


Abstract
Enhancer of zeste homolog 2 (EZH2) is a key epigenetic regulator that catalyzes the trimethylation of H3K27 and is modulated by post-translational modifications (PTMs). However, the precise regulation of EZH2 PTMs remains elusive. We, herein, report that EZH2 is acetylated by acetyltransferase P300/CBP-associated factor (PCAF) and is deacetylated by deacetylase SIRT1. We identified that PCAF interacts with and acetylates EZH2 mainly at lysine 348 (K348). Mechanistically, K348 acetylation decreases EZH2 phosphorylation at T345 and T487 and increases EZH2 stability without disrupting the formation of polycomb repressive complex 2 (PRC2). Functionally, EZH2 K348 acetylation enhances its capacity in suppression of the target genes and promotes lung cancer cell migration and invasion. Further, elevated EZH2 K348 acetylation in lung adenocarcinoma patients predicts a poor prognosis. Our findings define a new mechanism underlying EZH2 modulation by linking EZH2 acetylation to its phosphorylation that stabilizes EZH2 and promotes lung adenocarcinoma progression.
INTRODUCTION
The Polycomb group (PcG) proteins containing polycomb repressive complex 1 (PRC1) and PRC2 are discovered by its essential role in regulating body formation during Drosophila melanogaster development (1). Enhancer of zeste homolog 2 (EZH2) is the core catalytic subunit of PRC2 that includes EZH2, EED, SUZ12 and RbAp46/48 (2–4). EZH2, as a methyltransferase, mediates H3K27 trimethylation and functions in X-chromosome inactivation, stem cell maintenance and cancer progression (5–8). EZH2 is known frequently overexpressed in cancer patients and enhanced EZH2 level often correlates with the poor prognosis of patients (9–12). Aberrant expression of EZH2 functions as a transcriptional repressor that silences tumor suppressor genes, e.g. p16INK4A, p14ARF, ADRB2 and DAB2IP (13–16).
EZH2 and H3K27me3 have been the central molecules in epigenetic control of gene expression. However, it remains not completely clear that how EZH2 itself is precisely regulated in terms of protein stability and enzymatic activity. It has been reported that p130, RB and the microRNA miR-101 negatively regulate EZH2 gene expression (17–19). Post-translational modifications (PTMs) of EZH2 is critical for its role in silencing target genes and the regulation of tumor progression. EZH2-S21 phosphorylation by AKT inhibits its methyltransferase activity (20). EZH2-T345 phosphorylation by CDK1 and CDK2 is important for EZH2-mediated epigenetic gene silencing and also enhances its binding to the lncRNA HOTAIR (21,22). EZH2-T487 phosphorylation by CDK1 inhibits EZH2 methyltransferase activity and inhibits breast cancer cell migration and invasion (23). Other PTMs of EZH2 except phosphorylation include ubiquitination and O-GlcNAcylation (24,25). The findings greatly enlarged our understanding on the PTMs of EZH2. However, the molecular mechanisms underlying these EZH2 PTMs on its stability and biological functions or if other types of PTM exist in EZH2 remain mysterious and require further investigations.
Acetylation is an important form of PTMs that control gene expression, consisting of histone and non-histone acetylation (26,27). Non-histone protein acetylation has been recently reported as an evolutionarily conserved modification that regulates diverse biological functions including the regulation of cancer progression (28,29). It is interesting and important that if EZH2 can be acetylated and which acetyltransferase may acetylate EZH2 and what are the biological consequence of EZH2 acetylation. So far, there are no answers for these questions. In the present study, we provide the first evidence that EZH2 interacts with and is acetylated by acetyltransferase P300/CBP-associated factor (PCAF). We outlined the general picture of effects of EZH2 acetylation by demonstrating that acetylation of EZH2 affects its phosphorylation, stability and capacity in repression of the target genes. We also report that acetylated EZH2 promotes tumor cell migration and invasion and is correlated with the poor prognosis in lung adenocarcinoma patients.
MATERIALS AND METHODS
Cell culture, transfection and treatment
Human embryonic kidney cell line HEK-293T and HeLa cells were cultured in DMEM, human lung adenocarcinoma cell line H1299 was cultured in RPMI1640 and both were supplemented with 10% (vol/vol) fetal bovine serum (FBS), 100 units/ml penicillin and 100 mg/ml streptomycin, at 37°C with 5% (vol/vol) CO2. Transfections were performed using Lipofectamine 2000 according to the manufacturer's instruction. The histone deacetylase (HDAC) inhibitor Trichostatin A (TSA) (Sigma, St Louis, MO, USA) was added at a final concentration of 3 μM for 12 h before harvest. The class III sirtuin (SIRT) inhibitor nicotinamide (Sigma, St Louis, MO, USA) treatment was at 5 mM for 12 h before harvest. Cycloheximide (Sigma, St Louis, MO, USA) was used at a final concentration of 100 g/ml for the indicated times.
Plasmids
Human full-length FLAG-EZH2, HA-P300, FLAG-PCAF, FLAG-SIRT1 and FLAG-SIRT2 were generous gifts from Dr Wei-Guo Zhu (Peking University Health Science Center, Beijing, China). Human full-length HA-CBP, FLAG-CBP, FLAG-P300, HA-PCAF, HA-hMOF and FLAG-hMOF were kindly provided by Dr Jianyuan Luo (Peking University Health Science Center, Beijing, China). Human full-length Myc-EZH2 was kindly provided by Dr Haojie Huang (Mayo Clinic College of Medicine, USA). The FLAG-EZH2 mutants were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA, USA). GFP-EZH2 expression plasmids were constructed by subcloning the EZH2 cDNA fragments into pEGFP-C3 vector (BD, New Jersey, USA). To generate the GST-fusion proteins of EZH2, the sequence for the N-terminal (1-522), C-terminal (523-746), SET (610-746) and Cys-Rich (523-609) domains were amplified by polymerase chain reaction (PCR) and subcloned into pGEX-4T-1 vector (GE Healthcare, USA). The N-terminal (1-492), HAT (493-658), ADA (659-695) and BROMO (696-832) domains of PCAF were subcloned into pGEX-4T-1 vector for production of GST-PCAF fusion proteins. The full-length His-PCAF was amplified by PCR and subcloned into pET-22b (+) vector.
Antibodies
The following antibodies were used: Acetylated-Lysine, EZH2, H3 and PCAF (Cell Signaling Technology, Boston, MA, USA; Catalog #9441, 5246 and 9717, respectively), FLAG and HA (Sigma F1804 and H3663, St Louis, MO, USA), H3K27me3 and EED (Abcam ab6002 and ab4469, Cambridge, MA, USA), pEZH2 T345 (Active Motif 61241, Carlsbad, CA, USA), pEZH2 T487 (Millipore MABS160, Billerica, MA, USA). The rabbit polyclonal antibody against the acetylated EZH2 at lysine 348 residue was produced with a synthetic acetylated human EZH2 peptide: ERIKTPP(AcK)RPGGRR (Kang Wei Shi Ji, Beijing, China).
Co-immunoprecipitation and western blotting
Cells were collected and lysed in NP40 buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% NP40, 1 mM ethylenediaminetetraacetic acid (EDTA), 10 mM sodium butyrate) containing protease inhibitors for 30 min on ice. For co-immunoprecipitations, lysates were incubated with the relevant antibody (5–10 μg) overnight at 4°C. Then, 50 μl protein A/G agarose beads were added and the reaction mixtures were incubated for 2 h at 4°C. After washed with NP40 buffer for three times, the immunoprecipitated complexes were subjected to sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with the indicated antibodies.
In vitro acetylation and deacetylation assay
For the in vitro acetylation assays, 2 μg of full-length His-PCAF and 5 μg GST-fusion EZH2 fragments were incubated in 50 μl acetyltransferase assay buffer (50 mM Tris–HCl, pH 8.0, 10% glycerol, 0.1 mM EDTA and 1 mM dithiothreitol) with 20 μM acetyl CoA at 30°C for 2 h. Deacetylation assays were started using the acetylated proteins described above as substrates. Then immunopurified FLAG-SIRT1 by anti-FLAG resin (M2 beads) (Sigma, St Louis, MO, USA) and 1 mM NAD+ were added into the reaction and incubated for 1 h at 30°C. Then, SDS-PAGE loading buffer was added to stop the reaction. The proteins were resolved by SDS-PAGE and gels were stained with Coomassie blue. The acetylated EZH2 was detected using anti-acetylated-lysine or anti-AcK348-EZH2 antibodies.
GST pull-down assay
For the GST pull-down assays, the cell lysates were pre-cleared with Glutathione-Sepharose 4B beads (GE Healthcare, Sweden) and GST, and then were incubated with beads containing GST fusion proteins overnight at 4°C. After washed with NP40 buffer for three times, the beads were boiled in SDS-PAGE loading buffer and detected by western blot.
Immunofluorescence
Cells were cultured on glass coverslips, fixed with 4% paraformaldehyde for 15 min at room temperature and were permeabilized with 0.1% NP40 for 15 min. Then, cells were blocked with 5% bovine serum albumin in phosphate buffered saline (PBS) and incubated overnight at 4°C with indicated primary antibody. After washed with PBS three times, cells were incubated for 1 h at 4°C with appropriate secondary antibody. The coverslips were stained with 4′,6-Diamidino-2-phenylindole (DAPI) and mounted. Then the immunofluorescence images were captured under a confocal laser-scanning microscope (Carl Zeiss LSM780, Germany).
Identification of EZH2 acetylation sites by mass spectrometry
To identify in vivo acetylation sites of EZH2, HEK-293T cells were co-transfected with FLAG-EZH2 and FLAG-PCAF. After treated with 3 μM TSA plus 5 mM nicotinamide for 12 h, the cell lysates were co-immunoprecipitated with an anti-FLAG antibody. The immunoprecipitated FLAG-EZH2 was subjected to SDS-PAGE. And the bands corresponding to EZH2 were subjected to in-gel trypsin digestion. The labeled peptides were analyzed by LC-MS/MS with nano-LC combined with Orbitrap Q Exactive mass spectrometer (Thermo Scientific, Grand Island, NY, USA). The MS/MS spectra from each LC-MS/MS run were searched against the selected database (In-house database) using an in-house Proteome Discoverer 1.4 software (Thermo Scientific, Grand Island, NY, USA).
In vitro kinase assay
GST-fusion EZH2 fragments were incubated with recombinant cyclin B and CDK1 (New England Biolabs, Ipswich, MA, USA) in 50 μl kinase buffer (New England Biolabs, Ipswich, MA, USA) for 1 h at 30°C in the presence of 200 μM adenosine triphosphate. Reaction mixtures were resolved by SDS-PAGE and analyzed by western blot with anti-EZH2-T345-P and anti-EZH2-T487-P antibodies.
RNA isolation and quantitative RT-PCR
Total cellular RNA was extracted using Trizol reagent (Invitrogen, Grand Island, NY, USA). The cDNA was synthesized using the SuperScript kit (Invitrogen, Grand Island, NY, USA). Primers used are listed in Supplementary Table S1. All mRNAs were normalized to the level of GAPDH gene expression found in the same sample.
Chromatin immunoprecipitation (ChIP) assay
Stable H1299 cells ectopically expressing FLAG-tagged wild-type EZH2 (wt EZH2) and acetylation-mimetic K348Q mutant were crosslinked with 1% formaldehyde and subjected to chromatin immunoprecipitation (ChIP) assays. Briefly, cells were lysed and then sonicated to obtain DNA fragments (500–700 bp in size). The soluble chromatin was immunoprecipitated with indicated antibodies and supplemented with magna ChIP™ protein A and protein G beads (Millipore, Billerica, MA, USA). Reversing the cross-links was carried out at 65°C for 8 h. DNA was purified with a Qiagen DNA extraction kit. Then, the purified DNA was analyzed by real-time RT-PCR. The primers used are listed in Supplementary Table S1.
In vitro histone methyltransferase assay
HEK-293T cells were co-transfected with FLAG-EZH2, FLAG-SUZ12 and FLAG-EED. Then the PRC2 complexes containing different forms of EZH2 were immunopurified by an anti-FLAG Ab conjugated resin (M2 beads). The in vitro HMTase assay was performed using the EpiQuik™ Histone Methyltransferase Activity Kit (H3-K27) according to the manufacturer's instruction (Epigentek, P-3005, Farmingdale, NY, USA). The in vitro HMTase assay was also performed using recombinant histone H3 protein (New England Biolabs, Ipswich, MA, USA) as substrate. Briefly, 50 μl of reaction mixture containing immunoprecipitated protein, 2 μg recombinant histone H3 protein as substrate and 20 μM unlabeled SAM (New England Biolabs, Ipswich, MA, USA) as the methyl donor in methyltransferase activity buffer (50 mM Tris–HCl at pH 8.0, 100 mM NaCl and 10 mM dithiothreitol) was incubated for 2 h at 30°C. The reaction mixtures were subjected to SDS-PAGE, followed by western blot with anti-H3K27me3 antibody.
Cell migration assay
Cell suspension containing 1 × 105 cells/ml was prepared in serum free media and 100 μl cells in suspension were added into the insert. Six-hundred microliter RPMI1640 containing 10% (v/v) FBS was added to the lower wells. Then migration was performed at 37°C for 6 h. Non-migratory cells were removed using cotton-tipped swabs. Subsequently, the migrated cells through the inserts were fixed, stained with crystal violet and counted.
Cell invasion assay
For the cell invasion assay, 100 μl cells in suspension containing 1.0 × 105 cells/ml was added in the inside of each insert coated with Matrigel. RPMI1640 containing 20% FBS was placed in the lower well. After 48 h, the top Matrigel was removed and the cells that invaded to the lower surface were stained with crystal violet and counted.
Tumor tissues
Lung cancer tissue specimens were obtained from the Department of Thoracic Surgery Sino-Japan Friendship Hospital, Beijing China (with Permit Number: ZRLW-5 ZRLW-7). Tissue specimens were sectioned at the Department of Pathology Peking University Health Science Center, Beijing China. The clinicopathological characteristics of patients with lung adenocarcinoma was shown in Supplementary Table S2 (n = 40).
Immunohistochemical staining
Tissue sections were deparaffinized in xylene and rehydrated. Antigen retrieval was processed in pH 6.0 sodium citrate. The sections were incubated in 3% H2O2 for 30 min to quench endogenous peroxidases and then incubated with primary antibody at 4°C overnight. Then PV6001 2-step plus Poly-HRP anti-rabbit IgG detection system (Zhong Shan Jin Qiao, Beijing, China) was applied. The streptavidin-biotin-peroxidase method was used for detection. The diaminobenzidine was applied as substrate (ChemMate Detection Kit, DAKO, Glostrup, Denmark).
Assessment of immunohistochemistry
All immunohistochemical staining were investigated independently by two pathologists. The assessment was classified into four grades: low reactivity marked as 1+, faint reactivity as 2+, moderate reactivity as 3+, and strong reactivity as 4+. We defined that 3+ and 4+ expression are high expression and the others are low expression.
RESULTS
EZH2 is acetylated by the acetyltransferase PCAF in vitro and in vivo
Post-translational modifications of EZH2 including phosphorylation, ubiquitination, O-glcNAcylation have been reported to be important for its function in tumor and neurodegenerative diseases (21–25). However, there was no report about the relationship between post-translational acetylation and EZH2. Here we aimed to explore whether the function of EZH2 is regulated by acetylation. First, we found that EZH2 can be immunoprecipitated by an anti-acetylated-lysine antibody followed by western blot analysis, suggesting that EZH2 is an acetylated protein (Figure 1A). Due to the SV40 T antigen increases the global histone acetylation in cells (30), we co-transfected FLAG-EZH2 with a variety of acetyltransferases not only in HEK-293T cells but also in human lung adenocarcinoma H1299 and HeLa cells. FLAG-EZH2 was immunoprecipitated with an anti-FLAG antibody and immunoblotted with an anti-acetylated-lysine antibody. We demonstrated that EZH2 is acetylated by acetyltransferase PCAF, but not by CBP, P300 and hMOF in vivo (Figure 1B and Supplementary Figure S1A–C). Importantly, we found that the endogenous acetylation level of EZH2 was significantly increased with the presence of FLAG-PCAF in H1299 cells (Figure 1C). To determine the acetylated region of EZH2, we prepared two EZH2 GST-fusion proteins including the N-terminal (1-522) and the C-terminal (523-746) domains and then applied them for in vitro acetylation assays. In an in vitro experiment using purified EZH2 and His-PCAF we demonstrated that N-terminal (1-522) of EZH2 is a bona fide substrate of PCAF, whereas weak acetylation signals at the C-terminal region were also detected (Figure 1D). Therefore, we identified for the first time that EZH2 can be acetylated by PCAF both in vitro and in vivo.
Figure 1.
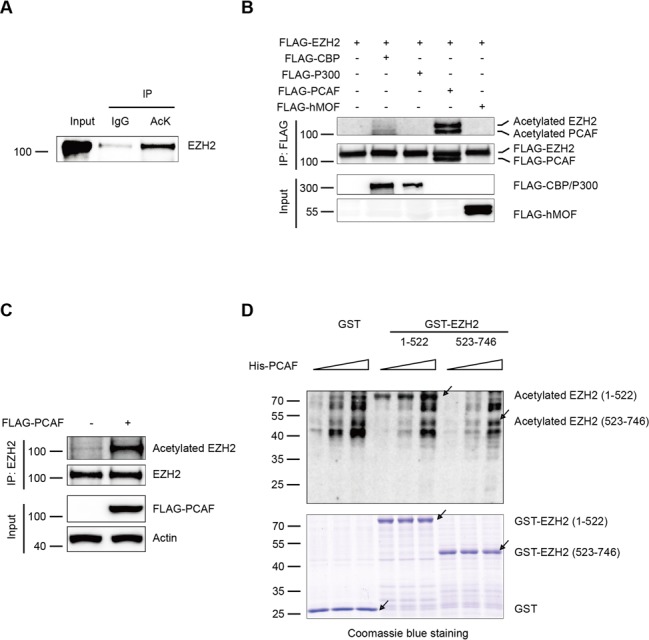
EZH2 is acetylated by the acetyltransferase PCAF both in vivo and in vitro. (A) Endogenous EZH2 is acetylated. Human lung adenocarcinoma H1299 cells were treated with 3 μM TSA plus 5 mM nicotinamide for 12 h. The cell lysates were immunoprecipitated with an anti-acetylated-lysine (AcK) antibody (Ab) or normal IgG, followed by western blot with an anti-EZH2 Ab. (B and C) EZH2 is acetylated by PCAF in vivo. FLAG-EZH2 was co-transfected with cDNAs of different acetyltransferases in H1299 cells. After transfection for 48 h, the cell lysates were subjected to co-immunoprecipitation with an anti-FLAG Ab and immunoblotted with the AcK Ab (B). H1299 cells were transfected with FLAG-PCAF. After transfection for 48 h, the cell lysates were co-immunoprecipitated with an anti-EZH2 Ab and immunoblotted with the AcK Ab (C). (D) EZH2 is acetylated by PCAF in vitro. The various amounts GST-fusion proteins of EZH2, N-terminal domain (1-522) and C-terminal domain (523–746) were affinity-purified and measured by in vitro acetylation assays. The reaction mixtures were subjected to SDS-PAGE and immunoblotted with the AcK Ab. The purified GST fusion proteins were examined by Coomassie blue staining. Arrows indicate proteins with correct molecular masses.
EZH2 interacts with PCAF both in vitro and in vivo
Given that EZH2 complexes with PCAF we continued to scrutinize the interaction between EZH2 and PCAF. EZH2 was co-immunoprecipitated endogenously with PCAF in H1299 and HEK-293T cells (Figure 2A and Supplementary Figure S1D), and then FLAG-PCAF was co-immunoprecipitated by GFP-EZH2 and reciprocally GFP-EZH2 was co-immunoprecipitated by FLAG-PCAF (Figure 2B). These data indicated that EZH2 interacts with PCAF both endogenously and exogenously in cells. To map which domain of EZH2 binds to PCAF, we constructed three GST-fusion proteins containing EZH2 individual domains and expressed them in Escherichia coli (Figure 2C upper). GST pull-down assays using cell lysates containing FLAG-PCAF showed that PCAF interacts with EZH2 mainly at the N-terminal (1-522) and the SET (610-746) domains of EZH2, but not the Cys-Rich (523-609) domain (Figure 2C lower). Reciprocally, four GST-fusion proteins containing PCAF individual domains were expressed in E. coli (Figure 2D upper) and GST pull-down assays were performed using cell lysates containing FLAG-EZH2. Data indicated that the PCAF N-terminal, HAT and ADA domains but not the BROMO domain bind to EZH2 (Figure 2D lower). To rule out the possibility that an adapter protein mediates the interaction between EZH2 and PCAF, we constructed, expressed and purified the full-length His-PCAF fusion protein, then incubated with purified EZH2 GST-fusion proteins. Results showed that PCAF interacts directly with EZH2 mainly at the N-terminal (1-522) and the SET (610-746) domains of EZH2, but not the Cys-Rich (523-609) domain (Figure 2E). Furthermore, endogenous EZH2 and PCAF were found to be co-localized in the nucleus (Figure 2F). Collectively, these findings demonstrated that EZH2 interacts with PCAF in a domain-dependent manner.
Figure 2.
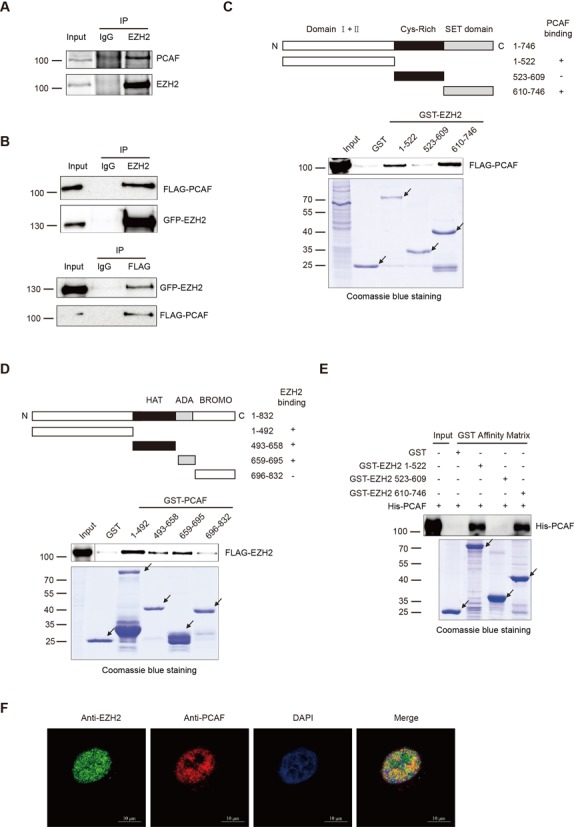
EZH2 interacts with PCAF (A and B) EZH2 interacts with PCAF in vivo. H1299 cells were immunoprecipitated with control IgG or an anti-EZH2 Ab, followed by immunoblotted with an anti-PCAF Ab (A). HEK-293T cells were co-transfected with GFP-EZH2 and FLAG-PCAF. The cell lysates were immunoprecipitated with control IgG or an anti-EZH2 Ab, and detected with an anti-FLAG Ab (top panel) or vice versa (bottom panel) (B). (C) PCAF interacts with EZH2 in GST pull-down assays. Top panel: a schematic diagram of EZH2 and domain constructs fused with GST. Bottom panels: HEK-293T cells were transfected with FLAG-PCAF. Cell lysates were incubated with GST or the three GST-fusion domains of EZH2 respectively. Protein-bound glutathione-agarose beads were washed with lysis buffer and subjected to SDS-PAGE, immunoblotted with an anti-FLAG Ab. The amount of different purified GST-EZH2 fusion domains was visualized by Coomassie blue staining. Arrows indicate proteins with correct molecular masses. (D) EZH2 interacts with PCAF in GST pull-down assays. Top panel: a schematic diagram of PCAF and PCAF domain constructs fused with GST. Bottom panels: HEK-293T cells were transfected with FLAG-EZH2. The cell lysates were incubated with GST or the four GST-fusion domains of PCAF respectively. Protein mixture of GST pull-down was subjected to SDS-PAGE and immunoblotted with an anti-FLAG Ab. Arrows indicate proteins with correct molecular masses. (E) PCAF directly interacts with EZH2 in GST pull-down assays. The full-length His-PCAF fusion protein was incubated with GST or the three GST-fusion domains of EZH2 respectively. Protein-bound Glutathione-agarose beads were subjected to SDS-PAGE, then immunoblotted with an anti-PCAF Ab. The amount of different purified GST-EZH2 fusion domains were visualized by Coomassie blue staining. Arrows indicate proteins with correct molecular masses. (F) Co-localization of endogenous EZH2 with PCAF. Endogenous EZH2 (green) and PCAF (red) were stained with specific Abs in HeLa cells. Nuclei were stained with DAPI (blue) and subsequently visualized by confocal microscopy. Scale bars: 10 μm.
EZH2 is acetylated at K348 in vivo
To identify which lysines in EZH2 can be acetylated in vivo, we co-transfected cells with FLAG-EZH2 and FLAG-PCAF and treated with 3 μM TSA plus 5 mM nicotinamide for 12 h before harvesting. Co-immunoprecipitation was performed, followed by SDS-PAGE. Bands containing FLAG-EZH2 were excised and analyzed by LC-MS/MS. Eight lysines (K10, K245, K309, K318, K344, K348, K472 and K735) of EZH2 were identified acetylated in cells (Figure 3A and Supplementary Figure S1E). To pinpoint which acetylation site within EZH2 is the authentic one, we made a lysine (K) to arginine (R) mutation (mimics of acetylation-deficient EZH2) for individual lysine of the eight acetylated sites identified in vivo. Meanwhile, we also made a mutant with all the eight lysines (K) mutated to arginines (R) (EZH2-8KR) to examine the role of EZH2 acetylation. Co-transfection of EZH2 mutants with FLAG-PCAF and then co-immunoprecipitations were performed as indicated. Interestingly, only K348R displayed a remarkable decrease in acetylation compared to the wild-type (wt) EZH2 (Figure 3B). Other mutants did not show significant decrease in acetylation (Supplementary Figure S1F). However, the EZH2-8KR displayed very low acetylation (Supplementary Figure S1G). These data indicated that Lysine 348 is the major acetylation site of EZH2 by PCAF.
Figure 3.
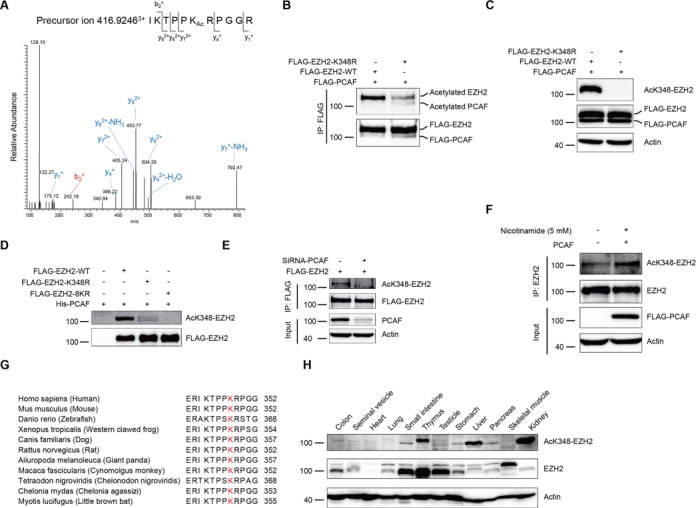
K348 is the major acetylation site of EZH2 (A) EZH2 is acetylated at lysine 348. The spectrum of the charged ion (m/z 416.9246) showed that lysine 348 is acetylated (lower case Ac) in the peptide 343-IKTPPKRPGGR-353. The b ions are the fragmentation ions containing the amino terminus of the peptide. The y ions are the fragmentation ions containing the carboxy terminus of the peptide. (B) Mutation of K348 decreases EZH2 acetylation. FLAG-PCAF was co-transfected in HEK-293T cells with FLAG-EZH2-wt or EZH2 K348R mutant, an acetylation-deficient mimic. Cell lysates were co-immunoprecipitated with an anti-FLAG Ab, followed by western blot with the AcK Ab. (C and D) Characterization of the home-made AcK348-EZH2 specific Ab. The lysates from (B) were subjected to SDS-PAGE and immunoblotted with the anti-AcK348-EZH2 Ab (C). The in vitro acetylation assay was performed using His-PCAF and different purified mutants of FLAG-EZH2 by anti-FLAG resin (M2 beads). Then the reaction mixtures were subjected to SDS-PAGE and immunoblotted with the AcK348-EZH2 specific Ab (D). (E) H1299 cells were transfected with FLAG-EZH2. Endogenous PCAF was knocked down with small interfering RNA (siRNA). Cell lysates were co-immunoprecipitated with an anti-FLAG Ab, followed by western blot with the anti-AcK348-EZH2 Ab. (F) Endogenous EZH2 is acetylated at lysine 348. HEK-293T cells were transfected with FLAG-PCAF and treated with 5 mM nicotinamide for 12 h. Cell lysates were co-immunoprecipitated with an anti-EZH2 Ab, followed by western blot with the anti-AcK348-EZH2 Ab. (G) Alignment of sequences from EZH2 homologs of other species for amino acid residues adjacent to human EZH2-K348. (H) EZH2-K348 acetylation is distributed in multiple tissues. A variety of tissues were prepared from C57BL6 mouse and the levels of endogenous EZH2 and acetylayed EZH2 at K348 were determined by western blot with an anti-EZH2 Ab or the anti-AcK348-EZH2 Ab respectively. Remarkably, thymus, liver and kidney showed high levels of EZH2-K348 acetylation.
To demonstrate that acetylation of EZH2 does exist in cells and tissues, we generated a rabbit polyclonal antibody specifically recognizing the acetylated EZH2-K348 (AcK348-EZH2). The AcK348-EZH2 antibody recognized EZH2-wt, but not the K348R with the presence of FLAG-PCAF (Figure 3C). The specificity of the AcK348-EZH2 antibody was also verified as it recognized the K348-acetylated peptide, but not the unmodified peptide (Supplementary Figure S1H). Moreover, in an in vitro acetylation assay using His-PCAF and different purified mutants of FLAG-EZH2, we demonstrated that the AcK348-EZH2 antibody only recognized EZH2-wt, but not the K348R and the eight lysine mutants 8KR (Figure 3D). Furthermore, the acetylation level of EZH2 was decreased when endogenous PCAF was knocked down with small interfering RNA (siRNA) (Figure 3E). Importantly, the endogenous K348 acetylation was found significantly increased with the presence of FLAG-PCAF and nicotinamide as probed by the AcK348-EZH2 antibody (Figure 3F), indicating that the AcK348-EZH2 antibody recognizes the endogenous EZH2-K348 acetylation. To uncover how conservative EZH2-K348 is among other species during evolution, we made an amino acid sequence alignment of EZH2 with ten other species and found that EZH2-K348 is highly conserved in those species examined (Figure 3G), suggesting that EZH2-K348 acetylation may play an important role among different species. In addition, we observed that K348-EZH2 acetylation existed in multiple mouse tissues at various levels (Figure 3H and Supplementary Figure S1I). Taken together, we demonstrated that K348 is the major in vivo acetylation site of EZH2, which is conserved during evolution.
EZH2 is deacetylated by SIRT1
Acetylation is a dynamic process and acetylated proteins, e.g. p53 and FOXO3a can be reversed by specific deacetylases (31,32). To this end, we found that cells treated with the class III SIRT inhibitor nicotinamide greatly enhanced the level of acetylated FLAG-EZH2 with the presence of PCAF, whereas the class I and II HDAC inhibitor TSA displayed no effect on the deacetylation of EZH2 (Figure 4A), indicating that EZH2 is deacetylated mainly by the class III SIRT deacetylases. Importantly, nicotinamide also increased EZH2 acetylation without PCAF overexpression in H1299, HeLa and HEK-293T cells (Figure 4B and Supplementary Figure S1J, K). Next, we selected SIRT1 and SIRT2 from SIRT family to examine if they interact with and deacetylate EZH2. We co-transfected Myc-EZH2 with FLAG-SIRT1 or FLAG-SIRT2, and found that Myc-EZH2 was co-immunoprecipitated by FLAG-SIRT1, not by FLAG-SIRT2 (Figure 4C). Reciprocally, FLAG-SIRT1 was co-immunoprecipitated by Myc-EZH2 (Figure 4D). These findings suggested that EZH2 selectively interacts with SIRT1 not SIRT2, which is in agreement with the previous report (33). We therefore focused on the role of SIRT1 in deacetylation of EZH2. In cells, EZH2 acetylation was completely blocked by SIRT1 and was drastically restored upon addition of nicotinamide (Figure 4E). Note that the self-acetylation of PCAF was also enhanced in the presence of nicotinamide (Figure 4E). Furthermore, in an in vitro deacetylation assay we confirmed that EZH2 is a bona fide substrate of SIRT1 (Figure 4F). Taken together, these results strongly demonstrated that SIRT1 interacts with and deacetylates EZH2 both in vivo and in vitro.
Figure 4.
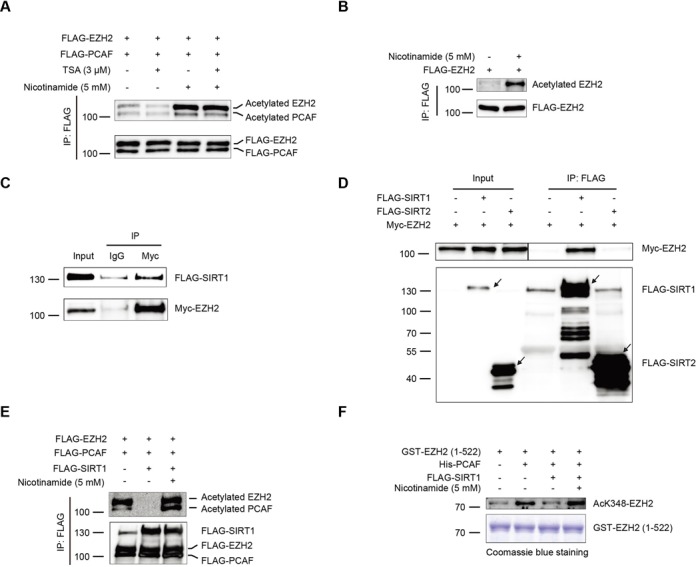
SIRT1 interacts with and deacetylates EZH2 (A and B) EZH2 acetylation is increased in the presence of nicotinamide. H1299 cells were co-transfected with FLAG-EZH2 and FLAG-PCAF and then treated with 3 μM TSA or 5 mM nicotinamide for 12 h. Lysates were co-immunoprecipitated with an anti-FLAG Ab and immunoprecipitates were immunoblotted with the AcK Ab (A). H1299 cells were transfected with FLAG-EZH2 and then treated with 5 mM nicotinamide for 12 h. Lysates were co-immunoprecipitated with an anti-FLAG Ab, then immunoblotted with the AcK Ab (B). (C and D) EZH2 interacts with SIRT1 in vivo. HEK-293T cells were co-transfected with Myc-EZH2, Myc-EZH2 plus FLAG-SIRT1 or Myc-EZH2 plus FLAG-SIRT2. Forty-eight hours post transfection, lysates were co-immunoprecipitated with an anti-FLAG Ab, followed by a western blot with an anti-Myc Ab (C). HEK-293T cells were co-transfected with Myc-EZH2 and FLAG-SIRT1. The lysates were co-immunoprecipitated with an anti-Myc Ab, followed by western blot with an anti-FLAG Ab (D). (E and F) EZH2 is deacetylated by SIRT1. HEK-293T cells were transfected with FLAG-EZH2, FLAG-PCAF and FLAG-SIRT1 as indicated. Lysates were co-immunoprecipitated with an anti-FLAG Ab and detected by the AcK Ab (E). The in vitro deacetylation reaction mixtures were subjected to SDS-PAGE and immunoblotted with the anti-AcK348-EZH2 Ab. The purified GST fusion proteins were detected by Coomassie blue staining (F).
Acetylation of EZH2 affects its phosphorylation and stability
Given that EZH2 phosphorylation at T345 and T487 are important for its function (21–23,34), it is necessary to clarify whether EZH2 acetylation affects its phosphorylation. To this end, we generated an EZH2-8KQ mutant with all the eight lysines (K) mutated to glutamine (Q), a mimic of hyperacetylated EZH2. Lysates from cells transfected with different mutants were analyzed by western blot with the anti-EZH2-T345-P and anti-EZH2-T487-P antibodies. Intriguingly, phosphorylation of EZH2-8KQ at T345 and T487 were markedly decreased compared to the EZH2-wt (Figure 5A). In contrast, the acetylation-deficient mimic EZH2-8KR retained a similar phosphorylation level to the EZH2-wt (Figure 5A). Furthermore, to pinpoint which acetylation site is critical to the decrease of EZH2 phosphorylation at T345 and T487, we generated eight mutants with all the single lysine (K) mutated to glutamine (Q). By transfection of these eight EZH2 acetylation mutants in cells we found that phosphorylation at T345 and T487 of EZH2-K348Q mutant was markedly reduced compared to the EZH2-wt, while other mutants did not show changes in EZH2 phosphorylation (Figure 5B and Supplementary Figure S2A for quantification), indicating that EZH2 acetylation at K348 may regulate the decrease of EZH2 phosphorylation at T345 and T487. The importance of EZH2-K348 acetylation on EZH2 phosphorylation was re-confirmed in H1299 cells (Figure 5C). Oppositely, phosphorylation of EZH2-K348R at T345 and T487 were increased compared with EZH2-wt in the presence of PCAF (Figure 5D). To avoid any possible cellular background interference, we set up an in vitro phosphorylation assay using purified CDK1, EZH2-wt, EZH2-K348Q and EZH2-K348R proteins. Consistent with the in vivo results, we found that phosphorylation of EZH2-K348Q at T345 and T487 by CDK1 were greatly decreased compared to the EZH2-wt (Figure 5E) and phosphorylation of EZH2-K348R by CDK1 at T345 and T487 were increased compared with EZH2-wt after acetylated by PCAF in vitro (Figure 5F). Furthermore, phosphorylation of EZH2 at T345 and T487 were reduced when cells treated with nicotinamide (Figure 5G). However, neither T345A nor T487A phosphorylation-deficient mutants affected the EZH2-K348 acetylation (Supplementary Figure S2B), suggesting that EZH2 phosphorylation at T345 and T487 does not affect its acetylation at K348. We also examined different PTMs status of EZH2 in lung adenocarcinoma patients by immunohistochemical analysis (IHC). Importantly, we found that there exists a negative correlation between K348 acetylated EZH2 and T487 phosphorylated EZH2 in a preliminary experiment, while not the same tendency was found for the T345 phosphorylation of EZH2 (Supplementary Figure S2C). Collectively, these data clearly demonstrated that EZH2-K348 acetylation impairs its phosphorylation at T345 and T487, but not vice versa.
Figure 5.
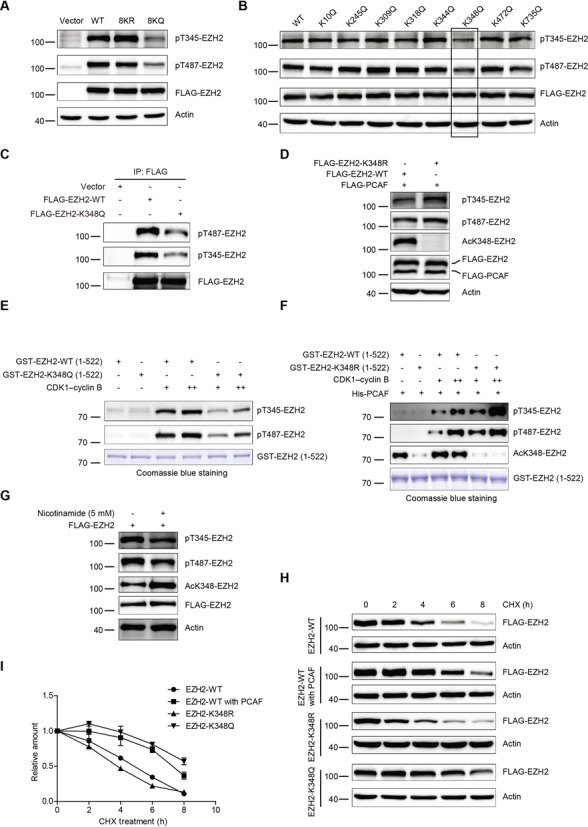
EZH2 acetylation regulates its phosphorylation and stability (A) HEK-293T cells were transfected respectively with EZH2-wt, EZH2-8KR and EZH2-8KQ. Cell lysates were immunoblotted and probed with the anti-EZH2-T345-P and anti-EZH2-T487-P Abs. (B) HEK-293T cells were transfected with FLAG-EZH2 constructs with various mutations, followed by SDS-PAGE and analyzed by western blot and probed with the anti-EZH2-T345-P and anti-EZH2-T487-P Abs. (C) H1299 cells were transfected with empty vector, EZH2-wt or EZH2-K348Q mutant. Lysates were co-immunoprecipitated with an anti-FLAG Ab, followed by immunoblotted with indicated Abs. (D) EZH2 hypoacetylated mimic EZH2-K348R raises the phosphorylation at T345 and T487. EZH2-wt or the acetylation-deficient mutant EZH2-K348R were co-transfected with PCAF and analyzed by western blot with indicated Abs. (E) GST-fusion proteins containing EZH2-wt and EZH2-K348Q N-terminal domain (1-522) were affinity-purified and measured by in vitro kinase assay. Reaction mixtures were resolved by SDS-PAGE and analyzed by western blot with the anti-EZH2-T345-P and anti-EZH2-T487-P Abs. The purified GST fusion proteins were detected by Coomassie blue staining. (F) GST-fusion proteins containing EZH2-wt and EZH2-K348R N-terminal domain (1-522) were affinity-purified and measured firstly by in vitro acetylation assay, then by in vitro kinase assay. Reaction mixtures were resolved by SDS-PAGE and analyzed by western blot with the indicated Abs. The purified GST fusion proteins were detected by Coomassie blue staining. (G) H1299 cells were transfected with FLAG-EZH2 and then treated with 5 mM nicotinamide for 12 h, followed by SDS-PAGE and western blot with indicated Abs. (H and I) K348 acetylation increases the protein stability of EZH2. HEK-293T cells were co-transfected with FLAG-PCAF plus FLAG-EZH2-wt or FLAG-EZH2-wt, FLAG-EZH2-K348R and FLAG-EZH2-K348Q alone. Thirty-six hours post transfection, cells were treated with 100 mg/ml cycloheximide for the indicated times, followed by western blot with an anti-FLAG Ab (H). The bands of FLAG-EZH2 proteins were quantified by software Image J and plotted (I). Results presented are expressed as mean ± SD from three independent experiments.
Given that phosphorylation at T345 and T487 led to EZH2 unstable, we therefore examined whether acetylation at K348 alters EZH2 stability. To answer this interesting question, cells were transfected with FLAG-EZH2-wt, FLAG-EZH2-K348R, FLAG-EZH2-K348Q or FLAG-EZH2-wt plus FLAG-PCAF, and then treated with cycloheximide (CHX). Results demonstrated that post addition of CHX for 8 h ∼90% of EZH2-wt, 65% of EZH2-wt plus PCAF, 87% of EZH2-K348R and 55% of EZH2-K348Q are degraded (Figure 5H), indicating that PCAF-primed acetylation significantly prevents EZH2 from degradation. Furthermore, the half-lives of EZH2-wt, EZH2-wt plus PCAF, EZH2-K348R and the EZH2-K348Q were measured and corresponded to 5, 7, 4 and 8 h separately (Figure 5I). Therefore, we demonstrated that K348 acetylation affects phosphorylation at T345 and T487 and is required for maintenance of EZH2 stability.
EZH2 K348 acetylation is important for silencing its target genes
To uncover the role of EZH2-K348 acetylation on its target gene expression, we established H1299 cells stably expressing FLAG-EZH2-wt, FLAG-EZH2-K348R and EZH2-K348Q separately (Figure 6A). Cells were analyzed for mRNA expression of two known EZH2 target genes HOXA10 and DAB2IP. Results showed that expression of the target genes were significantly repressed by EZH2-K348Q compared to the EZH2-wt (Figure 6B and Supplementary Figure S3A). However, the acetylation-deficient mimic EZH2-K348R retained a similar expression level to the EZH2-wt (Figure 6B and Supplementary Figure S3A). Furthermore, EZH2-K348Q occupied the promoter regions of the target genes to a high extent than the EZH2-wt as determined by ChIP assays (Figure 6C and Supplementary Figure S3B). EZH2-K348Q was found to increase H3K27me3 binding capacity to the target gene promoters compared to EZH2-wt in ChIP assays (Figure 6D and Supplementary Figure S3C). However, EZH2-K348R enhanced neither occupation nor H3K27me3 binding ability on the target gene promoters compared to EZH2-wt (Figure 6C, D and Supplementary Figure S3B, C).
Figure 6.
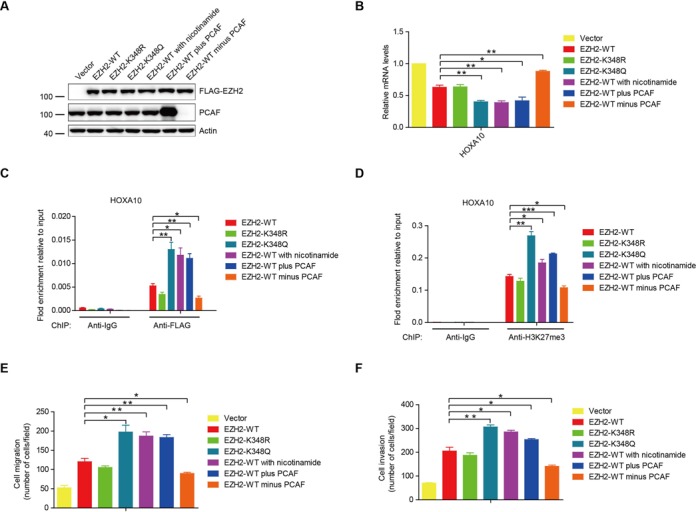
EZH2-K348 acetylation enhances its ability in silencing target gene transcription and promotes lung cancer cell migration and invasion (A) The H1299 cells stably expressed different EZH2 mutants or H1299 cells stably expressed FLAG-EZH2-wt were made. H1299 cells stably expressing FLAG-EZH2-wt were treated with nicotinamide for 12 h. H1299 cells were transfected with HA-PCAF or with PCAF small interfering RNA (siRNA). Cell lysates from these treatments were prepared and subjected to SDS-PAGE and immunoblotted with indicated Abs. (B) HOXA10 mRNA expression levels in stable H1299 cells were analyzed by qPCR. (C) Stable H1299 cells were subjected to ChIP assay with control IgG or an anti-FLAG Ab. The binding of EZH2 on promoters of HOXA10 were analyzed by qPCR. (D) ChIP analyses on HOXA10 promoters were done in H1299 stable cells using control IgG or anti-H3K27me3 Ab and were analyzed by qPCR. (E) Cell migration assay. Quantification of cell migration of stable H1299 cells. (F) Cell invasion assay. Quantification of cell invasion on Matrigel of stable H1299 cells.
When H1299 cells stably expressing FLAG-EZH2-wt were treated with nicotinamide or overexpressed with PCAF in order to increase acetylation of EZH2, the mRNA expression of two known EZH2 target genes HOXA10 and DAB2IP were greatly decreased compared to EZH2-wt. However, knockdown PCAF with small interfering RNA (siRNA) increased the expression of HOXA10 and DAB2IP (Figure 6B and Supplementary Figure S3A). Cells treated with nicotinamide or overexpressing with PCAF enhanced the EZH2-wt occupancy and H3K27me3 binding capacity to the target gene promoters by ChIP assays. In contrast, knockdown of PCAF decreased the ability of EZH2-wt in enhancing the occupancy and H3K27me3 binding capacity to the target gene promoters (Figure 6C, D and Supplementary Figure S3B, C). Then, we examined the AcK348-EZH2 antibody in ChIP assays. Unfortunately, the AcK348-EZH2 antibody is not good enough in ChIP assays, because no difference in quantity between PCR product from AcK348-EZH2 antibody and normal IgG was found even we treated the cells with nicotinamide (Supplementary Figure S3D). These data suggested that the acetylated EZH2 may stay longer on the target gene promoters due to its stability maintained by acetylation. However, EZH2 acetylation does not affect its cellular localization and interaction with SUZ12 and EED (Supplementary Figure S4A–E). These data indicated that EZH2-K348 acetylation functionally enhances its silencing effects on the target genes.
EZH2-K348 acetylation promotes cell migration and invasion in lung cancer cells
Given the high expression level of EZH2 in various tumors (9,10,12), we next investigated the effect of EZH2 acetylation on cell migration and invasion. We observed that expression of EZH2-K348Q notably increased its ability in promoting the migration and invasion of H1299 cells compared to the EZH2-wt (Figure 6E and F). Cells ectopically expressing EZH2-wt treated with nicotinamide or overexpressed with PCAF in order to increase the acetylation of EZH2 showed a similar effect to EZH2-K348Q in promoting the cell migration and invasion. Furthermore, knockdown of PCAF decreased the ability of EZH2-wt in promoting the migration and invasion of H1299 cells. However, ectopic expression of EZH2-K348R did not significantly increase the cell migration and invasion compared to the EZH2-wt (Figure 6E and F). These results demonstrated that acetylation at K348 enhances the ability of EZH2 in promoting lung cancer cell migration and invasion.
Elevated EZH2-K348 acetylation in lung adenocarcinomas predicts poor overall survival in the patients
It was known that EZH2 is dysregulated in lung cancer (11,12,35), we thereby examined the EZH2 acetylation status in a cohort of 40 lung adenocarcinoma patients by IHC using the AcK348-EZH2 antibody. We found that acetylated EZH2 is widely distributed in different cell types of normal lung tissue (Figure 7A–D). Importantly, EZH2-K348 acetylation was found significantly higher in lung adenocarcinoma than the adjacent normal lung tissue (Figure 7I). In a Kaplan–Meier analysis we found that elevated EZH2-K348 acetylation corresponds to a shorter overall survival in lung adenocarcinomas (Figure 7E–H and J). However, we observed no correlation between EZH2 acetylation levels with patients’ sex, age and based on the primary tumor site, the regional lymph node involvement and the distant metastatic spread (TNM) classification. These findings suggested that elevated EZH2-K348 acetylation may predict a poor outcome for lung adenocarcinoma patients.
Figure 7.
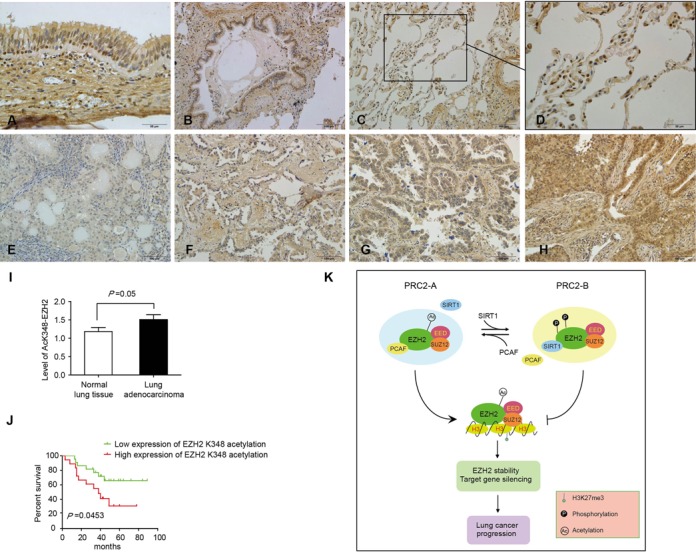
High expression of EZH2-K348 acetylation predicts poor overall survival in lung adenocarcinoma patients (A) Pseudo-stratified ciliated columnar epithelium tissue: EZH2-K348 is widely distributed in the nuclei of the epithelial cells and especially higher in the nuclei of cone cells. (B) Terminal bronchiole: EZH2-K348 showed in epithelial cells. (C) Alveolar tissue. (D) Magnification of alveolar tissue: EZH2-K348 is mainly located in type II alveolar. (E–H) Representative images of EZH2-K348 acetylation in immunohistochemical staining with 1+ (low reactivity) (E), 2+ (faint reactivity) (F), 3+ (moderate reactivity) (G) and 4+ (strong reactivity) (H). (I) The level of EZH2-K348 acetylation is significantly higher in lung adenocarcinoma tissue than the adjacent normal lung tissue as determined by IHC. Presented is the average of IHC intensity ± SD from two groups of patient samples. (J) Two groups of lung adenocarcinoma patients with high (3+ and 4+) or low (1+ and 2+) EZH2 K348 acetylation were determined by Kaplan-Meier analysis with a log-rank at P = 0.05. (K) A working model depicts how PRC2 complex is regulated by EZH2 acetylation. In this model, acetylation and deacetylation of EZH2 make PRC2 in two states: PCAF-loaded or SIRT1-loaded. By switching between the two states PRC2 regulates target gene transcription. In PRC2-A, a switch-on state, EZH2 interacts with acetyltransferase PCAF and is acetylated by PCAF at K348. Although four acetyltransferases have been examined we do not exclude other acetyltransferases may acetylate EZH2. Consequently, EZH2-K348 acetylation reduces the degree of phosphorylation at T345 and T487 caused by CDK1 and this leads to EZH2 stabilized due to an unidentified mechanism. EZH2-K348 acetylation also enhances the occupancy on target gene promoters and suppresses gene transcription. This could be result of increased EZH2 stability. In PRC2-B, a switch-off state, EZH2 interacts with deacetylase SIRT1 and EZH2 acetylation is relieved by SIRT1. However, we do not exclude other SIRT family members may also deacetylate EZH2. Therefore, a generic biological role of EZH2-K348 acetylation is to limit the over-phosphorylation of EZH2 at T345 and T487 and to maintain the stability and methylase activity of EZH2. As a paradigm, EZH2-K348 acetylation promotes lung adenocarcinoma progression.
DISCUSSION
In this study, acetylation was identified as a novel modulation of EZH2 PTMs. EZH2 within PRC2 is acetylated by PCAF and deacetylated by SIRT1. We thereby propose a model that PRC2 exists in two types of state by recruiting PCAF and SIRT1 respectively (Figure 7K). In PRC2-A state, EZH2 is acetylated by PCAF at K348 and PRC2 is on; whereas in PRC2-B state, EZH2 is deacetylated by SIRT1 and PRC2 is off. EZH2-K348 acetylation restricts its phosphorylation at T345 and T487 and increases EZH2 stability and the target gene occupancy. According to this model, EZH2 targeted gene silencing depends on which one is dominant spatially and temporally: PCAF or SIRT1. The dynamic switch between PCAF and SIRT1 controls level of EZH2 acetylation and hence determines PRC2 on or off.
Both phosphorylation and acetylation are modulators of protein interactions and functions (36). Cross-talk between phosphorylation and lysine acetylation has been described for the transcription factor p53, FOXO1 and STAT1 (37–39). It has been reported that EZH2 phosphorylation at T345 and T487 by CDK1 makes EZH2 less stable during mitosis (34). Interestingly we observed that the phosphorylation level at sites T345 and T487 of EZH2-K348Q hyperacetylated mimic mutant was significantly decreased compared to that of the EZH2-wt. Given that phosphorylation at sites T345 and T487 makes EZH2 less stable, decreased phosphorylation at sites T345 and T487 in EZH2-K348Q mutant suggests that acetylation raise the protein stability of EZH2. In agreement, our experiment did prove that acetylation increased the stability of EZH2. Co-transfection of EZH2 and PCAF in cells increased the half-life of EZH2 (Figure 5H). When cells were transfected with the EZH2 hyperacetylated mimic mutant K348Q, the half-life of EZH2-K348Q is longer than the EZH2-wt (Figure 5I). EZH2 can be degraded via the ubiquitin-mediated protein degradation pathway (24,34). However, EZH2 acetylation does not affect its level of ubiquitination in vivo as we found in this report (Supplementary Figure S5). Therefore, it is of interest to understand how EZH2-K348 acetylation enhances its stability without changing its ubiquitination. In addition, the acetylation at K348 may cause a local conformational change that affects the access of CDK1 to phosphorylate T345 and/or T487 within EZH2. This hypothesis warrants future investigations by means of structural biology.
EZH2 functions mainly as a silencer of tumor suppressor genes in tumorigenesis (7,35,40). Post-translational modifications of EZH2 have also been found to be important for its function in silencing its target genes. For example, phosphorylation at T345 and T487 of EZH2 by CDK1 is important for the EZH2-mediated epigenetic gene silencing in breast and prostate cancer cells (21,23). As report here we found that two EZH2 target genes including HOXA10 and DAB2IP were greatly repressed by EZH2-K348 acetylation compared to EZH2-wt (Figure 6B and Supplementary Figure S3A). To elucidate why EZH2 acetylation enhances its function in silencing target genes, we performed ChIP assays and found that EZH2-K348 acetylation increases the binding capacity of EZH2 and H3K27me3 with their target loci (Figure 6C, D and Supplementary Figure S3B, C). However, the acetylation of EZH2 does not change its interaction with other members of PRC2 complexes SUZ12 and EED (Supplementary Figure S4E). Furthermore, EZH2 acetylation does not affect its localization and HMTase activity (Supplementary Figure S4A–D). Previous report found that phosphorylation at T487 decreased the function of EZH2 as a transcriptional repressor. In this report we found that EZH2-K348 acetylation attenuated the phosphorylation at T487. We therefore speculate that EZH2-K348 acetylation may enhance target gene silencing mainly through inhibition of phosphorylation at T487. The specificity of target genes between K348 acetylated EZH2 and unacetylated EZH2 may be different, but it may depend on different physiological condition, such as cancer progression. It was known that phosphorylation at sites T345 and T487 of EZH2 executed opposite role in mediating epigenetic gene silencing effects in prostate and breast cancer cells (21,23). However, we identified that EZH2-K348 acetylation decreased the level of phosphorylation at both T345 and T487 in lung cancer cells with concomitant promotion of lung cancer cell motility, suggesting that EZH2 phosphorylation at sites Thr 345 and Thr 487 may play a distinct role in different cellular context.
EZH2 has been considered a potential novel therapeutic target for human cancers (35,41). In non-small cell lung carcinomas, high expression of EZH2 promotes aggressive tumor behavior and knockdown of EZH2 inhibits lung cancer cell migration and invasion (11,42). Consistent with our observation, overexpression of EZH2-wt increases migration and invasion of the human lung adenocarcinoma H1299 cells. Interestingly, EZH2 K348 acetylation enhances H1299 cell migration and invasion compared to EZH2-wt. Furthermore, the level of EZH2 acetylation at K348 is increased in lung cancer tissues compared to the normal lung tissues. Importantly, we found that high level of EZH2-K348 acetylation in lung adenocarcinomas predicts a poor outcome for the patients (Figure 7J), suggesting that EZH2-K348 acetylation may be of prognostic value for the lung adenocarcinoma patients. Thus, these results indicate a gain-of-function of EZH2 acetylation in human lung cancer progression.
Our findings suggest that EZH2 acetylation at K348 restricts the over-phosphorylation by CDK1 at T345 and T487 and maintains EZH2 stability. Acetylated EZH2 exerts a gain-of-function in cells and promotes lung cancer progression. EZH2 acetylation witnesses a new insight for the complexity of EZH2 PTMs and suggests a dynamic switch of PCR2 states between PCAF-loaded and SIRT1-loaded. However, the generic role of EZH2 acetylation in vivo should be investigated in the future using models of EZH2-K348Q transgenic and EZH2-K348R knock-in mice.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Ministry of Science and Technology of China [2015CB553906, 2013CB910501, 2013ZX09401004-006]; National Natural Science Foundation of China [81230051, 30830048, 31170711, 81321003]; 111 Project of the Ministry of Education; Beijing Natural Science Foundation [7120002]; Peking University [BMU20120314, BMU20130364]; Leading Academic Discipline Project of Beijing Education Bureau (to H.Z).
Conflict of interest statement. None declared.
REFERENCES
- 1.Kennison J.A. The Polycomb and trithorax group proteins of Drosophila: trans-regulators of homeotic gene function. Annu. Rev. Genet. 1995;29:289–303. doi: 10.1146/annurev.ge.29.120195.001445. [DOI] [PubMed] [Google Scholar]
- 2.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 3.Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller J., Hart C.M., Francis N.J., Vargas M.L., Sengupta A., Wild B., Miller E.L., O'Connor M.B., Kingston R.E., Simon J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 5.Plath K., Fang J., Mlynarczyk-Evans S.K., Cao R., Worringer K.A., Wang H., de la Cruz C.C., Otte A.P., Panning B., Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 6.Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon J.A., Lange C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 8.Shen X., Kim W., Fujiwara Y., Simon M.D., Liu Y., Mysliwiec M.R., Yuan G.C., Lee Y., Orkin S.H. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell. 2009;139:1303–1314. doi: 10.1016/j.cell.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varambally S., Dhanasekaran S.M., Zhou M., Barrette T.R., Kumar-Sinha C., Sanda M.G., Ghosh D., Pienta K.J., Sewalt R.G., Otte A.P., et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 10.Kleer C.G., Cao Q., Varambally S., Shen R., Ota I., Tomlins S.A., Ghosh D., Sewalt R.G., Otte A.P., Hayes D.F., et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huqun Ishikawa, R., Zhang J., Miyazawa H., Goto Y., Shimizu Y., Hagiwara K., Koyama N. Enhancer of zeste homolog 2 is a novel prognostic biomarker in nonsmall cell lung cancer. Cancer. 2012;118:1599–1606. doi: 10.1002/cncr.26441. [DOI] [PubMed] [Google Scholar]
- 12.Behrens C., Solis L.M., Lin H., Yuan P., Tang X., Kadara H., Riquelme E., Galindo H., Moran C.A., Kalhor N., et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin. Cancer Res. 2013;19:6556–6565. doi: 10.1158/1078-0432.CCR-12-3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H., Tu S.W., Hsieh J.T. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J. Biol. Chem. 2005;280:22 437–22 444. doi: 10.1074/jbc.M501379200. [DOI] [PubMed] [Google Scholar]
- 14.Yu J., Cao Q., Mehra R., Laxman B., Yu J., Tomlins S.A., Creighton C.J., Dhanasekaran S.M., Shen R., Chen G., et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell. 2007;12:419–431. doi: 10.1016/j.ccr.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Kotake Y., Cao R., Viatour P., Sage J., Zhang Y., Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agherbi H., Gaussmann-Wenger A., Verthuy C., Chasson L., Serrano M., Djabali M. Polycomb mediated epigenetic silencing and replication timing at the INK4a/ARF locus during senescence. PLoS One. 2009;4:e5622. doi: 10.1371/journal.pone.0005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varambally S., Cao Q., Mani R.S., Shankar S., Wang X., Ateeq B., Laxman B., Cao X., Jing X., Ramnarayanan K., et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohrer L.R., Chen S., Hallstrom T.C., Huang H. Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: a potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology. 2010;151:5136–5145. doi: 10.1210/en.2010-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cha T.L., Zhou B.P., Xia W., Wu Y., Yang C.C., Chen C.T., Ping B., Otte A.P., Hung M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 21.Chen S., Bohrer L.R., Rai A.N., Pan Y., Gan L., Zhou X., Bagchi A., Simon J.A., Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat. Cell Biol. 2010;12:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaneko S., Li G., Son J., Xu C.F., Margueron R., Neubert T.A., Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei Y., Chen Y.H., Li L.Y., Lang J., Yeh S.P., Shi B., Yang C.C., Yang J.Y., Lin C.Y., Lai C.C., et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Y.L., Chou R.H., Shyu W.C., Hsieh S.C., Wu C.S., Chiang S.Y., Chang W.J., Chen J.N., Tseng Y.J., Lin Y.H., et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol. Med. 2013;5:531–547. doi: 10.1002/emmm.201201783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu C.S., Lo P.W., Yeh Y.H., Hsu P.H., Peng S.H., Teng Y.C., Kang M.L., Wong C.H., Juan L.J. O-GlcNAcylation regulates EZH2 protein stability and function. Proc. Natl. Acad. Sci. U.S.A. 2014;111:1355–1360. doi: 10.1073/pnas.1323226111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latham J.A., Dent S.Y. Cross-regulation of histone modifications. Nat. Struct. mol. Biol. 2007;14:1017–1024. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- 27.Yang X.J., Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao D., Zou S.W., Liu Y., Zhou X., Mo Y., Wang P., Xu Y.H., Dong B., Xiong Y., Lei Q.Y., et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell. 2013;23:464–476. doi: 10.1016/j.ccr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y., Li F., Lv L., Li T., Zhou X., Deng C.X., Guan K.L., Lei Q.Y., Xiong Y. Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res. 2014;74:3630–3642. doi: 10.1158/0008-5472.CAN-13-3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valls E., de la Cruz X., Martinez-Balbas M.A. The SV40 T antigen modulates CBP histone acetyltransferase activity. Nucleic Acids Res. 2003;31:3114–3122. doi: 10.1093/nar/gkg418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaziri H., Dessain S.K., Ng Eaton E., Imai S.I., Frye R.A., Pandita T.K., Guarente L., Weinberg R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 32.Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y., Tran H., Ross S.E., Mostoslavsky R., Cohen H.Y., et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 33.Kuzmichev A., Margueron R., Vaquero A., Preissner T.S., Scher M., Kirmizis A., Ouyang X., Brockdorff N., Abate-Shen C., Farnham P., et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1859–1864. doi: 10.1073/pnas.0409875102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu S.C., Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J. Biol. Chem. 2011;286:28511–28519. doi: 10.1074/jbc.M111.240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chase A., Cross N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011;17:2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber S.L., Bernstein B.E. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- 37.Brooks C.L., Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kramer O.H., Knauer S.K., Greiner G., Jandt E., Reichardt S., Guhrs K.H., Stauber R.H., Bohmer F.D., Heinzel T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009;23:223–235. doi: 10.1101/gad.479209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morey L., Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010;35:323–332. doi: 10.1016/j.tibs.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Crea F., Paolicchi E., Marquez V.E., Danesi R. Polycomb genes and cancer: time for clinical application. Crit. Rev. Oncol. Hematol. 2012;83:184–193. doi: 10.1016/j.critrevonc.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Xu C., Hou Z., Zhan P., Zhao W., Chang C., Zou J., Hu H., Zhang Y., Yao X., Yu L., et al. EZH2 regulates cancer cell migration through repressing TIMP-3 in non-small cell lung cancer. Med. Oncol. 2013;30:713. doi: 10.1007/s12032-013-0713-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.