs were expressed in HST08 on a large scale as described (13,14,17). Total RNAs were isolated from E. coli using the Tris–HCl-saturated phenol extraction method, quantitated with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Rockford, IL, USA) and analyzed by denaturing urea (8 M) polyacrylamide (8%) gel electrophoresis (PAGE). All images were acquired with ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA). Intensities of bands were used to provide a rough estimation of relative levels of recombinant ncRNAs present in the total RNAs.
Purification of recombinant ncRNAs
Purification of recombinant ncRNAs was conducted with a NGC QUEST 10PLUS CHROM fast protein liquid chromatography (FPLC) System (Bio-Rad). Separation of recombinant ncRNAs from total RNAs was achieved on a UNO Q6 anion-exchange column (Bio-Rad), which was first equilibrated with Buffer A (10 mM sodium phosphate, pH 7.0) at a flow rate 5.0 ml/min for 0.5 min, followed by a gradient elution at the same flow rate, 0–56% Buffer B (Buffer A + 1 M sodium chloride) in 0.5 min, 56% Buffer B for 2 min, 56–65% Buffer B in 10 min, and then 100% Buffer B for 2 min, 100–0% Buffer B in 0.5 min and 100% Buffer A for 5 min. FPLC traces were monitored at 260 nm using a UV/Vis detector. Peak areas were employed to estimate the relative levels of recombinant ncRNAs within the total RNAs, which agrees with those determined by urea–PAGE analyses. After analyzed on a denaturing PAGE gel, the fractions containing pure ncRNAs were pooled. Recombinant ncRNAs were precipitated with ethanol, reconstituted with nuclease-free water, and then desalted and concentrated with Amicon ultra-2 ml centrifugal filters (30 kDa; EMD Millipore, Billerica, MA, USA). The only exception was OnRS/MGA that was reconstituted with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4) buffer. The quantity of purified ncRNAs was determined using NanoDrop and the quality was validated by PAGE analysis before other experiments.
Deep sequencing of small RNAs and data analysis
A549 cells were transfected with 50 nM FPLC-purified OnRS/miR-124 and tRNA/mir-34a (OnRS) and ES-2/GFP cells were transfected with OnRS/Neg and OnRS/GFP-siRNA using Lipofectamine 2000 (Life Technologies). Total RNAs were isolated using a Direct-zol RNA extraction kit (Zymo Research, Irvine, CA) at 48 h post-transfection, and small RNA libraries were generated using the Illumina Truseq™ Small RNA Preparation kit (Illumina, San Diego, CA, USA) according to the instructions. The purified cDNA library was used for cluster generation on Illumina's Cluster Station and then sequenced on Illumina GAIIx following vendor's instructions. Raw sequencing reads (40 nt) were obtained using Illumina's Sequencing Control Studio software version 2.8 (SCS v2.8) following real-time sequencing image analysis and base-calling by Illumina's Real-Time Analysis version 1.8.70 (RTA v1.8.70). The extracted sequencing reads were used for the standard sequencing data analysis by following a proprietary pipeline script, ACGT101-miR v4.2 (LC Sciences, Houston, TX, USA) (20,21). Cells were treated in triplicate and sequenced separately.
Reverse transcription quantitative real-time PCR (RT-qPCR)
Cells were transfected with various doses of recombinant ncRNAs and harvested at particular time points. Total RNAs were isolated using Direct-zol RNA isolation kit (Zymo Research), and RNA concentrations were determined using NanoDrop 2000 spectrophotometer. RT was conducted with NxGen M-MuLV reverse transcriptase (Lucigen, Middleton, WI, USA), and qPCR analysis was carried out on a CFX96 Touch real-time PCR system (Bio-Rad) using quantitative RT-PCR Master mix (New England Biolabs), as described (17,22). Levels of miRNAs were normalized to U74, and mRNA levels were normalized to PPIA. Gene specific primers were shown in Supplementary Table S1. Each experiment was conducted in triplicate and each sample was measured 2–3 times. Similar results were obtained when the study was repeated.
Western blots
A549 cells were transfected with 100 nM OnRS/miR-124 or OnRS/Neg and harvested after 48 h. Cell lysates were prepared with Radioimmunoprecipitation assay (RIPA) lysis buffer (Rockland Immunochemical Inc., Limerick, PA, USA) consisting of complete protease inhibitor cocktail (Roche, Nutley, NJ, USA). Protein concentrations were determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Whole-cell proteins (40 μg per lane) were separated on 10% SDS-PAGE gel, and electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad). Membranes were then incubated with selective antibody against P-STAT-3, STAT-3 (Cell Signaling Technology, Danvers, MA, USA) or GAPDH (Santa Cruz Biotech Inc., Texas, TX, USA) and subsequently with peroxidase anti-rabbit (Jackson ImmunoResearch Inc., West Grove, PA, USA) or anti-mouse IgG (Cell Signaling). The membranes were then incubated with ECL substrates (Bio-Rad), and images were acquired with ChemiDoc MP Imaging System (Bio-Rad). Cells were treated in triplicate and the same results were obtained when the whole study was repeated.
Apoptosis assay
The apoptosis assay was performed by using a FACS Annexin V assay kit (Trevigen, Inc., Gaithersburg, MD, USA) following the manufacturer's protocol. Briefly, A549 cells were transfected with 100 nM recombinant ncRNAs, harvested at 72 h post-transfection, incubated with Annexin V-FITC conjugate and propidium iodide solution, and then the samples were analyzed on a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA). Data analysis was performed using Flowjo (Ashland, OR, USA). Cells were treated in triplicate and similar results were obtained when the whole experiment was repeated.
MTT assay
A549 cells were transfected with 20 or 100 nM chimeric RNAs. At 72 h post-transfection, cell viability was determined using MTT as we described previously (23). Cells were treated in triplicate and similar results were obtained when the whole study was repeated.
Real-time cell growth analysis
A549 cells were seeded 40,000/well on an 8-well E-Plate and treated with 20 or 100 nM recombinant ncRNA 24 h later. Cell growth was monitored using an iCELLigence system (ACEA Biosciences, San Diego, CA, USA). Similar results were obtained when the whole experiment was repeated for three times.
In vitro knockdown of GFP
ES-2/GFP cells were seeded 8000 cells/well on a 24-well plate and transfected with 5 or 15 nM FPLC-purified chimeric RNAs at 24 h later. The fluorescence was monitored with an Olympus IX81 microscope (Olympus, Center Valley, PA, USA) at 24, 48 and 72 h post-transfection. All images were acquired using the same settings at the same time. At the end of the study, total RNAs were isolated from the cells and subject to RT-qPCR evaluation of GFP mRNA and siRNA levels. Cells were treated in triplicate, and similar results were obtained when the whole experiment was repeated.
In vivo knockdown of GFP
All animal procedures were approved by the Institutional Animal Care and Use Committee at UC Davis. Six- to seven-week-old male GFP-transgenic (C57BL/6-Tg(CAG-EGFP)1Osb/J) mice (24) (The Jackson Laboratory, Bar Harbor, ME, USA) were injected i.v. with 75 μg FPLC-purified OnRS/Neg (N = 3) or OnRS/GFP-siRNA (N = 4) after formulated with in vivo-jetPEI (Polyplus-transfection Inc., New York, NY, USA) each day for consecutive 3 days. Three days after the last injection, mice were sacrificed and liver tissues were collected. Frozen sections (8 μm) were prepared after embedded in Tissue-Tek O.C.T. (Sakura Finetek, Torrance, CA, USA) and examined directly using a Zeiss Axio Observer.z1 Microscope coupled to a Zeiss LSM 710 Scanning Device (Zeiss, Oberkochen, Germany). Different batches of sections (8 μm) were fixed with 4% paraformaldehyde (Sigma–Aldrich, St. Louis, MO, USA) and stained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Sigma–Aldrich). GFP fluorescence and DAPI-stained nuclei images were recorded with confocal microscope sequentially and then merged together.
In addition, liver tissues were subject to RNA isolation and RT-qPCR analyses for GFP mRNA and siRNA levels against 18S and U74 was used as control, respectively. Gene specific primers were presented in Supplementary Table S1.
Malachite green (MG) aptamer binding assays
Absorbance scanning was performed from 550 to 700 nm using a SpectraMax Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) after 32 μg purified ncRNAs or 80 μg total RNAs were incubated with 10 μM MG in 100 mM KCl, 5 mM MgCl2 and 10 mM HEPES (pH 7.4) buffer in a total volume of 100 μl. Fluorescent intensity was determined at 630/650 nm (excitation/emission) with the same SpectraMax Microplate Reader for purified ncRNAs (32 μg) or total RNAs (80 μg) in the absence and presence of MG (10 μM). To establish the linearity of MGA-bound MG fluorescent intensity vis-à-vis MG and MGA concentrations, the intensities of fluorescence were examined when 2.08 μM OnRS/MGA5 was exposed to 0–10 μM MG and 10 μM was incubated with 0–5.2 μM OnRS/MGA5, respectively, in 10 mM HEPES (pH 7.4) buffer in a total volume of 100 μl. Each assay was carried out in triplicate, and all experiments were repeated at least once that showed similar results.
Serum RNase activity assay
Serum specimens from IRB-approved, prospectively-collected UC Davis Pancreas Registry bank were utilized. The serum has been processed uniformly within 4 h of blood collection, aliquoted and stored in a −80°C freezer till usage with minimal freeze-thaw cycle. A total of 20 patients’ serum from 10 pancreatic ductal adenocarcinoma (PDAC) (5 early-stage PDAC (American Joint Committee of Cancer, Stages 1 & 2)) and 10 benign/normal pancreas cases (five chronic pancreatitis and five normal pancreases) were selected. The PDAC cases consisted of four males and six females, benign/normal of five males and five females. Age ranges were 51–80 (mean = 67 years old) in the PDAC and 37–85 (mean = 60 years old) in benign/normal groups. The selected serums were from a period spanning March 2012 to September 2014. A normal pooled human serum sample (Fisher Scientific Inc., Waltham, MA, USA), human recombinant RNase A (Novoprotein, Summit, NJ, USA) and human recombinant angiogenin (R&D Systems, Minneapolis, MN, USA) were used for method development.
To evaluate the change in MGA-bound MG fluorescent intensity in relationship to incubation time (0–30 min) after exposure to human serum, 2.08 μM OnRS/MGA5 was incubated with 0.4 or 2.0 μl normal pooled human serum in 10 mM HEPES (pH 7.4) buffer in a total volume of 90 μl, and then fluorescence was determined after the addition of 10 μl MG to a final 10 μM concentration. To assess the protection of MGA by in vivo-jetPEI, 2.08 μM OnRS/MGA5 and 1.0 μl pool human serum were incubated for 0–60 min. To determine the dose response, various volumes of the pooled human serum (0.01–10 μl) or concentrations of recombinant human RNase A (0–10−4 μg/μl) and angiogenin (0–10−2 μg/μl) were incubated with 2.08 μM OnRS/MGA5 for 10 min. To define the influence of RNase inhibitor, the pooled human serum (0, 1, 2 and 5 μl) was incubated with 2.08 μM OnRS/MGA5 for 10 min, with or without 0.4 U/μl RNase inhibitor (Lucigen, Middleton, WI, USA). Each incubation reaction was carried out in triplicate, and all experiments were repeated at least once that offered consistent findings.
Based upon the linearity of fluorescent intensity over MG and MGA concentrations, incubation time and quantity of human sera, 0.4 μl patient serum sample was incubated with 2.08 μM OnRS/MGA5 in 10 mM HEPES (pH 7.4) buffer in a total volume of 90 μl at 37°C for 5 min and then 10 μl MG was added to give 10 μM final concentration for the determination of fluorescence at 630/650 nm (excitation/emission). Serum RNase activity was calculated as the change in fluorescent intensity over time and quantity of serum sample, i.e., △A.U./min/μl. Each patient sample was assayed twice with <10% variations.
Statistical analysis
Values were expressed as mean ± SD. According to the number of groups and variances, data were analyzed with unpaired Student's t-test, or one-way or two-way ANOVA (GraphPad Prism, San Diego, CA, USA). Difference was considered as significant for P-value less than 0.05 (P < 0.05).
RESULTS
An OnRS is developed to achieve high-yield production of recombinant RNAi agents
Motivated by the concept of ‘prodrug’ and the idea to deploy biological RNAs to perform RNA actions, we intended to bioengineer tRNA fusion pre-miRNA (tRNA/mir) agents in common strains of bacteria (Figure 1a) on a large scale, i.e. milligrams of recombinant ncRNAs from 1 l bacterial culture. Thus, a series of plasmids were created and employed to transform E. coli. Surprisingly, we found that the levels of recombinant pre-miRNA chimeras expressed/accumulated in HST08 E. coli were largely variable when the same tRNA scaffold was used. The majority of tRNA/pre-miRNA chimeras were unfortunately not accumulated or at a negligible level (Figure 1a). Use of other E. coli strains still offered no or even lower levels of target ncRNA chimeras (data not shown). Nevertheless, given the findings that tRNA/mir-34a was accumulated to a high level in bacteria (e.g. ∼15–20% of total RNAs) and chimeric tRNA/mir-34a was stable and selectively processed to mature hsa-miR-34a in various types of human carcinoma cells (unpublished data), we hypothesized that tRNA/mir-34a might be developed as an OnRS over the tRNA scaffold towards a consistent high-level production of target miRNAs (Figure 1b).
We thus took up the challenge to assemble miR-124 using the OnRS (tRNA/mir-34a) platform, noticing that miR-124 differs much from miR-34a in size (20 versus 22 nt) and arm of origin (3′ versus 5′). We replaced the 22-nt miR-34a-5p with 20-nt miR-124-3p and substituted their complementary sequences accordingly (Supplementary Table S1), which indeed offered a high-level expression of OnRS/miR-124 chimera in HST08 E. coli (Figure 1b). Recombinant OnRS/miR-124 was then readily purified to a high degree of homogeneity (>99%) using the anion-exchange FPLC method (Figure 1c). Likewise, the OnRS was able to assemble other miRNAs (e.g. 21-nt miR-27b and 22-nt miR-22, etc.; unpublished data) and a 22-nt scrambled RNA sequence (chimeric RNA was named OnRS/Neg and used as a control in the following studies; Supplementary Table S1), which were all consistently produced in HST08 E. coli at high yields and on a large scale, i.e. >15% of OnRS/miRNAs in total RNAs and >1.5 mg of FPLC-purified OnRS/miRNAs from 0.5 l bacterial culture at all times.
We further evaluated if we could utilize this OnRS-based approach to produce milligrams of functional siRNA agents in 1 l E. coli culture. A 22-nt GFP siRNA (25) was chosen as a model siRNA to assemble chimeric OnRS/GFP-siRNA (Supplementary Table S1). In contrast to a minimal level of accumulation in bacteria when tRNA/mir-155 was utilized as a carrier, the use of OnRS (tRNA/mir-34a) provided a consistent high-level expression of OnRS/GFP-siRNA (Figure 1b) and facilitated the FPLC purification of recombinant ncRNAs (Figure 1d). As a result, we were able to produce 1.5–2.0 mg, >98% pure OnRS/GFP-siRNA from 0.5 l bacterial culture every time. These results indicate that target miRNA/siRNA agents can be assembled by using OnRS-based platform to offer a consistent high-level production of chimeric ncRNAs in bacteria.
Target miRNAs/siRNAs are selectively released from chimeric ncRNAs in human cells while tRNA scaffold is processed to tRNA fragments (tRFs)
Next we assessed if mature miR-124 could be selectively produced from OnRS/miR-124 in human cells. An unbiased deep sequencing study was conducted after the preparation of small RNAs library from human lung carcinoma A549 cells at 48 h post-transfection with OnRS/miR-124 and OnRS (tRNA/mir-34a). The data showed that OnRS/miR-124 was selectively processed to large numbers (5613 ± 975 reads) of 20-nt miR-124 in A549 cells (Figure 2a). In contrast, there was 0 ± 1 reads of mature miR-124 identified in A549 cells treated with tRNA/mir-34a (OnRS) that actually offered 22-nt miR-34a. Other miR-124 isoforms including those of 21 nt in length, as well as some passenger strands, were also noted whereas at much lower levels (Supplementary Table S2). In addition, OnRS/miR-124 had no or relatively much smaller influence on other cellular miRNAs including the hsa-mir-34a-p3 fragment (Figure 2a and Supplementary Table S3), while the tRNA scaffold was degraded to tRFs that actually exhibited similar patterns between OnRS/miR-124- and tRNA/mir-34a-treated cells (Figure 2c and Supplementary Table S2).
Figure 2.
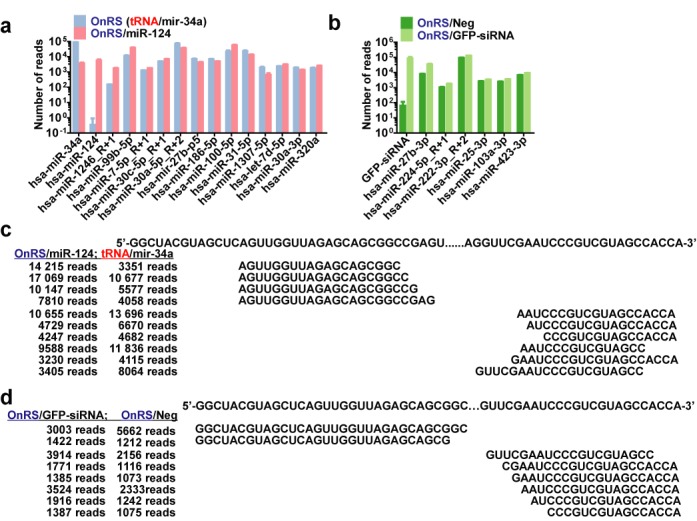
Fate of recombinant ncRNAs in human cells. (a and b) Unbiased deep sequencing study revealed that OnRS-carried miR-124 and GFP-siRNA were precisely processed to target small RNAs, leading to 3 orders of magnitude increase in miR-124 in A549 cells and GFP siRNA in ES-2/GFP cells, respectively. Note the presence of miRNA and siRNA isoforms as well as corresponding passenger strands and other small RNAs at much lower levels (Supplementary Tables S2 and S4). In contrast, the levels of other cellular miRNAs showed no or minor changes (Supplementary Tables S3 and S5). Values are mean ± SD of triplicated treatments that were sequenced separately. (c and d) Mapping major cellular tRFs derived from OnRS/miR-124 versus OnRS (tRNA/mir-34a) in A549 cells and OnRS/GFP-siRNA versus OnRS/Neg in ES-2/GFP cells, respectively. Shown are the mean numbers of reads of triplicated treatments. 3000 and 1000 reads was used as a cut off for the A549 and ES-2/GFP cells, respectively. See the Supplementary Tables S2 and S4 for a complete list of tRFs derived from each ncRNA.
Likewise, we conducted the unbiased deep sequencing analyses of cellular small RNAs in human ES-2/GFP cells at 48 h post-transfection with FPLC-purified OnRS/GFP-siRNA and OnRS/Neg. The data showed that GFP-siRNA levels were about 1000-fold higher in ES-2/GFP cells treated with chimeric OnRS/GFP-siRNA than the control OnRS/Neg (Figure 2b), which was mainly attributable to the increase in 22-, 23- and 21-nt isoforms and accompanied by lower levels of passenger strands (Supplementary Table S4). It was also noted that OnRS/Neg was indeed processed to a number of scrambled RNAs at 22–23 nt in length, but at much lower levels (Supplementary Table S4), which might be related to a lower stability of the scrambled RNAs or insufficient processing. Nevertheless, there were no or minimal differences in the levels of other cellular miRNAs between OnRS/GFP-siRNA- and OnRS/Neg-treated cells (Supplementary Table S5). Furthermore, the tRF patterns were also similar between OnRS/GFP-siRNA and OnRS/Neg-treated cells (Figure 2d and Supplementary Table S4), despite that overall tRF levels were much lower in ES-2/GFP cells than A549 cells. Together, these results support the utility of OnRS for ‘stealth delivery’ of target miRNAs and siRNAs into human cells beyond high-yield production of the chimeric ncRNAs in bacteria and the use of OnRS/Neg as a control to assess OnRS/siRNA activities.
OnRS-carried miR-124 is biologically/pharmacologically active in controlling target gene expression and cancer cellular processes
Then we evaluated the bioactivities of OnRS-carried miR-124, as miR-124 is known to regulate a number of target genes such as the oncogenic signal transducer and activator of transcription 3 (STAT3), enhance apoptosis, and inhibit cell proliferation (26–28). Consistent with deep sequencing data, selective stem–loop RT-qPCR analyses showed that mature miR-124 levels were around 1000-fold higher in A549 cells from day 1 to 4 after transfection with OnRS/miR-124, compared with OnRS/Neg (Figure 3a). Increase in miR-124 in OnRS/miR-124-treated A549 cells led to a 60% reduction of STAT3 protein levels (Figure 3b), and 1- to 2-fold greater degrees of early and late apoptosis as well as necrosis (Figure 3c). Consequently, OnRS/miR-124 exhibited significantly greater antiproliferative activity than OnRS/Neg, as demonstrated by MTT assay and using a Real-Time Cell Analyzer (Figure 3d-e). These results demonstrate that chimeric OnRS/miR-124 is biologically/pharmacological active in regulating miR-124 target gene expression and controlling cancer cell growth after being processed to mature miR-124 in the cells.
Figure 3.
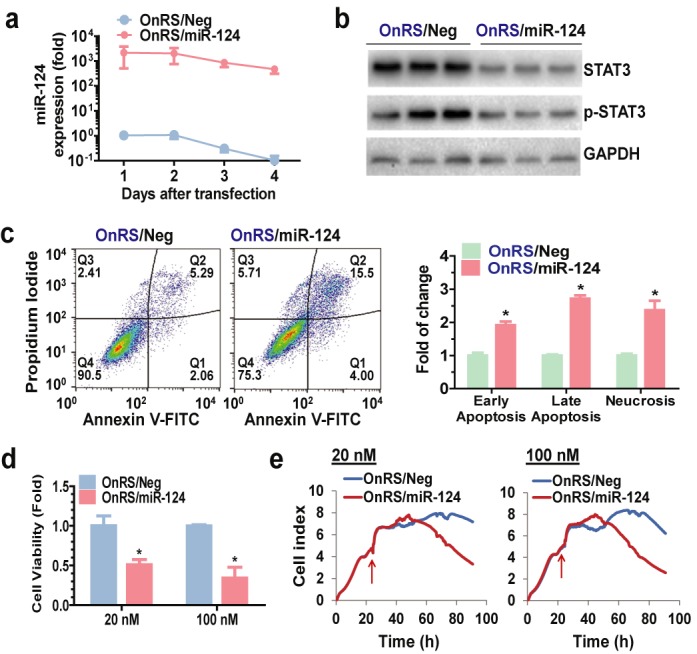
OnRS-carried miRNA is biologically/pharmacologically active in regulating target gene expression and controlling cellular processes in human cells. (a) RT-qPCR analysis revealed that mature miR-124 levels retained 3 orders of magnitude higher in A549 cells for 4 days since transfection with OnRS/miR-124, as compared with OnRS/Neg. (b) Western blots showed that OnRS/miR-124 was effective in reducing the protein expression level of miR-124 target gene STAT3 in A549 cells at 72 h post-transfection. (c) Flow cytometric analyses demonstrated that OnRS/miR-124 was effective in inducing apoptosis in A549 cells at 48 h post-transfection. Cells treated with OnRS/Neg were used as controls. (d) MTT assay showed that OnRS/miR-124 significantly suppressed the proliferation of A549 cells at 72 h post-treatment, as compared to OnRS/Neg. (e) Inhibition of A549 cell proliferation by OnRS/miR-124 was also demonstrated when cell growth was monitored using Icelligence Real-Time Cell analyzer. The arrow points to the time point of ncRNA treatment. Values are mean ± SD of triplicated treatments. *P < 0.01.
OnRS-carried GFP siRNA is effective in knocking down GFP expression in vitro and in vivo
We also assessed the effectiveness of OnRS-carried GFP siRNA using GFP-expressing ES-2 cells and GFP-transgenic mouse models. In ES-2/GFP cells, OnRS/GFP-siRNA significantly suppressed the GFP fluorescence intensity and mRNA levels at 72 h post-transfection (Figure 4a and b), which was associated with 3 orders of magnitude increase in GFP siRNA levels (Figure 4c). We then treated GFP-transgenic mice (24) with in vivo-jetPEI-formulated OnRS/GFP-siRNA. Compared to the GFP-transgenic mice treated with the same doses of in vivo-jetPEI-formulated OnRS/Neg, there was a remarkable reduction of hepatic GFP fluorescence intensity (Figure 4d and e) and mRNA levels (Figure 4f) in GFP-transgenic mice treated with OnRS/GFP-siRNA, which was linked to an over 3000-fold increase in GFP siRNA levels. These data indicate that chimeric GFP-siRNAs produced on large scale using the OnRS cargo are effective agents for in vitro and in vivo RNAi applications.
Figure 4.
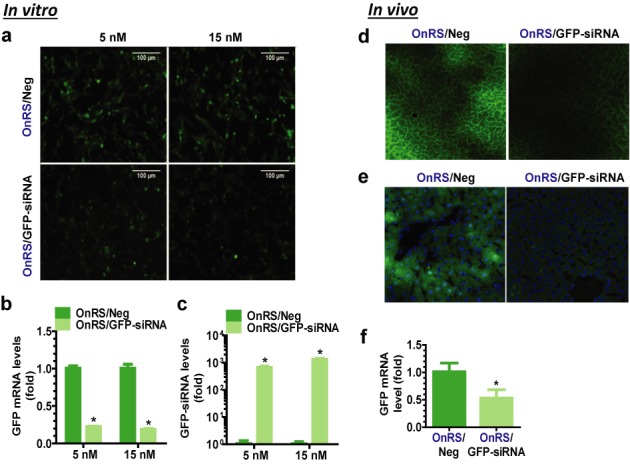
OnRS-carried siRNA is effective for RNAi in vitro and in vivo. GFP fluorescence intensity was sharply reduced in ES-2/GFP cells in vitro at 72 h after transfected with OnRS/GFP-siRNA (a), which was associated with (b) 70–80% lower GFP mRNA levels and (c) 1000-fold higher GFP siRNA levels. Following i.v. administration of OnRS/GFP-siRNA, hepatic GFP fluorescence was significantly suppressed in the GFP-transgenic mouse models in vivo, as demonstrated by microscopic examination of (d) non-fixed and (e) fixed liver slices, as well as (f) RT-qPCR analysis of hepatic GFP mRNA levels. Fixed liver slices were stained with DAPI, and GFP fluorescence and DAPI-stained nuclei (blue) images were merged together (e). Control ES-2/GFP cells (N = 3 per group) or GFP-transgenic mice (N = 3–4 per group) were treated with the same doses of OnRS/Neg. Values are mean ± SD. *P < 0.01, compared with OnRS/Neg treatment.
Utility of OnRS for high-level production of active RNA aptamer chimeras in bacteria
Encouraged by these findings, we further challenged the potential applications of OnRS to the production of functional RNA aptamers. A malachite green aptamer (MGA) (9) was chosen as a model aptamer and inserted at the 5′ and 3′ of miR-34a to offer OnRS/MGA5 and OnRS/MGA3, respectively (Figure 5a). Both chimeras were revealed to be expressed at surprisingly high levels in bacteria, i.e. over 50% of OnRS/MGA in total RNAs (Figure 5b). Thus, we could use FPLC (Figure 5c) to easily purify 5–6 mg OnRS/MGA from 15 to 20 mg total RNAs isolated from 0.5 l bacterial culture at all times.
Figure 5.
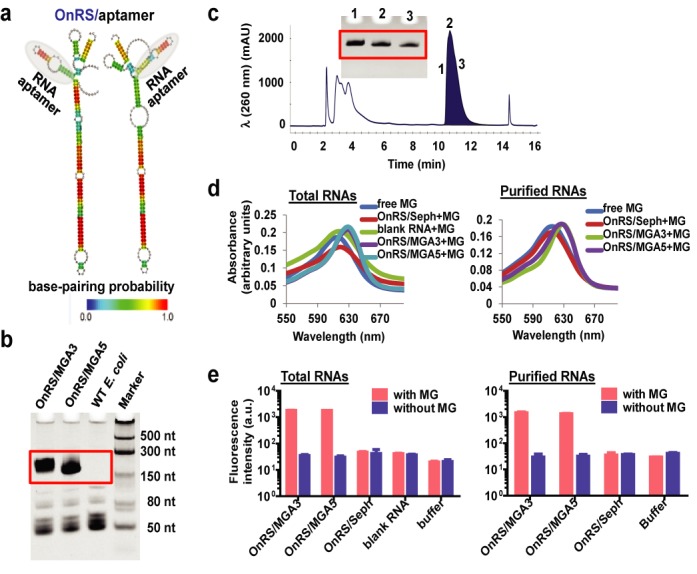
High-yield production of functional RNA aptamers in bacteria using OnRS. (a) Representative design of OnRS/aptamer forms where RNA aptamer was inserted at the 5′ or 3′ end of hsa-mir-34a. The heat color gradation indicates the base-pairing probability from 0 to 1. (b) A consistent high-level expression of OnRS-carried MGA in E. coli, i.e. over 50% of OnRS/MGA5 and OnRS/MGA3 in total RNAs. (c) Representative FPLC traces of OnRS/MGA5 during FPLC purification. Insert is urea–PAGE analysis of the collected RNA fractions (1, 2 and 3) eluted at 10.6 min. (d) Binding to OnRS/MGA5 and OnRS/MGA3 led to a shift of the wavelength of MG maximum absorbance from 618 to 630 nm. The same shift was observed when FPLC-purified OnRS/MGA and total RNAs isolated from OnRS/MGA-expressing bacteria were used. The sephadex aptamer (OnRS/Seph) and corresponding total RNAs were used as additional controls. (e) Strong and selective fluorescence was shown when MG bound to OnRS/MGA5 or OnRS/MGA3. The same results were obtained when using FPLC-purified OnRS/MGA and OnRS/MGA-containing total RNAs.
Consistent with the reported property of MGA (9), we found that the wavelength of MG maximum absorbance was shifted from 618 to 630 nm upon binding the label-free, chimeric OnRS/MGA sensor (Figure 5d). Interestingly, the use of FPLC-purified OnRS/MGA or total RNAs isolated from OnRS/MGA-expressing bacteria gave the same shift in wavelength, whereas a sephadex aptamer (OnRS/Seph) and corresponding total RNAs did not, indicating the selectivity of MGA–MG interactions. The function of OnRS-carried MGA was further demonstrated by a strong fluorescent intensity at 630/650 nm (excitation/emission) upon binding MG (Figure 5e). In contrast, label-free OnRS/MGA itself did not exhibit any fluorescence, and only minimal basal-level MG fluorescent intensity was shown in the absence or presence of non-MGA-containing total RNAs and HPLC-purified OnRS/Seph (Figure 5e), supporting the specificity of MGA-bound-MG fluorescence. These results demonstrate that OnRS is also powerful for high-yield production of functional RNA aptamers of interests.
Application of label-free, OnRS-carried malachite green aptamer sensor to determine serum RNase activities among human pancreatic cancer patients
Given the unique property of MGA-bound MG in exhibiting the fluorescence, we further developed an OnRS/MGA-based RNase activity assay and employed the label-free chimeric OnRS/MGA to investigate and compare serum RNase activities between human PDAC and benign (including chronic pancreatitis)/normal patients because pancreatic cancer patients were shown to have much higher serum RNase activities (29). The intensity of the fluorescence increased with MG concentrations and nearly plateaued at 10 μM MG when OnRS/MGA concentration was fixed at 2.08 μM (or 0.16 μg/μl), while a good linear range was shown for 0.04–5.2 μM OnRS/MGA when MG concentration was fixed at 10 μM (Figure 6a). As expected, the intensity of OnRS/MGA-bound MG fluorescence was decreased over time (Figure 6b) when OnRS-carried MGA was cleaved by the RNases in a normal pooled human serum sample, and the response was dependent upon the doses of human sera (Figure 6c) while sera themselves did not have any significant fluorescence. Indeed, use of in vivo-jetPEI provided good protection against the decrease in fluorescent intensity over time, and addition of RNase inhibitor completely blocked the cleavage of OnRS/MGA by serum RNases. These data indicate that OnRS/MGA may be utilized to directly determine RNase activities.
Figure 6.
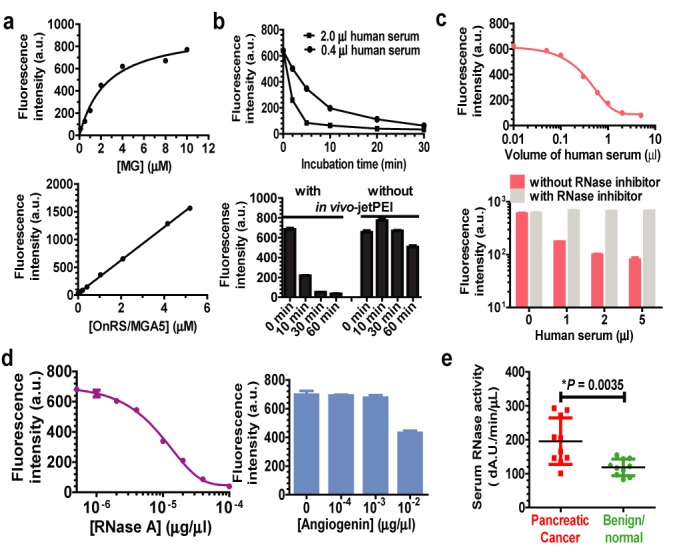
Application of label-free, OnRS-carried MGA sensor to the determination of serum RNase activities in pancreatic cancer patients. (a) Change in MGA-bound-MG fluorescent intensity with the increase in MG and MGA concentrations. Corresponding MGA and MG concentrations were fixed at 1.6 μg/ml and 10 μM, respectively. (b) The fluorescent intensity was decreased over time when incubated with human serum, and in vivo-jetPEI formulated OnRS/MGA was protected from cleavage by serum RNases. (c) Dose response was obvious for the exposure to human serum RNases and the intensity of OnRS/MGA-bound MG fluorescence, and addition of RNase inhibitor completely blocked the cleavage of OnRS/MGA by serum RNases. (d) OnRS/MGA was much more susceptible to human RNase A (10 min incubation) than angiogenin (RNase 5; 30 min incubation). (e) Human pancreatic cancer patients showed significantly higher serum RNase activities than benign/normal patients, as determined by the decrease in MGA-bound MG fluorescence intensity (△A.U./min/μl). N = 10 in each group. OnRS/MGA5 was used in this study.
To define the role of RNase A (the major form RNase in human serum) in the cleavage of chimeric ncRNAs, we directly compared the susceptibility of OnRS-carried MGA to cDNA-expressed RNase A and angiogenin (RNase 5). As manifested by the degrees of change in the intensity of MGA-bound MG fluorescence, 2.08 μM OnRS/MGA was completely cleaved by 5.0 × 10−5 μg/μl RNase A in 10 min, whereas only 40% OnRS/MGA was degraded by 500-fold higher concentration (0.01 μg/μl) of angiogenin in 30 min. Since RNase A is the major form of RNase in human serum (30), this assay would mainly indicate pancreas-derived RNase A activity in human serum. Therefore, we utilized OnRS/MGA to evaluate the RNase activities in a set of serum samples prospectively collected from PDAC and benign/normal patients. The data showed that serum RNase activities (△A.U./min/μl) were significantly (P < 0.01) higher in PDAC (196 ± 22) than benign/normal (118 ± 8) patients. These results implicate that chimeric MGA sensor produced using the OnRS platform could be useful for determination of RNase activities.
DISCUSSION
A general approach has been established for a consistent, cost-effective production of multiple to tens of milligrams of chimeric ncRNAs in 1 l culture of a common strain of E. coli, bearing various types of small RNAs of interests. The OnRS used in this platform is based upon the tRNA fusion pre-miRNA-34a that is resistant to nucleolytic digestion in bacteria and thus accumulated to significantly high level (e.g. >15% of total RNAs) for an easy purification with the anion-exchange FPLC method. The miR-34a-5p/miR-34a-3p duplex within the OnRS cargo may be replaced by any target double-stranded small RNAs such as siRNA or miRNA/miRNA* duplex (Figure 1b) to achieving high-yield production of corresponding chimeric siRNA or miRNA agents, as exemplified by successful production of >1.5 mg of OnRS/miR-124, OnRS/GFP-siRNA and control OnRS/Neg chimeras from 0.5 l bacterial culture in this report. In addition, single-stranded small RNAs such as RNA aptamers can be sprouting at particular sites on OnRS to offer the aptamer chimeras (Figure 5a), which are nicely demonstrated by the assembling of OnRS/MGA5 and OnRS/MGA3 sensors. The robustness and versatility of OnRS-based platform is also supported by successful production of other target RNA agents (e.g. miR-27b, miR-22 and a vascular endothelial growth factor (VEGF) aptamer, etc.; unpublished data), whereas its application to bioengineer other types of biological RNAs such as catalytic RNAs (ribozymes) for biotransformation and guide RNAs (gRNAs) for genome editing warrants further investigations.
Chimeric OnRS/miRNAs and OnRS/siRNAs are expected to act as ‘pro-drugs’ for the ‘delivery’ of target RNAi agents into the cells. Indeed they were selectively processed to large numbers of target miRNAs and siRNAs in human cells, as determined by unbiased deep sequencing studies (Figure 2a and b). So was the scrambled RNA from OnRS/Neg (Supplementary Table S4). The presence of small RNA isoforms differing in 1- or 2-nt at 5′ or 3′ within chimeric ncRNA- and vehicle-treated human cells (Supplementary Tables S2 and S4) may indicate the flexibilities of endoribonucleases in producing small RNAs from pre-miRNAs or shRNAs (31–33). As a result, selective stem-loop RT-qPCR assays revealed a three orders of magnitude increase in miR-124 in A549 cells and GFP siRNA in ES-2/GFP cells, respectively. The results are also in good agreement with our findings on the stability of tRNA/mir-34a chimera in human cells (unpublished data), i.e. the increases in target small RNAs levels were associated with higher levels of OnRS chimeras lasting as long as 4 days post-treatment, which highlights a favorable stability of OnRS chimeras within human cells. On the other hand, there were no or diminutive changes in the levels of other cellular miRNAs, and the tRNA-derived tRFs exhibited similar patterns in the same human cell lines (Figure 2c and d). Nevertheless, the levels of individual or total tRFs identified in ES-2/GFP cells were much lower than A549 cells, which is presumably due to the differences in generating, degrading, excreting and/or retaining tRFs in different types of cells. In addition, while the target miRNAs/siRNAs, tRFs and other small RNAs derived from chimeric ncRNA agents in human cells were fully elucidated (Supplementary Tables S2 and S4), the roles of specific ribonucleases such as Dicer in the processes remain undefined.
The bioactivities of miRNAs (Figure 3) and siRNAs (Figure 4) released from the OnRS cargo are clearly demonstrated by the selective suppression of corresponding target gene expression in vitro and in vivo, using the OnRS/Neg as a critical control. The transcription factor STAT3, a known miR-124 target gene (27,28), plays an important role in many cellular processes such as cell proliferation and apoptosis. Reduction of STAT3 protein expression levels by miR-124 may at least partially provide an explanation for the enhanced apoptosis and repressed proliferation of A549 cells (Figure 3). On the other hand, the suppression of GFP mRNA expression levels in GFP-expressing ES-2/GFP cells and GFP-transgenic mouse liver tissues by OnRS-delivered GFP siRNA explains the lower GFP fluorescent intensities (Figure 4). While the advantages and disadvantages of using recombinant DNA agents, synthetic and recombinant RNAs to achieve RNAi are undoubtedly subjects of debate, the OnRS-based technology offers a new opportunity to readily and cost-effectively produce multi-milligrams of chimeric miRNA and siRNA agents in a research laboratory setting and allows one to utilize biological RNAs to perform RNA actions in vitro and in vivo. Nevertheless, the relative selectivity, efficiency and safety of OnRS-carried RNAi agents, as compared to existing agents or methods, await more extensive evaluation.
The utility of OnRS was further extended to consistent high-yield production of RNA aptamers. A priori it is unknown whether aptamer activity would still be present in the tRNA (13–15) and 5S rRNA (16) scaffolds, although ribozyme activity was observed in the context of the tRNA scaffold when Hammerhead ribozyme sequences were inserted together with the target RNA to be produced (15). The OnRS-resembled RNA aptamer MGA sensor indeed interacted with MG to produce a specific strong fluorescence at 630/650 nm (excitation/emission; Figure 5e), as it was originally discovered (9), which was further employed for the determination of serum RNase activities in clinical samples (Figure 6e). The RNase activity assay using label-free MGA sensor developed in this study is different from current methods. The Kunitz RNase activity assay (29,34–36) is based upon the ultraviolet absorbance of label-free nucleic acids at 260 nm or nucleosides at 280 nm, which is relatively less selective and sensitive. Recent and current RNase activity assays including those commercially-available kits rely on isotope- or fluorophore-labeled RNAs or antibodies (37–40), and thus offer greater sensitivities to determine very low levels of RNase activities or indicate RNase protein levels. However, human serum is comprised of much higher RNase activities. Without extensive dilutions (e.g. 1:1000) of the serum sample that might affect the RNase activity assay including linear range, larger quantities (e.g. >10 μg) of labeled synthetic RNA agents are needed, and thus the assays become costly. After careful examination of the linearity in relation to MG and OnRS/MGA concentrations as well as the quantity of human serum and incubation time, we were able to establish a fluorescence-based RNase assay using label-free OnRS/MGA sensor. Consistent with previous findings (29,36,38), our assays revealed a significantly higher serum RNase activity in PDAC patients which may be attributable to the major form RNase A released from the cancerous pancreases.
In summary, we presented a novel OnRS-based general approach for a consistent high-yield production of chimeric RNAs in common E. coli strains that carry functional small RNAs of interests such as miRNAs, siRNAs and RNA aptamers. This approach is proven robust and versatile and shall have broad applications to engineer chimeric ncRNAs, which may be utilized as in vitro and in vivo research tools and further developed as diagnostic and therapeutic agents.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [1U01CA175315 to A.M.Y.]; National Major Projects from the Ministry of Science and Technology of China [2012ZX09506001-004 to S.Z.]. Funding for open access charge: NIH [1U01CA175315].
Conflict of interest statement. None declared.
REFERENCES
- 1.Dykxhoorn D.M., Lieberman J. Knocking down disease with siRNAs. Cell. 2006;126:231–235. doi: 10.1016/j.cell.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burnett J.C., Rossi J.J. RNA-based therapeutics: current progress and future prospects. Chem. Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kole R., Krainer A.R., Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulrich H., Trujillo C.A., Nery A.A., Alves J.M., Majumder P., Resende R.R., Martins A.H. DNA and RNA aptamers: from tools for basic research towards therapeutic applications. Combinat. Chem. High Throughput Screen. 2006;9:619–632. doi: 10.2174/138620706778249695. [DOI] [PubMed] [Google Scholar]
- 5.Kelnar K., Peltier H.J., Leatherbury N., Stoudemire J., Bader A.G. Quantification of therapeutic miRNA mimics in whole blood from non-human primates. Anal. Chem. 2014;86:1534–1542. doi: 10.1021/ac403044t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling H., Fabbri M., Calin G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013;12:847–865. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi M., Yamada N., Hatakeyama H., Murata M., Sato Y., Minakawa N., Harashima H., Matsuda A. In vitro optimization of 2′-OMe-4′-thioribonucleoside-modified anti-microRNA oligonucleotides and its targeting delivery to mouse liver using a liposomal nanoparticle. Nucleic Acids Res. 2013;41:10659–10667. doi: 10.1093/nar/gkt823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gebert L.F., Rebhan M.A., Crivelli S.E., Denzler R., Stoffel M., Hall J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014;42:609–621. doi: 10.1093/nar/gkt852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babendure J.R., Adams S.R., Tsien R.Y. Aptamers switch on fluorescence of triphenylmethane dyes. J. Am. Chem. Soc. 2003;125:14716–14717. doi: 10.1021/ja037994o. [DOI] [PubMed] [Google Scholar]
- 10.Beckert B., Masquida B. Synthesis of RNA by in vitro transcription. Methods Mol. Biol. 2011;703:29–41. doi: 10.1007/978-1-59745-248-9_3. [DOI] [PubMed] [Google Scholar]
- 11.Huang F., He J., Zhang Y., Guo Y. Synthesis of biotin-AMP conjugate for 5′ biotin labeling of RNA through one-step in vitro transcription. Nat. Protoc. 2008;3:1848–1861. doi: 10.1038/nprot.2008.185. [DOI] [PubMed] [Google Scholar]
- 12.Huang L., Jin J., Deighan P., Kiner E., McReynolds L., Lieberman J. Efficient and specific gene knockdown by small interfering RNAs produced in bacteria. Nat. Biotechnol. 2013;31:350–356. doi: 10.1038/nbt.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponchon L., Dardel F. Recombinant RNA technology: the tRNA scaffold. Nat. Methods. 2007;4:571–576. doi: 10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- 14.Ponchon L., Beauvais G., Nonin-Lecomte S., Dardel F. A generic protocol for the expression and purification of recombinant RNA in Escherichia coli using a tRNA scaffold. Nat. Protoc. 2009;4:947–959. doi: 10.1038/nprot.2009.67. [DOI] [PubMed] [Google Scholar]
- 15.Nelissen F.H., Leunissen E.H., van de Laar L., Tessari M., Heus H.A., Wijmenga S.S. Fast production of homogeneous recombinant RNA–towards large-scale production of RNA. Nucleic Acids Res. 2012;40:e102. doi: 10.1093/nar/gks292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y., Stepanov V.G., Strych U., Willson R.C., Jackson G.W., Fox G.E. DNAzyme-mediated recovery of small recombinant RNAs from a 5S rRNA-derived chimera expressed in Escherichia coli. BMC Biotechnol. 2010;10:85. doi: 10.1186/1472-6750-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M.M., Wang W.P., Wu W.J., Huang M., Yu A.M. Rapid production of novel pre-MicroRNA agent hsa-mir-27b in Escherichia coli using recombinant RNA technology for functional studies in mammalian cells. Drug Metab. Dispos. 2014;42:1791–1795. doi: 10.1124/dmd.114.060145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato K., Hamada M., Asai K., Mituyama T. CENTROIDFOLD: a web server for RNA secondary structure prediction. Nucleic Acids Res. 2009;37:W277–W280. doi: 10.1093/nar/gkp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamada M., Yamada K., Sato K., Frith M.C., Asai K. CentroidHomfold-LAST: accurate prediction of RNA secondary structure using automatically collected homologous sequences. Nucleic Acids Res. 2011;39:W100–W106. doi: 10.1093/nar/gkr290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer C., Grey F., Kreklywich C.N., Andoh T.F., Tirabassi R.S., Orloff S.L., Streblow D.N. Cytomegalovirus microRNA expression is tissue specific and is associated with persistence. J. Virol. 2011;85:378–389. doi: 10.1128/JVI.01900-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei Z., Liu X., Feng T., Chang Y. Novel and conserved micrornas in Dalian purple urchin (Strongylocentrotus nudus) identified by next generation sequencing. Int. J. Biol. Sci. 2011;7:180–192. doi: 10.7150/ijbs.7.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bi H.C., Pan Y.Z., Qiu J.X., Krausz K.W., Li F., Johnson C.H., Jiang C.T., Gonzalez F.J., Yu A.M. N-methylnicotinamide and nicotinamide N-methyltransferase are associated with microRNA-1291-altered pancreatic carcinoma cell metabolome and suppressed tumorigenesis. Carcinogenesis. 2014;35:2264–2272. doi: 10.1093/carcin/bgu174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Y.Z., Zhou A., Hu Z., Yu A.M. Small nucleolar RNA-derived microRNA hsa-miR-1291 modulates cellular drug disposition through direct targeting of ABC transporter ABCC1. Drug Metab. Dispos. 2013;41:1744–1751. doi: 10.1124/dmd.113.052092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okabe M., Ikawa M., Kominami K., Nakanishi T., Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 25.Boudreau R.L., Davidson B.L. Generation of hairpin-based RNAi vectors for biological and therapeutic application. Methods Enzymol. 2012;507:275–296. doi: 10.1016/B978-0-12-386509-0.00014-4. [DOI] [PubMed] [Google Scholar]
- 26.Cao X., Pfaff S.L., Gage F.H. A functional study of miR-124 in the developing neural tube. Genes Dev. 2007;21:531–536. doi: 10.1101/gad.1519207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai B., Li J., Wang J., Luo X., Ai J., Liu Y., Wang N., Liang H., Zhang M., Chen N., et al. microRNA-124 regulates cardiomyocyte differentiation of bone marrow-derived mesenchymal stem cells via targeting STAT3 signaling. Stem Cells. 2012;30:1746–1755. doi: 10.1002/stem.1154. [DOI] [PubMed] [Google Scholar]
- 28.Hatziapostolou M., Polytarchou C., Aggelidou E., Drakaki A., Poultsides G.A., Jaeger S.A., Ogata H., Karin M., Struhl K., Hadzopoulou-Cladaras M., et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell. 2011;147:1233–1247. doi: 10.1016/j.cell.2011.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddi K.K., Holland J.F. Elevated serum ribonuclease in patients with pancreatic cancer. Proc. Natl. Acad. Sci. U.S.A. 1976;73:2308–2310. doi: 10.1073/pnas.73.7.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akagi K., Murai K., Hirao N., Yamanaka M. Purification and properties of alkaline ribonuclease from human serum. Biochim. Biophys. Acta. 1976;442:368–378. doi: 10.1016/0005-2787(76)90311-7. [DOI] [PubMed] [Google Scholar]
- 31.Gu S., Jin L., Zhang Y., Huang Y., Zhang F., Valdmanis P.N., Kay M.A. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castellano L., Stebbing J. Deep sequencing of small RNAs identifies canonical and non-canonical miRNA and endogenous siRNAs in mammalian somatic tissues. Nucleic Acids Res. 2013;41:3339–3351. doi: 10.1093/nar/gks1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dueck A., Ziegler C., Eichner A., Berezikov E., Meister G. microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012;40:9850–9862. doi: 10.1093/nar/gks705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kunitz M. Crystalline desoxyribonuclease; isolation and general properties; spectrophotometric method for the measurement of desoxyribonuclease activity. J. Gen. Physiol. 1950;33:349–362. doi: 10.1085/jgp.33.4.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crook E.M., Mathias A.P., Rabin B.R. Spectrophotometric assay of bovine pancreatic ribonuclease by the use of cytidine 2′:3′-phosphate. Biochem. J. 1960;74:234–238. doi: 10.1042/bj0740234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson L.M. Serum RNase in the diagnosis of pancreatic carcinoma. Proc. Natl. Acad. Sci. U.S.A. 1979;76:2630–2634. doi: 10.1073/pnas.76.6.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potenza N., Salvatore V., Migliozzi A., Martone V., Nobile V., Russo A. Hybridase activity of human ribonuclease-1 revealed by a real-time fluorometric assay. Nucleic Acids Res. 2006;34:2906–2913. doi: 10.1093/nar/gkl368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kottel R.H., Hoch S.O., Parsons R.G., Hoch J.A. Serum ribonuclease activity in cancer patients. Br. J. Cancer. 1978;38:280–286. doi: 10.1038/bjc.1978.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vlassov A., Florentz C., Helm M., Naumov V., Buneva V., Nevinsky G., Giege R. Characterization and selectivity of catalytic antibodies from human serum with RNase activity. Nucleic Acids Res. 1998;26:5243–5250. doi: 10.1093/nar/26.23.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakata D. Increased N-glycosylation of Asn(8)(8) in serum pancreatic ribonuclease 1 is a novel diagnostic marker for pancreatic cancer. Sci. Rep. 2014;4:6715. doi: 10.1038/srep06715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.