

Abstract
Background
Ovarian cancer is a deadly female reproductive cancer. Understanding the biological mechanisms underlying ovarian cancer could help lead to quicker and more accurate diagnosis and more effective treatments. Both changes in microRNA(miRNA) expression and miRNA/mRNA dysregulation have been associated with ovarian cancer. With the availability of whole-genome miRNA and mRNA sequencing we now have new potentials to study these associations. In this study, we performed a comprehensive analysis of miRNA and mRNA expression in ovarian cancer using an integrative network approach combined with association analysis.
Results
We developed an integrative approach to construct a network that illustrates the complex interplay among miRNA and gene expression from a systems perspective. Our method is composed of expanding networks from eQTL associations, building network associations in eQTL analysis, and then combine the networks into an integrated network. This integrated network takes account of miRNA expression quantitative trait loci (eQTL) associations, miRNAs and their targets, protein-protein interactions, co-expressions among miRNAs and genes respectively. Applied to the ovarian cancer data set from The Cancer Genome Atlas (TCGA), we created an integrated network with 167 nodes containing 108 miRNA-target interactions and 145 from protein-protein interactions, starting from 44 initial eQTLs. This integrated network encompassed 26 genes and 14 miRNAs associated with cancer. In particular, 11 genes and 12 miRNAs in the integrated network are associated with ovarian cancer.
Conclusion
We demonstrated an integrated network approach that integrates multiple data sources at a systems level. We applied this approach to the TCGA ovarian cancer dataset, and constructed a network that provided a more inclusive view of miRNA and gene expression in ovarian cancer. This network included four separate types of interactions among miRNAs and genes. Simply analyzing each interaction component in isolation, such as the eQTL associations, the miRNA-target interactions or the protein-protein interactions, would create a much more limited network than the integrated one.
Keywords: microRNA, ovarian cancer, cancer genomics, integrated network, expression Quantitative Trait Loci (eQTL) analysis
Background
Ovarian cancer is the deadliest reproductive cancer in women, accounting for 5% of female cancer deaths. It is estimated that there will be 21,980 new cases and 14,270 deaths from ovarian cancer in 2014 [1]. The overall 5 year relative survival is less than 44%, but when diagnosed in the earliest stages the 5 year survival is over 90%. [2]. Therefore, a better understanding of the biological mechanisms of ovarian cancer is crucial to create earlier diagnosis and more effective treatment.
MicroRNAs (miRNAs) are small (~22 nucleotides) non-coding RNAs that regulate gene expression by targeting complementary mRNA, which triggers a translational blockade or degradation[3]. miRNAs have been increasingly recognized as an important participant of gene regulation activity [4] that affect various cellular processes [5] and contribute to disease pathogenesis in a wide variety of diseases [6] including cancer [7]. Changes in miRNA expression and miRNA dysregulation have been found in cancer, including ovarian cancer[8-13]. In another recent study, miRNAs were described as regulators of tumor phenotype [14]. Early studies show that miRNA expression profiles can be used to classify cancer phenotypes [15]. A study by The Cancer Genome Atlas (TCGA) found three miRNA subtypes in high grade serous ovarian cancer[16]. By exploiting the miRNA expression profiles and their effects on gene expression and other phenotypes in cancer, more profound markers for prognosis can be identified and utilized.
Genetic variation has also been determined to have functional effects on cancer through miRNA expression as well. Various studies have indicated the genetic variants such as single nucleotide polymorphisms (SNPs) are associated with cancer risk in individuals [17]. Particularly, eQTL analysis [18-25] provides a powerful tool to study how genetic variation affects quantitative traits, including measurable phenotypes like gene or miRNA expression. For instance, miRNA eQTL analysis [6,26,27] has also been performed to find genetic loci that introduce differences in miRNA expression.
In general, eQTL analysis analyzes the effect of genetic variation on gene expression by utilizing correlation or regression analysis between the genotypes of a genetic variant and a particular trait [25]. Such analysis assumes that genetic variants are independent, and that the expressions of different genes are independent as well. Nonetheless, many genetic variants are correlated, and the genes can be co-regulated as well. To address the correlation or interactions among these genetic variants or genes, new machine learning methods such as multi-task learning methods [28,29] have been designed to consider all the genes during regression while considering the network structure among genetic variants and genes [30-32]. New methods have also been developed to seek for other relationships, such as redundancy among eQTLs [33] where multiple genetic variants have the same effect on a particular trait and are thus exchangeable.
However, it is still unclear to what extent that changes in miRNA expression, an epigenetic marker, affect gene expression and bring about fluctuations in biological pathways and networks in ovarian cancer. It has recently been determined that some miRNAs promote epithelial-to-mesenchymal transition (EMT) and in turn, EMT transition promotes cell mobility and invasion of cancerous cells [14]. In efforts to understand such complex effects of miRNA expression on gene expression, we develop an integrated network approach that combines expression quantitative trait locus (eQTL) analysis with network analysis using various types of relationships and interactions among miRNAs and genes.
In order to understand the effects discussed above, we perform a genome wide eQTL analysis between miRNA expression and gene expression in ovarian cancer. The suggested method can help identify a set of miRNA eQTLs that have significant impact on gene expression. Using these miRNA eQTLs and their perturbed genes as seed nodes for network expansion, we created an integrated network that utilizes multiple types of networks such as miRNA targets and protein-protein interactions. In parallel, we used another graph guided machine learning model for eQTL analysis that incorporates correlations among miRNAs and genes simultaneously [30]. With these various types of relationships combined, we are able to generate an integrated network that not only captures different types of interactions among these miRNAs and genes, but also includes important cancer genes embedded in a network view. Such an integrative approach thus generates insightful results that could shed light on the interaction of miRNA, gene expression and their contribution to the development and progression of ovarian cancer.
Methods
We developed an integrative approach that combines the strength of eQTL analysis with network analysis, toward the goal of building an integrated network to understand the relationship between miRNA expression and gene expression. The integrated network is composed of various types of relationships among miRNAs and genes, including miRNA-gene eQTL associations, miRNAs and their targets, protein-protein interactions, and correlation networks of both miRNA and gene expression.
The overall workflow of our integrative approach is illustrated in Figure 1. Our approach consisted of five phases, including data preprocessing, eQTL analysis, network association, network expansion and network integration. Prior to analysis, we performed data pre-processing in which, we collected and normalized the data. The next phase of our workflow was the eQTL analysis between miRNA and gene expression. Here, we used Matrix eQTL to perform the analysis. In the network expansion phase, we used TarBase [34] to identify targets based on the eQTLs generated from Matrix eQTL [35]. Next, we found protein-protein interactions connected with the identified miRNA targets. In order to select the most confident interactions, we chose the interactions with a p-value ≤ 0.05. From there, we expanded the network using the selected protein-protein interactions using DAPPLE [36], generating the initial integrated network. Simultaneously, in the network association phase we used MtLasso2G [30] to discover network associations between miRNAs to build miRNA correlation networks. Finally, we merged the eQTL expanded network and the miRNA correlation networks to create the integrated network.
Figure 1.
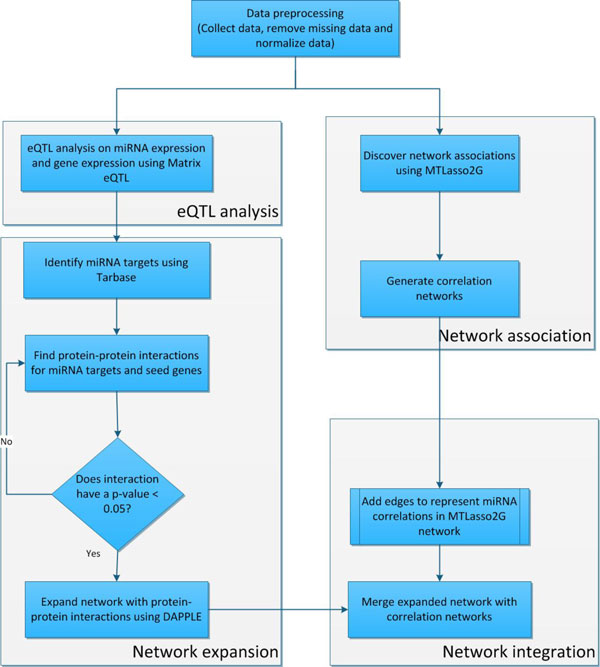
The overall workflow of our integrative approach. The workflow to create the integrated network consisted of four phases; eQTL analysis, network association, network expansion and network integration. We began with data pre-processing in which we collected data and normalized it. The first phase of the workflow started with an eQTL analysis between miRNA and gene expression data using Matrix eQTL. Once the eQTL analysis was complete, we proceeded to the network expansion phase. In the network expansion phase, we used TarBase to identify miRNA targets based on the eQTLs found in Matrix eQTL. Next, we found protein-protein interactions for the identified targets. To select the most confident interactions, we chose the interactions with a p-value ≤ 0.05. Then we expanded the network with the selected protein-protein interactions using DAPPLE, creating the first integrated network. Simultaneously, we used MtLasso2G to discover network associations between miRNAs, creating miRNA correlation networks. We then combined the eQTL expanded network with the correlation networks to create the final integrated network.
Data preprocessing
We downloaded miRNA isoform and mRNA expression data for 480 samples of serous ovarian cancer from the TCGA data portal [37]. Both data sets were generated on the Illumina HiSeq platform. To pre-process the data, we removed samples with missing data and extracted quantifications for 183 miRNAs and 13,536 genes. To reduce the variation between samples, we used between-sample quantile normalization to normalize both the miRNA and gene expression data.
Network expansion
To help exploit the relationships between miRNA and gene expression, we performed an eQTL analysis between the miRNA expression and gene expression profiles. The eQTL analysis was performed using Matrix eQTL, an R package [35]. Matrix eQTL was chosen because of its efficient computational time with large datasets [35] and its proven efficiency in many large studies [22,23]. We performed cis eQTL analysis, whereby we only tested miRNA and gene pairs that were within 1 MB of each other. To test for associations between the miRNA and genes, we used a linear regression model provided by Matrix eQTL. To correct for multiple comparisons, we utilized a multi-test correction based on false discovery rate (FDR) and chose the significant eQTLs with a FDR cutoff at < 0.01 for further analysis.
Using the eQTLs identified in Matrix eQTL as seed nodes for an initial network, we searched for linked targets of the miRNAs to expand the network. In order to identify miRNA targets, we used TarBase to generate a list of the miRNAs and their corresponding gene targets[34]. TarBase is a manually curated database of all experimentally verified miRNA targets. In generating the list, we discovered that some miRNAs may have multiple designated names associated with the 3' untranslated region (UTR) and the 5' untranslated region. In this case, we used both variations to keep it consistent with our data. For example, if there was an ambiguous miRNA name used in the TarBase search, it was replaced by both the -3' and -5' version of the name.
We used the miRNA target genes gathered from TarBase, in combination with the perturbed genes from the eQTLs, to further extend the network using protein-protein interactions. We chose the Disease Associated Protein-Protein Link Evaluator (DAPPLE)[36] for this step of network expansion since it's been well evaluated in finding candidate genes through expanding networks on signals from genome wide association studies [36,38,39]. By using DAPPLE on the miRNA target genes and the miRNA perturbed genes in the eQTL associations, we were able to find protein-protein interactions for miRNA targets and the miRNA eQTL perturbed genes. To perform the link evaluation, DAPPLE uses a statistical model to extend a network from an input seed gene list. The protein-protein interactions were derived from the InWeb database [40].
At each network expansion step, DAPPLE selects a new edge from the existing node set with the highest probability based on its significance against random permutations of the protein-protein interaction network. During the permutations, the network topology is conserved. For our analysis, we performed 1000 random permutations of the network. As part of our selection criteria, we only selected the interactions that contained a seed gene with a corrected p-value of ≤ 0.05. This allowed us to keep those interactions with a high-confidence for further analysis. We then overlapped the selected protein-protein interactions, the eQTLs and the miRNA-target interactions to create an initial integrated network.
Network associations
In parallel with the network expansion, we used a two-graph guided multitask Lasso (MtLasso2G) to perform a second eQTL analysis [30]. MtLasso2G is an orthologous approach capable of determining network associations. Unlike Matrix eQTL, which we used to perform linear regression for each pair of miRNA and gene independently for eQTL analysis, MtLasso2G uses a sparse learning model that incorporates the relationships among miRNAs and genes. Here, we used a correlation expression network among miRNAs, and another co-expressed network among genes that potentially captured the structure of co-regulation networks. The MtLasso2G model accomplished eQTL analysis by adding two regularization terms based on the two graphs on features (i.e. miRNA expression) and labels (i.e. gene expression), which are encoded as miRNA co-expression network and gene co-expression network in our analysis respectively. The model can then be learned by minimizing the following objective function in a linear system:
| (1) |
| (2) |
The two graphs, G1 = (V1, E1) and G2 = (V2, E2) represent the correlation networks among genes and miRNAs respectively. V1 and V2 are the nodes in G1 and G2. E1 and E2 are the edges representing the correlations among the corresponding vertices. w(·) is the edge weight. Individual edges in G1 and G2 are represented as em,l and ef,g respectively. The correlation between ym and yl is denoted as rm,l, and rf,g denotes the correlation between yf and yg. The learned β matrix from the model represents the association between each feature and label. Those nonzero terms stand for the identified eQTLs. As the MtLasso2G model captures the correlation structures among the miRNAs and genes, it produces not only a set of eQTLs, but also network associations. These network associations include a subnetwork capturing miRNA correlations, and another subnetwork representing the correlations among the perturbed genes.
Network integration
We finally created an integrated network by merging the expanded network extended using DAPPLE, and the network associations generated from the MtLasso2G model. Specifically, we added representations of the correlation graphs from MtLasso2G to the expanded network from DAPPLE as following. If one miRNA was present in the current integrated network, we added highly correlated miRNAs. If both genes were already present in the current integrated network, we added an edge to the network.
The edges between miRNAs and perturbed genes from network associations in MtLasso2G are added to the integrated network as well. By merging the networks generated from DAPPLE and MtLasso2G as stated above, we created the final integrated network which was then visualized using Cytoscape[41]. Integrating the networks in this manner allowed us to conserve many of the pre-determined relationships found early on in the analysis.
Results and discussion
Our approach generates an integrated network (Figure 2) that incorporates the comprehensive relationship between miRNA and gene expression. We create this network from multiple sources of data and analysis including miRNA eQTL and associated genes, miRNAs and their target genes, protein-protein interactions among miRNA eQTL perturbed genes and miRNA target genes. This network thus captures the complexity of the interplay among miRNAs and genes in ovarian cancer, and point to a list of candidate miRNAs and genes for future studies.
Figure 2.
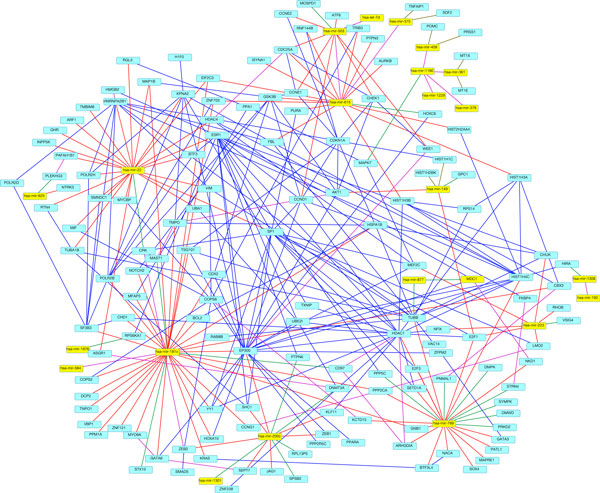
The integrated network incorporating multiple data sources. This network incorporated eQTLs, protein-protein interactions from DAPPLE, and miRNA-target interactions from TarBase. miRNAs are shown in yellow nodes, and genes are shown in light blue nodes. eQTLs are shown as green edges, protein-protein interactions are shown as blue edges, miRNA-target interactions are shown as red edges, MtLasso2G correlation interactions are in purple, and MtLasso2G eQTLs are in gold.
Our method is scalable and applicable to integrating many genomic datasets. Here, as a demonstration of our approach, we apply it to the miRNA and gene expression data in ovarian cancer from TCGA [37]. After pre-processing, we use the expression of 183 miRNAs, and the expression of 13,536 genes from 480 samples.
We identified 44 miRNA eQTLs with a FDR < 0.01 using Matrix eQTL. There were 16 unique miRNAs, and 44 unique genes represented in the eQTLs. We found 310 unique target genes using TarBase, of which 244 could be used as inputs for DAPPLE. Figure 3 shows the miRNA-taget and eQTL network. There were 236 direct connections found using DAPPLE, and 145 that contained a seed gene with a corrected p-value of <0.05. We added 9 correlated miRNAs, and 18 correlated gene edges to the network based on the MtLasso2G correlation graph.
Figure 3.
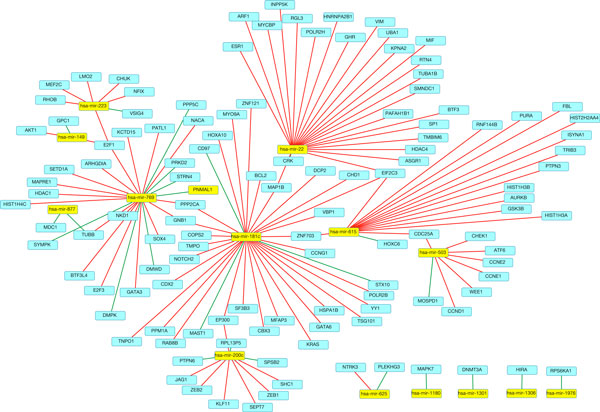
The miRNA-target and eQTL network. This network contains the eQTLs as well as the target genes from TarBase for the miRNAs. The initial integrated network is formed when the protein-protein interactions for the genes in this network are added.
With MtLasso2G, we found 48 eQTLs, and were able to add 8 of them to the network, composed of the miRNA-target interactions found in TarBase, the eQTLs from Matrix eQTL, and the MtLasso2G correlations. This process created the final integrated network including 167 nodes and 277 edges shown in Figure 2. Table 1 summarizes the number of cancer genes found in each part of the analysis. Of the 145 gene nodes in this integrated network, 26 were found to be cancer related, and 11 of those were associated with ovarian cancer[42,43], as illustrated in Table 2.
Table 1.
Cancer associated genes in the network components and the integrated network.
Table 2.
List of cancer and ovarian cancer associated genes in the integrated network.
| Genes Associated with Cancer | Genes Associated with Ovarian Cancer |
|---|---|
| AKT1 | AKT1 |
| BCL2 | BCL2 |
| CCND1 | CCND1 |
| CD97 | CDC25A |
| CDC25A | CDKN1A |
| CDKN1A | E2F1 |
| CDX2 | E2F3 |
| COPS2 | EP300 |
| E2F1 | GATA6 |
| E2F3 | KRAS |
| EP300 | SHC1 |
| GATA6 | |
| GSK3B | |
| JAG1 | |
| KRAS | |
| LMO2 | |
| MAPK7 | |
| MAPRE1 | |
| MIF | |
| NOTCH2 | |
| NTRK3 | |
| RHOB | |
| RPS6KA1 | |
| RTN4 | |
| SHC1 | |
| SOX4 |
One of the genes found in the integrated network, AKT 1, has been implicated in tumorigenesis, specifically in ovarian cancer[44,45]. In our analysis AT K1 was found in the DAPPLE extended network, the miRNA-target network and the MtLasso2G gene-correlation graph. Figure 4 shows a subnetwork involving AKT 1 of our integrated network. This subnetwork involves 5 miRNAs and 12 genes, connected by 31 edges. Three of the edges are eQTLs, and six edges were miRNA-target interactions. Another gene in the subnetwork was ESR1, which has been associated with breast cancer, and ESR1 expression has been suggested as a predictor of ovarian cancer survival[46-48].
Figure 4.
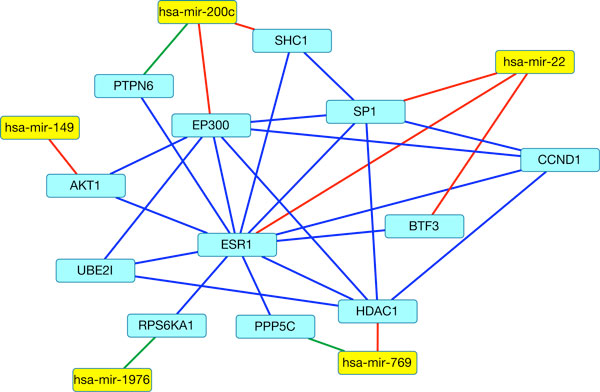
A subnetwork illustrating multiple data sources for network construction. This subnetwork illustrates how the multiple data sources are involved in the integrated network. If the network just contained the eQTLs in green, and the miRNA-target interactions in red, most of the connections would not exist. Adding the protein-protein interactions of the genes, allows us to visualize potential downstream effects.
Interestingly, in addition to capturing known ovarian cancer genes, this subnetwork provides a set of miRNAs and genes interacting with these known cancer genes. We propose that these interactive miRNAs and genes offer a new list of potential miRNA and gene markers for tumorigenesis, particularly in ovarian cancer.
Another interesting gene in the integrated network is E2F 1 (Figure 2), which was targeted by three miRNAs. E2F 1 has been studied as a predictor of drug resistance in ovarian cancer, and the down regulation of E2F 1 has been shown to inhibit growth of ovarian cancer [49,50]. Therefore, E2F 1 and their interactive partners are worthy of follow up studies for tumorigenesis as well.
We then analyzed the role of the miRNA eQTLs in different diseases including cancer and particularly ovarian cancer. Using multiple datasets (HMDD2, miRCancer, Phenomir, miRdSNP, miR2Disease, miRPD and Marchini) [51-57], we found that 19 of the 22 miRNAs have been implicated in some type of disease. Of these miRNAs, 14 were associated with a form of cancer, and 12 were specifically associated with ovarian cancer. Table 3 lists these cancer and ovarian cancer related miRNAs in our integrated network. Some of these miRNAs have demonstrated biological and clinical utility and could serve as disease biomarkers. For example, increased expression of hsa-miR-200c, one of the miRNAs found in our analysis, has been associated with increased survival, decreased chance of relapse and increased sensitivity to paclitaxel, a chemotherapy drug[57-59]. hsa-mir-22 has been identified as an marker for both survival and recurrence of ovarian cancer[60]. hsa-mir-22 has also been suggested as an inhibitor of cell movement and invasion in ovarian cancer[61]. Both hsa-mir-22 and hsa-mir-200c are found in the subnetwork, with hsa-mir-22 targeting ESR1.
Table 3.
Number and names of miRNAs that are disease associated, cancer associated and ovarian cancer associated respectively.
| miRNAs | Disease associated miRNA | Cancer associated miRNA | Ovarian Cancer associated miRNA | |
|---|---|---|---|---|
| Number | 22 | 19 | 14 | 12 |
| Names | hsa-let-7d | hsa-mir-1180 | hsa-mir-149 | hsa-mir-149 |
| hsa-mir-1180 | hsa-mir-1228 | hsa-mir-181c | hsa-mir-181c | |
| hsa-mir-1228 | hsa-mir-1301 | hsa-mir-192 | hsa-mir-192 | |
| hsa-mir-1301 | hsa-mir-149 | hsa-mir-200c | hsa-mir-200c | |
| hsa-mir-1306 | hsa-mir-181c | hsa-mir-22 | hsa-mir-22 | |
| hsa-mir-149 | hsa-mir-192 | hsa-mir-223 | hsa-mir-223 | |
| hsa-mir-181c | hsa-mir-200c | hsa-mir-361 | hsa-mir-375 | |
| hsa-mir-192 | hsa-mir-22 | hsa-mir-375 | hsa-mir-379 | |
| hsa-mir-1976 | hsa-mir-223 | hsa-mir-379 | hsa-mir-503 | |
| hsa-mir-200c | hsa-mir-361 | hsa-mir-409 | hsa-mir-584 | |
| hsa-mir-22 | hsa-mir-375 | hsa-mir-503 | hsa-mir-625 | |
| hsa-mir-223 | hsa-mir-379 | hsa-mir-584 | hsa-mir-877 | |
| hsa-mir-361 | hsa-mir-409 | hsa-mir-625 | ||
| hsa-mir-375 | hsa-mir-503 | hsa-mir-877 | ||
| hsa-mir-379 | hsa-mir-584 | |||
| hsa-mir-409 | hsa-mir-615 | |||
| hsa-mir-503 | hsa-mir-625 | |||
| hsa-mir-584 | hsa-mir-769 | |||
| hsa-mir-615 | hsa-mir-877 | |||
| hsa-mir-625 | ||||
| hsa-mir-769 | ||||
| hsa-mir-877 | ||||
Conclusions
Changes in miRNA and mRNA expression are known to be involved in both ovarian cancer development and progression. Pin-pointing the exact changes and the relationships that occur between them could lead to advances in how ovarian cancer is treated and diagnosed. Creating an integrated network involving eQTLs, miRNA targets, protein-protein interactions and correlation graphs is one way to explore these relationships. Integrating multiple data sources allows us to create a wider and more holistic view of the interactions in ovarian cancer. This can lead us to understanding more about the underlying mechanisms that affect progression in ovarian cancer.
Therefore, in this paper we developed a new method of constructing an integrated network by combining the strength of eQTL study and network analysis. As a demonstration, we applied our method to TCGA ovarian cancer data and constructed an integrated network of miRNA and gene interactions in ovarian cancer. Utilizing disparate data sources allowed us to find relationships that would not have been visible otherwise. The network information we incorporated included protein-protein interactions, eQTL associations and miRNA-target interactions. None of those interactions alone would have shown all of the connections, but combining all these network information together allows us to see the complex interplay between miRNAs and genes in ovarian cancer. Our integrated analysis found miRNAs and genes linked to cancer and specifically to ovarian cancer. Utilizing integrated approaches like this one, could lead to a more comprehensive understanding of the biology that drives ovarian cancer.
In this study we used multiple data sources including one miRNA target database (i.e. TarBase), a protein-protein interaction network (i.e. InWeb) and expression data from the TCGA data portal to construct an integrated network. Based on recent studies [15,17], it is known that miRNAs and genetic variation have a critical impact on cancer progression and to an extent, cancer phenotype. Our approach allows us to incorporate various data sources into an integrated network. The generated network contained miRNA to target gene relationships, eQTL associations, protein-protein interactions, the correlations or co-expressions among microRNAs and genes respectively.
In the future, we will extend our approach by incorporating multiple databases of a particular network type, such as the STRING database [62] for protein-protein interaction networks. We will also incorporate other types of network information. For example, we can include regulatory and signaling pathways accumulated in existing pathway databases. Using various data sources, we can exploit the relationships between different molecular components to help understand how ovarian cancer progresses. Although ovarian cancer is a very complex disease, our approach is a leading step for future investigative methods.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AQ and XS contributed to the design of the study. AQ performed the implementation and analysis of the project. AQ, XS, LT and BH performed the interpretation of the results and wrote the manuscript. All the authors read and approved the final manuscript.
Acknowledgements
We thank the anonymous reviewers for their constructive comments. This work was partially supported by a Faculty Research Grant from the University of North Carolina at Charlotte.
Declarations
Publication costs for this article were funded by the senior author's institute.
This article has been published as part of BMC Bioinformatics Volume 16 Supplement 5, 2015: Selected articles from the 10th International Symposium on Bioinformatics Research and Applications (ISBRA-14): Bioinformatics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/16/S5.
References
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: A Cancer Journal for Clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA. 1975-2009 (Vintage 2009 Populations) National Cancer Institute. Bethesda, MD; SEER Cancer Statistics Review. [Google Scholar]
- Yang H, Kong W, He L, Zhao JJ, O'Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, Cheng JQ. MicroRNA Expression Profiling in Human Ovarian Cancer: miR-214 Induces Cell Survival and Cisplatin Resistance by Targeting PTEN. Cancer Res. 2008;68:425–433. doi: 10.1158/0008-5472.CAN-07-2488. [DOI] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;15:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;15:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- LaCroix B, Gamazon ER, Lenkala D, Im HK, Geeleher P, Ziliak D, Cox NJ, Huang RS. Integrative analyses of genetic variation, epigenetic regulation, and the transcriptome to elucidate the biology of platinum sensitivity. BMC Genomics. 2014;15:292. doi: 10.1186/1471-2164-15-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2013;15:287–314. doi: 10.1146/annurev-pathol-012513-104715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley BS, Loftus JC, Mielke CJ, Dinu V. Differential expression of microRNAs as predictors of glioblastoma phenotypes. BMC Bioinformatics. 2014;15:21. doi: 10.1186/1471-2105-15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffa FM, Camps C, Winchester L, Snell CE, Gee HE, Sheldon H, Taylor M, Harris AL, Ragoussis J. microRNA-Associated Progression Pathways and Potential Therapeutic Targets Identified by Integrated mRNA and microRNA Expression Profiling in Breast Cancer. Cancer Res. 2011;71:5635–5645. doi: 10.1158/0008-5472.CAN-11-0489. [DOI] [PubMed] [Google Scholar]
- Miles GD, Seiler M, Rodriguez L, Rajagopal G, Bhanot G. Identifying microRNA/mRNA dysregulations in ovarian cancer. BMC Research Notes. 2012;5:164. doi: 10.1186/1756-0500-5-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadran S, Remacle F, Levine RD. miRNA and mRNA cancer signatures determined by analysis of expression levels in large cohorts of patients. PNAS. 2013;110(47):19160–19165. doi: 10.1073/pnas.1316991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinia S, Croce CM. Prognostic microRNA/mRNA signature from the integrated analysis of patients with invasive breast cancer. PNAS. 2013;110(18):7413–7417. doi: 10.1073/pnas.1304977110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Volinia S, Bonome T, Calind GA, Greshock J, Yang N, Liud CG, Giannakakis A, Alexiou P, Hasegawa K, Johnstone CN, Megraw MS, Adams S, Lassus H, Huang J, Kaur S, Liang S, Sethupathy P, Leminen A, Simossis VA, Sandaltzopoulos R, Naomoto Y, Katsaros D, Gimotty PA, DeMichele A, Huang Q, Butzow R, Rustgi AK, Weber BL, Birrer MJ, Hatzigeorgiou AG, Croce CM, Coukos G. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. PNAS. 2008;105(19):7004–7009. doi: 10.1073/pnas.0801615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh Aditya, Lee Christine, Joseph Peronne, Marchini Sergio, Baccarini Alessia, Kolev Valentin, Romualdi Chiara, Fruscio Robert, Shah Hardik, Wang Feng, Mullokandov Gavriel, Fishman David, D'Incalci Maurizio, Rahaman Jamal, Kalir Tamara, Redline Raymond W, Brown Brian D, Narla Goutham, DiFeo Analisa. microRNA-181a has a critical role in ovarian cancer progression through the regulation of the epithelial-mesenchymal transition. Nat Commun. 2014;01:2977. doi: 10.1038/ncomms3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Jun, Getz Gad, Miska Eric A, Alvarez-Saavedra Ezequiel, Lamb Justin, Peck David, Sweet-Cordero Alejandro, Ebert Benjamin L, Mak Raymond H, Ferrando Adolfo A, Downing James R, Jacks Tyler, Horvitz Robert H, Golub Todd R. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan Brid M, Robles Ana I, Harris Curtis C. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10(6):389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, Lyle R, Hunt S, Kahl B, Antonarakis SE, Tavare S, Deloukas P, Dermitzakis ET. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1(6):e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavare S, Deloukas P, Hurles ME, Dermitzakis ET. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315(5813):848–53. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat Rev Genet. 2009;10(9):595–604. doi: 10.1038/nrg2630. Sep; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Seo JH, Stranger B, McKenna A, Pe'er I, Laframboise T, Brown M, Tyekucheva S, Freedman ML. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152(3):633–41. doi: 10.1016/j.cell.2012.12.034. Jan 31; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedlander MR, Hoen PAC, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlof J, Ribeca P, Pulyakhina I, Esser D, Giger T, Tikhonov A, Sultan M, Bertier G, MacArthur DG, Lek M, Lizano E, Buermans HPJ, Padioleau I, Schwarzmayr T, Karlberg O, Ongen H, Kilpinen H, Beltran S, Gut M, Kahlem K, Amstislavskiy V, Stegle O, Pirinen M, Montgomery SB, Donnelly P, McCarthy MI, Flicek P, Strom TM. The Geuvadis Consortium; Lehrach H, Schreiber S, Sudbrak R, Carracedo A, Antonarakis SE, Hasler R, Syvanen AC, van Ommen GJ, Brazma A, Meitinger T, Rosenstiel P, Guigo R, Gut IG, Estivill X, Dermitzakis ET. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–11. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Morar N, Dixon AL, Lathrop GM, Abecasis GR, Moffatt MF, Cookson WO. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome Res. 2013;23(4):716–26. doi: 10.1101/gr.142521.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Quitadamo A, Lin F, Shi X. Methods for Population Based eQTL Analysis in Human Genetics. Tsinghua Science and Technology. 2014;19(6):624–634. [Google Scholar]
- Gamazon ER, Innocenti F, Wei R, Wang L, Zhang M, Mirkov S, Ramirez J, Huang RS, Cox NJ, Ratain MJ, Liu W. A genome-wide integrative study of microRNAs in human liver. BMC Genomics. 2013;14:395. doi: 10.1186/1471-2164-14-395. Jun 13; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Ziliak D, Im HK, LaCroix B, Park DS, Cox NJ, Huang RS. Genetic architecture of microRNA expression: implications for the transcriptome and complex traits. Am J Hum Genet. 2012;90(6):1046–63. doi: 10.1016/j.ajhg.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Seunghak, Xing Eric P. Statistical Estimation of Correlated Genome Associations to a Quantitative Trait Network. PLoS Genetics. 2009;5(8):e1000587. doi: 10.1371/journal.pgen.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Seunghak, Zhu Jun, Xing Eric P. Adaptive Multi-Task Lasso: with Application to eQTL Detection. NIPS. 2010.
- Chen X, Shi X, Xu X, Wang Z, Mills RE, Lee C, Xu J. A two-graph guided multi-task lasso approach for eQTL mapping. Proceedings of the 15th International Conference of Artificial Intelligence and Statistics (AISTATS), Journal of Machine Learning Research (JMLR) W&CP. 2012;22:208–217. [Google Scholar]
- S Lee, EP Xing. Leveraging Input and Output Structures For Joint Mapping of Epistatic and Marginal eQTLs. Bioinformatics, Special Issue of the Proceedings of the International Conference on Intelligent Systems for Molecular Biology (ISMB) 2012;28(12):i137–i146. doi: 10.1093/bioinformatics/bts227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Cheng, Xiang Zhang, Zhishan Guo, Yu Shi, Wei Wang. Graph Regularized Dual Lasso for Robust eQTL Mapping. Bioinformatics, Special Issue of the Proceedings of the International Conference on Intelligent Systems for Molecular Biology (ISMB) 2014;30(12):i139–i148. doi: 10.1093/bioinformatics/btu293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu J, Shi X. Finding alternative eQTLs by exploring sparse model space. Journal of Computational Biology. 2014;21(5):385–393. doi: 10.1089/cmb.2014.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos GL, Reczko M, Simossis VA, Sethupathy P, Hatzigeorgiou AG. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res. 2009. pp. D155–8. [DOI] [PMC free article] [PubMed]
- Shabalin AA. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28(10):1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tartar D, Cotsapas C, Daly MJ. Proteins Encoded in Genomic Regions Associated with Immune-Mediated Disease Physically Interact and Suggest Underlying Biology. PLoS Genetics. 2011;7(1):e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas: understanding genomics to improver cancer care. http://cancergenome.nih.gov/
- Raj Towfique, Shulman Joshua M, Keenan Brendan T, Chibnik Lori B, Evans Denis A, Bennett David A, Stranger Barbara E, De Jager Philip L. Alzheimer Disease Susceptibility Loci: Evidence for a Protein Network under Natural Selection. Am J Hum Genet. 2012;90(4):720–726. doi: 10.1016/j.ajhg.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Neale BM, Liu L. et al. Polygenic transmission and complex neuro developmental network for attention deficit hyperactivity disorder: genome-wide association study of both common and rare variants. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(5):419–30. doi: 10.1002/ajmg.b.32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tumer Z, Pociot F, Tommerup N, Moreau Y, Brunak S. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol. 2007;25(3):309–16. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas of Genetics and Cytogenetics in Oncology and Haematology. http://AtlasGeneticsOncology.org
- Wall DP, Pivovarov R, Tong M, Jung JY, Fusaro VA, DeLuca TF, Tonellato PJ. Genotator: a disease-agnostic tool for genetic annotation of disease. BMC Med Genomics. 2010;3:50. doi: 10.1186/1755-8794-3-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. PNAS. 2001;98(20):10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, Janikariman M, Olvera N, Stevens EV, She QB, Aghajanian C, King TA, de Stanchina E, Spriggs DR, Heguy A, Taylor BS, Sander C, Rosen N, Levine DA, Solit DB. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2012;2(1):56–67. doi: 10.1158/2159-8290.CD-11-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbier AK, Anderson H, Ghazoui Z, Lopez-Knowles E, Pancholi S, Ribas R, Drury S, Sidhu K, Leary A, Martin L, Dowsett M. ESR1 Is Co-Expressed with Closely Adjacent Uncharacterised Genes Spanning a Breast Cancer Susceptibility Locus at 6q25.1. PLoS Genet. p. e1001382. [DOI] [PMC free article] [PubMed]
- Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, Roberts JS, Rae JM, Van Poznak CH, Hayes DF, Chugh R, Kunju LP, Talpaz M, Schott AF, Chinnaiyan AM. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nature Genetics. 2013. pp. 1446–1451. [DOI] [PMC free article] [PubMed]
- Darb-Esfahani S, Wirtz1 RM, Sinn BV, Budczies J, Noske A, Weichert W, Faggad A, Scharff A, Sehouli J, Oskay-Ozcelik G, Zamagni C, De Iaco P, Martoni A, Dietel M, Denkert C. Estrogen receptor 1 mRNA is a prognostic factor in ovarian carcinoma: determination by kinetic PCR in formalin-fixed paraffin-embedded tissue. Endocrine-Related Cancer. 2009;16:1229–1239. doi: 10.1677/ERC-08-0338. [DOI] [PubMed] [Google Scholar]
- Reimer D, Sadr S, Wiedemair A, Stadlmann S, Concin N, Hofstetter G, Moller-Holzner E, Marth C, Zeimet AG. Clinical Relevance of E2F Family Members in Ovarian Cancer, An Evaluation in a Training Set of 77 Patients. Clin Cancer Res. 2007;13:144. doi: 10.1158/1078-0432.CCR-06-0780. [DOI] [PubMed] [Google Scholar]
- D'Souza T, Becker KG, Wood WH, Zhang Y, Wersto RP, Morin PJ. Non-Steroidal Anti-inflammatory Drugs Decrease E2F1 Expression and Inhibit Cell Growth in Ovarian Cancer Cells. PLoS ONE. 2013;8(4):e61836. doi: 10.1371/journal.pone.0061836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, Cui Q. An analysis of human microRNA and disease associations. PLoS One. 2008;3(10):e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B, Ding Q, Han H, Wu D. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics. 2013;29(5):638–644. doi: 10.1093/bioinformatics/btt014. [DOI] [PubMed] [Google Scholar]
- Ruepp A, Kowarsch A, Schmidl D, Buggenthin F, Brauner B, Dunger I, Fobo G, Frishman G, Montrone C, Theis FJ. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biology. 2010;11:R6. doi: 10.1186/gb-2010-11-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno AE, Li L, Kalabus JL, Pan Y, Yu A, Hu Z. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3'UTRs of human genes. BMC Genomics. 2012;13(1):44. doi: 10.1186/1471-2164-13-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- miRPD. http://mirpd.jensenlab.org/HTML/miRPD
- Marchini S, Cavalieri D, Fruscio R, Calura E, Garavaglia D, Nerini IF, Mangioni C, Cattoretti G, livio L, Beltrame L, Katsaros D, Scarampi L, Menato G, Perego P, Chiorino G, Buda A, Romualdi C, D'Incalci M. Association between miR-200c and the survival of patients with stage I epithelial ovarian cancer: a retrospective study of two independent tumour tissue collections. The Lancet Oncology. 2011;12(3):273–285. doi: 10.1016/S1470-2045(11)70012-2. [DOI] [PubMed] [Google Scholar]
- Prislei S1, Martinelli E, Mariani M, Raspaglio G, Sieber S, Ferrandina G, Shahabi S, Scambia G, Ferlini C. MiR-200c and HuR in ovarian cancer. BMC Cancer. 2013;13:72. doi: 10.1186/1471-2407-13-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cittelly DM, Dimitrova I, Howe EN, Cochrane DR, Jean A, Spoelstra NS, Post MD, Lu X, Broaddus RR, Spillman MA, Richer JK. Restoration of miR-200c to ovarian cancer reduces tumor burden and increases sensitivity to paclitaxel. Mol Cancer Ther. 2012;11(12):2556–2565. doi: 10.1158/1535-7163.MCT-12-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino KR, Rodriguez-Zas SL. Transcription Factor-MicroRNA-Target Gene Networks Associated with Ovarian Cancer Survival and Recurrence. PLoS ONE. 2013;8(3):e58608. doi: 10.1371/journal.pone.0058608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Liang S, Yu H, Zhang J, Ma D, Lu X. An inhibitory effect of miR-22 on cell migration and invasion in ovarian cancer. Gynecol Oncol. 2010;119(3):543–8. doi: 10.1016/j.ygyno.2010.08.034. [DOI] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013. pp. D808–15. [DOI] [PMC free article] [PubMed]