

Abstract
We performed genome-wide tests for association between haplotype clusters and each of 9 metabolic traits in a cohort of 5402 Northern Finnish individuals genotyped for 330 000 single-nucleotide polymorphisms. The metabolic traits were body mass index, C-reactive protein, diastolic blood pressure, glucose, high-density lipoprotein (HDL), insulin, low-density lipoprotein (LDL), systolic blood pressure, and triglycerides. Haplotype clusters were determined using Beagle. There were LDL-associated clusters in the chromosome 4q13.3-q21.1 region containing the albumin (ALB) and platelet factor 4 (PF4) genes. This region has not been associated with LDL in previous genome-wide association studies. The most significant haplotype cluster in this region was associated with 0.488 mmol/l higher LDL (95% CI: 0.361–0.615 mmol/l, P-value: 6.4 × 10−14). We also observed three previously reported associations: Chromosome 16q13 with HDL, chromosome 1p32.3-p32.2 with LDL and chromosome 19q13.31-q13.32 with LDL. The chromosome 1 and chromosome 4 LDL associations do not reach genome-wide significance in single-marker analyses of these data, illustrating the power of haplotypic association testing.
Introduction
The identification of genetic factors that influence quantitative traits such as low-density lipoprotein (LDL) has clinical importance. For example, higher LDL levels are associated with increased risk of cardiovascular health disease, and the discovery of the association between PCSK9 mutations and LDL led to the development of PCSK9 inhibitors as a novel class of LDL-reducing drugs.1 Notably, in the cohort study that found the LDL-associated PCSK9 mutations, each mutation was present in fewer than 2% of study individuals.2 The standard single-SNP analysis commonly employed in genome-wide association studies (GWAS) has low power for detecting such low frequency causal variants. By employing haplotypic analysis in combination with single-SNP analysis, we can improve power above that of single-SNP analysis alone for detecting causal variants with low minor allele frequency.3
The standard approach for association analysis of genome-wide single-nucleotide polymorphism (SNP) array data in population samples is to test each SNP individually for association with the trait. This approach can have high power to detect an ungenotyped causal variant when the causal variant is common and correlated with one or more genotyped variants on the array. However, the single-SNP approach has lower power to detect low frequency ungenotyped causal variants.3 To improve power for low frequency variants, one could impute them and test for association with the trait, but imputation of low frequency variants suffers from poor accuracy.4 Moreover, variants that are unique to the population of interest cannot be imputed, unless there exists a reference panel drawn from that population.
An alternative to single-SNP association analysis is to perform tests of association between haplotypes and the trait. When a new variant arises in a population, it occurs on a specific haplotype, which is transmitted with the variant from generation to generation. Over time, the haplotype background is shortened by recombinations around the new variant, but since rare variants are usually of relatively recent origin, the correlation between the haplotype in the local genomic region around the variant and the variant itself is usually still strong. A haplotype can therefore serve as a proxy for a rare variant. One would then expect a haplotypic test to have higher power than a single-marker test for detecting an ungenotyped low frequency causal variant. Indeed, a previous simulation study suggests that multi-marker tests can have higher power than single-marker tests,5 and a previous genome-wide haplotypic study found a gene cluster associated with coronary artery disease that was not found with genome-wide SNP testing.6
One haplotype association test with appealing properties is the Beagle haplotype cluster test.3, 7 Unlike window-based methods that define haplotypes in a window of a specified number of genotyped variants, the Beagle haplotype cluster test is not confined to windows, but at each genomic location, clusters together locally similar haplotypes. This local clustering avoids the need to define an arbitrary window length, allowing the effective window length for clustering to vary by genomic position, depending on the local linkage disequilibrium (LD) between genotyped variants. The Beagle haplotype cluster test has previously been applied to case–control data.7, 8 Here, we extend the methodology to analyze quantitative traits.
One disadvantage of the Beagle haplotype cluster test in case–control data is that the test is very sensitive to genotype errors. It is common for case and control data to be collected separately, with differences in DNA collection and storage. These differences result in differential genotype error, so that apparent haplotypes may arise at frequencies differing between cases and controls, resulting in spurious associations.8 In contrast, in population cohort samples one does not expect genotype quality to be correlated with trait values, so spurious associations due to genotype error are unlikely to occur. Nevertheless, care needs to be taken to ensure that such effects are not present.
In this study, we test for association between nine metabolic traits and haplotype clusters in data from the North Finland Birth Cohort. We compare the results with those from single-SNP analysis of the same data and with single-SNP analysis of the same traits in other populations.
Materials and Methods
Data
We analyze genotypes, metabolic traits, and other measurements from the Northern Finland Birth Cohort (NFBC), a sample of 5402 individuals from Northern Finland who were born in 1966. The data were downloaded from dbGaP (accession number phs000276). All measurements were taken when participants were 31 years old. The 5402 individuals were typed at 320 959 autosomal SNPs and 9581 X- chromosome SNPs, using an Illumina Infinium SNP array. Quantitative metabolic traits were body mass index (BMI), C-reactive protein (CRP), diastolic blood pressure (DBP), fasting glucose (Glucose), high-density lipoprotein (HDL), fasting insulin (insulin), low-density lipoprotein (LDL), systolic blood pressure (SBP), and triglycerides (TG) (Supplementary Table S1). Other measured variables were sex (male or female), oral contraceptive pill usage, pregnancy status, diabetes medication usage, whether weight was self-reported and fasting status (Supplementary Table S2). Methods of trait measurement were published previously.9
Before testing for association, we estimated haplotype phase using Beagle.10 Excluding 503 close relatives identified previously,11 we used Beagle version 4 revision r780 to phase genotypes, resulting in 4899 individuals phased at all 330 540 SNPs. We then used Beagle 3.3.2 with default settings to cluster the haplotypes at each SNP position.3 At each SNP position, the set of haplotype clusters is a partition of the set of haplotypes (that is, chromosomes), and each haplotype is a member of exactly one haplotype cluster. For each haplotype cluster at a SNP position, we defined a pseudomarker variable that gave the number (0, 1, or 2) of an individual's two haplotypes belonging to the cluster. A haplotype cluster at one SNP position can consist of the same set of haplotypes as a haplotype cluster at another SNP position. In this case, the two pseudomarkers defined by the haplotype clusters are perfectly correlated, and we say that the pseudomarkers represent the same haplotype cluster. Autosomal pseudomarkers were used to perform regressions in R, described later in this section.
To control for population structure, we used the first 10 eigenvectors as covariates in our regression analysis. To determine eigenvectors, we first used Plink to obtain a pruned set of SNPs in approximate linkage equilibrium with each other. The option used was ‘-indep-pairwise 50 5 0.5', which accomplished the following: for each window of 50 SNPs, with windows spaced 5 SNPs apart, Plink calculated the linkage disequilibrium measure r2 between each pair of SNPs in the window and removed one SNP of each pair that had r2 greater than 0.5.12 Using this pruned set of SNPs typed at 4899 individuals as input to the smartpca program from Eigensoft version 4.2, we obtained 10 eigenvectors. The smartpca program implements the Eigenstrat method.13 By default, it iteratively calculates 10 preliminary eigenvectors corresponding to 10 largest eigenvalues and removes any individual with an element of a preliminary eigenvector >6 SD values away from the mean of the elements of the preliminary eigenvector. After all iterations, 11 outlier individuals were removed.
Regression analyses
For each of the nine metabolic traits, and for each haplotype cluster, we regressed Trait onto Cluster, adjusting for Sex, Pills, and 10 eigenvectors. The regression model was
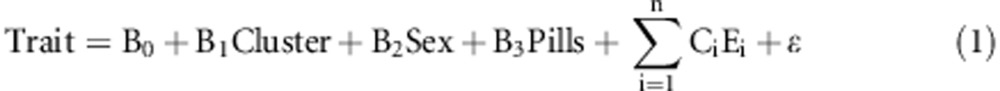 |
with E(ɛ)=0 and Var(ɛ)=σ2. Cluster is the number of an individual's two haplotypes that belong to the haplotype cluster being tested and takes values 0, 1, or 2. Sex takes value 0 for female and 1 for male. Pill is an indicator for taking oral contraceptive pills, encoded 0 for not taking and 1 for taking. The first 10 eigenvectors from Eigenstrat are Ei, i=1, …, 10. All regressions were performed in R using the lm function with P-values obtained from the summary function. Only autosomal pseudomarkers were tested. To avoid obtaining a significant P-value due to a non-normal trait, we tested only pseudomarkers representing haplotype clusters containing haplotypes from more than eight individuals.
For certain traits, we transformed the trait before performing regression. For CRP, BMI, TG, Insulin, and Glucose, the transformed trait was log10(trait). For CRP, values of 0 were changed to one-half of the detection limit (0.002 mg/l) before log transformation. Traits were winsorized so that values >4 SD above the mean were set to 4 SD above the mean, and values <4 SD below the mean were set to 4 SD below the mean. Regression was not performed on haplotype clusters that had only one haplotype belonging to the cluster.
We excluded 27 individuals taking diabetes medication and 199 pregnant women from the regression analyses for all nine traits. For BMI, we included only individuals with direct weight measurements, excluding 170 individuals whose weights were measured by mail inquiry. For TG, insulin, glucose, HDL, and LDL, we excluded 228 individuals not fasting at the time of measurement. Individuals with missing values for any variable in a regression model were excluded from the corresponding regression analysis (Supplementary Tables S1 and S2). After exclusions, the following numbers of individuals were included in the traits' regression analyses: 4418 people for TG, 4472 for HDL, 4406 for LDL, 4590 for CRP, 4409 for glucose, 4387 for insulin, 4530 for BMI, 4579 for SBP, and 4573 for DBP.
Significance
A haplotype cluster was considered significant if it contained haplotypes from more than eight individuals and had a P-value less than the Bonferroni-corrected threshold of 6.92 × 10−10, which accounts for the 72 262 826 total number of pseudomarkers tested in this study, across the genome and across traits. This Bonferroni correction based on the number of tested pseudomarkers is conservative due to LD and due to multiple pseudomarkers representing the same haplotype cluster. Clusters with eight or fewer individuals having haplotypes belonging to the cluster were not considered statistically significant because the significance of small clusters could be driven by non-normality of the trait.
To gauge the novelty of the HDL and LDL signals, we identified previous GWAS results for HDL and LDL. From the catalog of published genome-wide association studies (see Online Resources),14 we identified the HDL and LDL GWAS meta-analyses with the greatest number of individuals.15 These meta-analyses involved individuals of European descent, including NFBC individuals, with 99 900 individuals in the HDL analysis and 95 454 individuals in the LDL analysis.
To find genes near an association and order them by distance from the association, we identified gene transcripts in a trait-associated region using the UCSC Table Browser. We defined an association's position as the mean position of SNPs that correspond to pseudomarkers with smallest P-value in a trait-associated region. We defined each gene's distance from an association as the minimum distance from the association to a base in any of the gene's transcripts. cM positions were calculated from hg18 bp positions using linear interpolation and the HapMap genetic map.16 The LiftOver program with chain file hg18ToHg19.over.chain.gz was used to convert hg18 positions to hg19 positions to calculate a gene's distance from the association in hg19 base pairs (see Online Resources).
Defining a haplotype cluster
We used the cluster2haps.jar program (see Online Resources) to identify sequences of alleles that define the most significantly associated haplotype cluster. We identified a set of allele sequences that are found only on the haplotypes in the most significantly associated haplotype cluster.
Comparison of single-SNP tests and haplotype cluster tests
A single-SNP analysis previously performed on these data did not find two of the regions that haplotype cluster tests found to be associated with LDL.9 The main regression model in the previous SNP analysis differed slightly from the regression model in the haplotype cluster analysis. Different variables represented population structure, and a factor variable defined by combinations of Sex and Pills values was used instead of separate variables for Sex and Pills. Also, we recoded missing values for males in the original data to define Pills and Pregnant variables (Supplementary Table S2). To check that the absence of signal in the previous analysis was not due to differences in regression models, but due to usage of a SNP test instead of a haplotype cluster test, we performed a single-SNP analysis (Figure 1). For each genotyped SNP in each region found to be associated with LDL by haplotype cluster tests, we performed single-SNP tests using the model
Figure 1.

−log10(P-value) vs bp position plots for clusters significant at the Bonferroni threshold of 6.92 × 10−10. LDL-associated regions are chr1:g.53.73-57.23 Mb (upper left), chr4:g.71.50–76.48 Mb (upper right), and chr19:g.44.56-45.81 Mb (lower left). The HDL-associated region is chr16:g.56.86–57.03 Mb (lower right). Bp positions are from hg19. Hollow black circles represent results of haplotype cluster tests. Blue crosses represent results of our single SNP tests. The red, horizontal line indicates a –log10(P-value) corresponding to a Bonferroni threshold of 6.92 × 10−10.
 |
with E(ɛ)=0 and Var(ɛ)=σ2. This model is the same as the haplotype cluster model (Equation 1), except that the SNP variable (0, 1, or 2 for the number of minor alleles) has replaced the Cluster variable.
Results
Across all traits, multiple haplotype clusters containing haplotypes belonging to more than eight individuals were significantly associated with LDL or HDL at the conservative Bonferroni-corrected threshold of 6.92 × 10−10. HDL was associated with a region on chromosome 16. LDL was associated with regions on chromosome 1, chromosome 4, and chromosome 19 (Table 1, Figure 1). The smallest haplotype cluster P-value in each region was 1.7 × 10−15, 1.2 × 10−14, 6.5 × 10−14, and 8.7 × 10−12 respectively. The most significant cluster in each region had, respectively, 2438, 174, 168, and 174 individuals with haplotypes in the cluster.
Table 1. Genomic regions that haplotype cluster tests associate with traits.
| Trait | Genomic region | Build | Cytogenetic band | Smallest haplotype cluster P-value |
|---|---|---|---|---|
| HDL | chr16:g.55420000-55590000 | hg18 | 16q13 | 1.69 × 10−15 |
| chr16:g.56862499-57032499 | hg19 | |||
| LDL | chr1:g.53500000-57000000 | hg18 | 1p32.3-p32.2 | 1.21 × 10−14 |
| chr1:g.53727412-57227412 | hg19 | |||
| LDL | chr4:g.71640000–76700000 | hg18 | 4q13.3-q21.1 | 6.46 × 10−14 |
| chr4:g.71495001-76480976 | hg19 | |||
| LDL | chr19:g.49250000-50500000 | hg18 | 19q13.31-q13.32 | 8.66 × 10−12 |
| chr19:g.44558160-45808160 | hg19 |
Abbreviations: HDL, high-density lipoprotein; LDL, low-density lipoprotein. For each association, we give the region in which the values of –log10(P-value) are elevated above background level. These are the regions shown in Figure 1. The cytogenetic band containing a region and the P-value of the most significant haplotype cluster in the region are given.
No region on chromosome 4 has previously been reported in LDL GWAS (see Online Resources).14 In the chromosome 4 region, there were 80 markers that had a haplotype cluster with P-value<6.92 × 10−10 and 139 or more individuals with haplotypes in the cluster. The data are consistent with the chromosome 4 association being due to a single causal variant (Supplementary Material). The smallest P-value of 6.4 × 10−14 corresponded to a haplotype cluster found at four consecutive markers. For this haplotype cluster, the estimated effect size of having one haplotype in the cluster was 0.488 mmol/l (18.8 mg/dl) with a 95% CI of 0.361–0.615 mmol/l (13.9–23.8 mg/dl). The haplotype cluster was characterized by five allele sequences at 88 markers. Each pair of allele sequences from the five allele sequences differed by exactly one allele. Of 186 haplotypes in the cluster, 182 haplotypes carried the same sequence while the four remaining haplotypes carried the remaining four sequences (Supplementary Table 3). The most common allele sequence carried by the haplotypes in the cluster was associated with 0.502 mmol/l higher LDL (95% CI: 0.370–0.633 mmol/l, P-value: 2.50 × 10−14) in a regression model that followed Equation 1, except with Cluster replaced by the number (0, 1, or 2) of an individual's haplotypes carrying the allele sequence. This allele sequence had a sample frequency of 182/(4899 × 2)=1.9%.
Using a leave-one-out analysis, we investigated whether the trait-cluster associations of Table 1 could be artifacts of genotype error. For each SNP in a trait-associated region, we removed the SNP from the data, repeated the analysis from phasing through haplotype cluster testing, and recorded the minimum haplotype cluster test P-value in the repeated analysis. We analyzed SNPs in an extended region beginning 2 Mb upstream and ending 2 Mb downstream of the trait-associated region. We used the same eigenvectors as in the original analysis. All other aspects of a repeated analysis were the same as in the original analysis. The largest (across left-out SNPs) minimum (across haplotype cluster tests for a left-out SNP) P-value was, respectively, 1.6 × 10−9, 2.6 × 10−11, 1.5 × 10−12, and 2.6 × 10−9 for the regions on chromosomes 16, 1, 4 and 19 (respective minimum P-values in the original analysis: 1.7 × 10−15, 1.2 × 10−14, 6.5 × 10−14, and 8.7 × 10−12). The P-values for the chromosome 1 and chromosome 4 regions are significant at the Bonferroni threshold of 6.92 × 10−10, which suggests that genotype error did not drive the chromosome 1 and 4 associations in the original analysis. When removed from the data, three left-out SNPs on chromosomes 16 and 19 resulted in minimum haplotype cluster test P-values above the Bonferroni threshold of 6.92 × 10−10. We examined the allele intensity plots for these three SNPs and did not observe poor clustering by genotype that would indicate genotype error (Supplementary Figure 1).
We investigated whether the trait-cluster associations of Table 1 could be artifacts of residual population structure. For each trait, we created a QQ plot of P-values on the –log10 scale. Since we would expect clusters that are not associated with a trait to have P-values distributed Uniform (0,1), we compared the trait's P-values to a Uniform (0,1) distribution, by plotting observed –log10(p(i)) versus expected –log10 (p(i)), where p(i) denotes the ith largest of n considered P-values. Expected –log10 (p(i)) was calculated as –log10 (i/n). We calculated the genomic inflaction factor as the median of the observed negative log10(p(i)) values divided by the median of the expected negative log10(p(i)) values.
Genomic inflation factors for all traits were close to 1, with values of 0.9953 (BMI), 0.9936 (DBP), 0.9857 (HDL), 0.9905 (LDL), 0.9897 (TG), 0.9993 (CRP), 0.9938 (glucose), 0.9976 (insulin), and 0.9950 (SBP). This is consistent with little confounding by population structure. QQ plots for all nine traits are given in Supplementary Figure 3. QQ plots for LDL including and excluding P-values of pseudomarkers in the three LDL-associated regions in Table 1 are given in Supplementary Figure 4, and QQ plots for HDL including and excluding P-values of pseudomarkers in the HDL-associated region in Table 1 are given in Supplementary Figure 5.
To further examine whether the association between LDL and the most significant haplotype cluster on chromosome 4q13.3-q21.1 could be an artifact of residual population structure, for this most significant haplotype cluster, we plotted the first two eigenvectors against each other, labeling haplotype cluster membership (Supplementary Figure 2). Points corresponding to individuals belonging to the haplotype cluster appear to be spread around the same area as points corresponding to individuals not in the haplotype cluster, consistent with population structure not confounding the LDL-haplotype cluster association.
We identified 39 genes in the LDL-associated region on chromosome 4q13.3-q21.1 and calculated their distances away from the chromosome 4 association (Table 2). Experimental evidence suggests a direct, mechanistic connection between some of the genes in this region and LDL. For instance, experimental evidence suggests that serum albumin, the protein product of ALB, enhances cholesterol efflux from cells to LDL.17 Experimental evidence also suggests that Platelet Factor 4, the protein product of PF4, binds cell surface LDL receptors, preventing serum LDL from entering cells.18
Table 2. Genes near the chromosome 4 association with LDL.
| Gene name | OMIM ID | hg19 Distance from association (kb) | cM Distance from association |
|---|---|---|---|
| RASSF6 | 612620 | 24 | 0.004 |
| AFM | 104145 | 140 | 0.023 |
| AFP | 104150 | 189 | 0.033 |
| ALB | 103600 | 223 | 0.044 |
| ANKRD17 | Not in OMIM | 386 | 0.061 |
| COX18 | 610428 | 575 | 0.070 |
| IL8 | 146930 | 96 | 0.140 |
| CXCL6 | 138965 | 192 | 0.162 |
| PF4V1 | 173461 | 209 | 0.170 |
| CXCL1 | 155730 | 225 | 0.240 |
| PF4 | 173460 | 337 | 0.296 |
| PPBP | 121010 | 343 | 0.305 |
| CXCL5 | 600324 | 351 | 0.315 |
| CXCL3 | 139111 | 392 | 0.371 |
| PPBPL2 | 611591 | 410 | 0.377 |
| ADAMTS3 | 605011 | 1076 | 0.378 |
| CXCL2 | 139110 | 453 | 0.389 |
| MTHFD2L | 614047 | 470 | 0.400 |
| LOC541467 | Not in OMIM | 512 | 0.435 |
| BC016361 | Not in OMIM | 634 | 0.508 |
| EPGN | Not in OMIM | 664 | 0.534 |
| EREG | 602061 | 721 | 0.642 |
| NPFFR2 | 607449 | 1491 | 0.730 |
| GC | 139200 | 1840 | 0.847 |
| SLC4A4 | 603345 | 2072 | 1.068 |
| BTC | 600345 | 1161 | 1.162 |
| PARM1 | Not in OMIM | 1348 | 1.503 |
| AK027257 | Not in OMIM | 1371 | 1.537 |
| DCK | 125450 | 2613 | 1.704 |
| MOBKL1A | 609282 | 2656 | 1.715 |
| GRSF1 | 604851 | 2804 | 1.736 |
| RUFY3 | 611194 | 2837 | 1.740 |
| UTP3 | 611614 | 2954 | 1.761 |
| IGJ | 147790 | 2978 | 1.816 |
| ENAM | 606585 | 2998 | 1.882 |
| RCHY1 | 607680 | 1894 | 1.988 |
| THAP6 | 612535 | 1930 | 1.991 |
| DKFZp313E1330 | Not in OMIM | 1930 | 1.991 |
| DKFZp686B2235 | Not in OMIM | 1930 | 1.991 |
The association's position and a gene's distance from the association are defined in Materials and Methods. Genes are listed in order of ascending cM distance from the association. cM was calculated from hg18 bp positions using linear interpolation and the HapMap genetic map.16 hg19 distances were obtained from hg18 bp positions using the liftOver program.
We also found regions on chromosomes 1 and 19 associated with LDL and a region on chromosome 16 associated with HDL. We calculated the distance between each association that we found and the closest significant SNP in a previous analysis and a previous meta-analysis of the phenotypes (Table 3). Although a previous single-SNP analysis of these data and a meta-analysis that included these data did not find LDL-associated SNPs on chromosome 4, these studies both found the LDL-associated region on chromosome 19 and the HDL-associated region on chromosome 16. In addition, the meta-analysis found the LDL-associated region on chromosome 1.9, 15 Plausible candidate genes lie within these regions that haplotype cluster tests associate with LDL or HDL. For LDL, the chromosome 1 region includes PCSK9 and the chromosome 19 region includes APOE. For HDL, the chromosome 16 region includes CETP.
Table 3. Prior GWAS results for LDL on chromosome 1 and chromosome 19, and HDL on chromosome 16.
| Trait | Source | SNP | Build | Position | Distance from association (kb) | Distance from association (cM) |
|---|---|---|---|---|---|---|
| LDL | Sabatti Table 2 | rs646776 | hg18 | chr1:g.109620 | 53446 | 55.3 |
| hg19 | chr1:g.119757 | 53417 | ||||
| LDL | Meta-Analysis Table 1 | rs2479409 | hg18 | chr1:g.55277 | −897 | −0.743 |
| hg19 | chr1:g.65414 | −897 | ||||
| LDL | Sabatti Table 2 | rs157580 | hg18 | chr19:g.50087 | −3 | −0.002 |
| hg19 | chr19:g.99087 | −4 | ||||
| LDL | Meta-Analysis Table 1 | rs4420638 | hg18 | chr19:g.50115 | 24 | 0.058 |
| hg19 | chr19:g.99115 | 24 | ||||
| HDL | Sabatti Table 2 | rs3764261 | hg18 | chr16:g.55551 | −12 | −0.025 |
| hg19 | chr16:g.115551 | −12 | ||||
| HDL | Meta-Analysis Table 1 | rs3764261 | hg18 | chr16:g.55551 | −12 | −0.025 |
| hg19 | chr16:g.115551 | −12 |
Abbreviations: HDL, high-density lipoprotein; LDL, low-density lipoprotein. For each associated chromosomal region and each of two previous GWAS studies,15, 19 we identified each study's genome-wide significant SNP that was closest to the association detected using haplotype cluster tests. An association's position is defined in Materials and Methods. cM was calculated from hg18 bp using linear interpolation and the HapMap genetic map.16 hg19 bp was calculated from hg18 bp using the liftOver program.
We hypothesized that the chromosome 1 and chromosome 4 regions were not identified in a previous single-SNP analysis of these data, and the chromosome 4 region was not identified in the LDL meta-analysis,9, 15 because haplotype cluster tests have higher power to detect these associations than single-SNP tests. For the chromosome 1 and chromosome 4 regions associated with LDL by haplotype cluster tests, we also performed single-SNP tests and compared the P-values with those of the haplotype cluster tests (Figure 1). In the chromosome 1 region, the smallest P-value for the single-SNP tests was 1.45 × 10−6, whereas the smallest P-value for the haplotype cluster tests was 1.21 × 10−14. In the chromosome 4 region, the smallest P-value for the single-SNP tests was 2.41 × 10−5, whereas the smallest P-value for the haplotype cluster tests was 1.21 × 10−14. Single-SNP tests do not identify the chromosome 1 and chromosome 4 regions as statistically significant at the commonly used 5 × 10−8 significance level.
Discussion
We analyzed nine metabolic traits in the North Finland Birth Cohort for association with haplotype clusters defined using Beagle. We found a novel association with LDL on chromosome 4q13.3-q21.1 containing the ALB and PF4 genes. The association is significant (P-value 1.21 × 10−14), even after using a conservative Bonferroni adjustment for the number of tests performed across the genome and across traits.
We performed checks to be confident that the novel chromosome 4 association is real. Although trait-correlated genotype errors are unlikely for these data, we performed a leave-one-out analysis, leaving out each SNP in the chromosome 4 region and recording the minimum P-value. The largest minimum P-value was still Bonferroni significant. A further possibility is that population structure is inducing a spurious association. Population structure induces spurious associations if mean trait values differ between different genetic subpopulations. A standard and effective approach for reducing the impact of population structure is to adjust for principal component eigenvectors, as we did in this study. The genomic inflation factor for each analyzed trait was close to 1, which is consistent with eigenvectors effectively adjusting for population structure. Departures from model assumptions can also sometimes cause spurious results. In this case, we assume approximate normality of the trait values. Outlier trait values were winsorized (adjusted toward the mean), reducing the risk that non-normality would induce spurious associations. Because the number of individuals with haplotypes in the chromosome 4 cluster is relatively large at 168, the central limit theorem ensures that the distribution of adjusted mean trait values will be approximately normal even if there is a fair degree of non-normality in the trait values.
We also found haplotype cluster associations with LDL on chromosome 1 and chromosome 19 and with HDL on chromosome 16. The leave-one-out analysis did not suggest that genotype error drove these associations, and QQ plots and genomic inflation factors did not indicate that residual population structure drove these associations. Two of these associations were also detected in the earlier single-SNP association testing of these data.9 The remaining association, of a chromosome 1 region with LDL, was not found in single-SNP analysis of these data, but has been found in a meta-analysis that included these data.9, 15 The two associations that were found using haplotype cluster analysis but not with single-SNP analysis in these data (the chromosome 4 and chromosome 1 regions with LDL) demonstrate the utility of haplotype cluster analysis as a complement to single-SNP analysis, particularly in data such as these where there does not yet exist a large, publicly-available, well-matched reference panel for imputation. In contrast, single-SNP association testing identified many associations that were not found by haplotype cluster testing. Across all nine traits, the previous single SNP analysis of these data identified 29 additional associated regions that are not close to the four regions identified by haplotype cluster tests.9 Hence, haplotype cluster testing should not be viewed as a replacement for single-SNP association testing.
Identification of the chromosome 1, chromosome 19, and chromosome 16 associations confirms that haplotype cluster tests can identify real associations, further lending credence to the novel chromosome 4 signal. Several genes, namely ALB and PF4, in the associated chromosome 4 region are good candidates for influencing LDL levels. Sequencing the chromosome 4 region of individuals belonging to the most significant, LDL-associated haplotype cluster on chromosome 4 could reveal nonsense or missense mutations in candidate genes that will identify the causal variants.
Acknowledgments
The work in the study was supported by research grants HG004960, HG005701, GM099568, GM075091, T32 GM007266, and T32 ES015459 from the National Institutes of Health, USA. The NFBC1966 Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with the Broad Institute, UCLA, University of Oulu, and the National Institute for Health and Welfare in Finland. This manuscript was not prepared in collaboration with investigators of the NFBC1966 Study and does not necessarily reflect the opinions or views of the NFBC1966 Study Investigators, Broad Institute, UCLA, University of Oulu, National Institute for Health and Welfare in Finland and the NHLBI.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Kohli P, Giugliano RP. Low-density lipoprotein lowering in 2013 by nonstatin agents: the discovery and development of promising novel targeted therapies. J Cardiovasc Pharmacol Ther. 2013;18:199–210. doi: 10.1177/1074248413480270. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Boerwinkle E, Mosley TH, Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- Browning BL, Browning SR. Efficient multilocus association testing for whole genome association studies using localized haplotype clustering. Genet Epidemiol. 2007;31:365–375. doi: 10.1002/gepi.20216. [DOI] [PubMed] [Google Scholar]
- Li L, Li Y, Browning SR, et al. Performance of genotype imputation for rare variants identified in exons and flanking regions of genes. PLoS One. 2011;6:e24945. doi: 10.1371/journal.pone.0024945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Herold C. Joint analysis of tightly linked SNPs in screening step of genome-wide association studies leads to increased power. Eur J Hum Genet. 2009;17:1043–1049. doi: 10.1038/ejhg.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregouet DA, Konig IR, Erdmann J, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- Browning SR. Multilocus association mapping using variable-length Markov chains. Am J Hum Genet. 2006;78:903–913. doi: 10.1086/503876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. Haplotypic analysis of Wellcome Trust Case Control Consortium data. Hum Genet. 2008;123:273–280. doi: 10.1007/s00439-008-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatti C, Service SK, Hartikainen AL, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet. 2009;41:35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. Identity-by-descent-based heritability analysis in the Northern Finland Birth Cohort. Hum Genet. 2013;132:129–138. doi: 10.1007/s00439-012-1230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. Frazer KA, Ballinger DG, et al. The International HapMap Consortium: A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, de la Llera-Moya M, Drazul-Schrader D, Phillips MC, Kellner-Weibel G, Rothblat GH. Serum albumin acts as a shuttle to enhance cholesterol efflux from cells. J Lipid Res. 2013;54:671–676. doi: 10.1194/jlr.M031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachais BS, Kuo A, Nassar T, et al. Platelet factor 4 binds to low-density lipoprotein receptors and disrupts the endocytic itinerary, resulting in retention of low-density lipoprotein on the cell surface. Blood. 2002;99:3613–3622. doi: 10.1182/blood.v99.10.3613. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.