

Abstract
Arsenic trioxide (As2O3) has been shown to induce apoptosis in hepatocellular carcinoma cells. However, the molecular mechanism of As2O3-induced apoptosis in the hepatocellular carcinoma cells remains poorly understood. Here, we investigated the impact of As2O3 exposure on the human hepatocellular carcinoma cell line HepG2 and examined the underlying mechanism of cell death. As2O3 induced apoptosis of HepG2 cells in a dose- and time-dependent manner and caused a massive production of reactive oxygen species (ROS). The antioxidant N-acetylcysteine (NAC) was able to prevent As2O3-induced cell death, implying an involvement of ROS in the induction of As2O3-triggered apoptosis. Furthermore, As2O3 initiated apoptosis by triggering of the mitochondria apoptotic pathway as indicated by inhibited Bcl-2 expression, a collapse of the mitochondrial membrane potential (MMP), release of cytochrome c and activation of the caspase cascade. However, these As2O3-induced events can be prevented by NAC. Taken together, these findings suggest that the As2O3 induced apoptosis through a ROS-mediated mitochondrial pathway and activation of caspases.
Keywords: Arsenic trioxide, hepatocellular carcinoma, reactive oxygen species, mitochondrial pathway, caspase
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer in the world. It is caused by a malignant transformation of hepatocytes, the major cell type in the liver, and accounts for 80% to 90% of primary liver cancer. The annual incidence of HCC is more than 1,000,000 newly diagnosed cases, and less than 5% of patients survive beyond five years. Due to its high mortality, HCC is a major health problem worldwide [1,2]. HCC treatment mainly consists of surgical resection, liver transplantation, and local ablative therapy. Systemic therapies, however, have not shown any survival benefit [2-4]. Moreover, prolonged treatment with chemotherapy drugs can lead to multidrug resistant tumor cells and these drugs have severe adverse effects [5,6]. Therefore, novel and effective therapeutics with lower toxicity are urgently needed for patients with HCC.
The medicinal effect of As2O3 is of great significance, though, it is also well known for its toxicity. It has been used to treat various diseases in both ancient China and Western societies [7-9]. As2O3 treatment of Chinese patients with acute promyelocytic leukemia (APL) has shown some success [10,11]. Subsequently, the effect of As2O3 on a variety of solid tumor cells such as hepatocellular carcinoma was investigated in vitro and vivo. Interestingly, the work of several labs suggested an important role for As2O3-induced apoptosis in the treatment of HCC cells [5,9,12]. However, the exact mechanism of action of As2O3-induced apoptosis in HCC cells remains unclear.
Here, we investigated the molecular mechanism of As2O3-induced apoptosis in HepG2 cells and examined mitochondrial apoptosis signaling pathways and ROS production in As2O3-induced cell death. The results suggest that As2O3 induced apoptotic cells death in HepG2 cells occurs via a ROS-mediated mitochondrial pathway and activation of caspases.
Materials and methods
Reagents and cell cultures
As2O3 was obtained from Sigma-Aldrich Chemical company, 2’,7’-Dichlorofluorescein diacetate (DCFH-DA), N-acetylcysteine (NAC), Annexin V-FITC Apoptosis Detection Kit, Hoechst 33258, Rhodamine-123 and 3-(4,5-dimethylthiazol-2-y-1)-2,5-diphenyltetrazolium bromide (MTT) were obtained from the Beyotime Institute of Biotechnology (Shanghai, China). Rabbit polyclonal antibodies against Bcl-2, Bax, cytochrome c, caspase-3, caspase-6, caspase-9, β-actin, were obtained from Abcam (Abcam, Cambridge, MA, USA).Horseradish peroxidase-conjugated secondary antibodies was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
As2O3 was prepared as a 10 mM stock solution in phosphate buffered and saline and stored at 4°C. The human hepatocellular carcinoma cell line HepG2 was obtained from the Chinese Academy of Sciences (Shanghai, China). All cells were grown in Dulbecco’s Modified Eagle Medium (DMEM; Gibco BRL, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco BRL, Grand Island, NY, USA) which was supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen, Carlsbad, CA, USA), in a humidified atmosphere of 5% CO2 at 37°C.
Cell viability assays
Cell viability was measured by MTT assay as previously described [13]. Briefly, 1×104 cells/well were plated in 96-well microplates and cultured for 24 h. The cells were subsequently treated with 2, 5, 15 μM As2O3 for 24, 48, and 72 h with or without pretreatment with 10 mM N-acetylcysteine (NAC) for 1 h, respectively. Cells were incubated with 20 μL MTT (5 mg/ml) for 4 h. Formazan crystals were dissolved in 150 μL DMSO, and the absorbance at 570 nm was measured in a microplate reader (Bio-Rad, USA). Each test was performed in triplicate.
Apoptosis assay
The percentage of apoptotic cells was measured by flow cytometry using an annexin V-FITC/PI kit (Beyotime) according to manufacturer’s instructions. Briefly, cells were treated for 24 h with the As2O3 concentrations mentioned above with or without 10 mM NAC pretreatment. The cells (1×106) were washed twice with ice-cold PBS and collected. Apoptosis rates were measured by double staining with annexin V-FITC and propidium iodide (PI) in binding buffer using a FACSCalibur flow cytometer (Becton Dickinson, CA, USA).
The nuclear morphology of HepG2 cells was analyzed by staining with Hoechst 33258 according to manufacturer’s instructions. Briefly, the As2O3-treated cells were washed with PBS, fixed with 4% formaldehyde for 30 min, then washed with PBS three times, and incubated with Hoechst 33258 (3 μg/ml) for 30 min at room temperature in the dark. Then they were washed three times with PBS and the nuclear morphology was examined using a fluorescence microscope (Olympus, Japan).
ROS production determinates
The level of intracellular ROS was quantified using a Reactive Oxygen Species Assay Kit. DCFH-DA, a fluorescent probe, is oxidized by reactive oxygen species in viable cells to 2’,7-dichlorofluorescein (DCF). Using the designated treatment, the same densities of cells were harvested by cell counting. The cells were then incubated with 100 μM DCFH-DA (dissolved in DMSO) for 30 min at 37°C. After washing three times with PBS, the relative levels of fluorescence were quantified by a multi-detection microplate reader (485 nm excitation and 535 nm emission).
Analysis of mitochondrial membrane potential (Δψm)
The mitochondrial membrane potential was measured by the incorporation of the cationic fluorescent dye rhodamine 123, which preferentially partitions into active mitochondria based on the highly negative mitochondrial membrane potential. Depolarization of mitochondrial membrane potential results in the loss of rhodamine 123 from the mitochondria and a decrease in intracellular fluorescence. After incubation in the normal medium with or without treatment, the cells were transferred to a serum-free medium containing 10 μM rhodamine 123 and incubated for 30 min at 37°C. The cells were then collected and the fluorescence intensity was analyzed by flow cytometry.
Western blot analysis
As2O3-treated HepG2 cells were harvested, washed twice with PBS, and lysed in 200 μl lysis buffer for 5 min on the ice. The cell lysates were centrifuged at 12,000×g for 5 min at 4°C. Protein concentration in the supernatant were measured using a Bradford Protein Assay. Samples with equal amounts of protein were separated through a 12% SDS-PAGE. After electrophoresis, the proteins were transferred onto a nitrocellulose membrane (Amersham Pharmacia Biotech, UK) using a semi-dry transfer system (Bio-Rad). The membrane was blocked with 5% not-fat milk in TBST f or 2 h, incubated with primary antibodies for 2 h at room temperature (diluted 1:2000 in TBST), washed and incubated with secondary antibodies (diluted 1:5000) in TBST at room temperature for 1 h, respectively. Lastly, the proteins were detected using enhanced chemiluminescence (ECL) detection reagents (Amersham Pharmacia).
Statistical analysis
All experiments were performed at least in triplicate, and data were expressed as mean ± S.E.M. Statistical comparisons were performed using one-way analysis of variance (ANOVA) and Student’s t-test. P<0.05 was considered statistically significant.
Results
Effect of As2O3 on HepG2 cell viability
Cell counts were performed manually by workers blinded to the treatment schedule as described. To identify the therapeutic potential of As2O3, HepG2 cells were cultured with a concentration of 2, 5, 15 μM As2O3 for 24, 48, and 72 h, respectively. As2O3 inhibited the viability of HepG2 cells. Cell viability significantly decrease when the with concentration was 2, 5, or 15 μM of As2O3 while being treated for 24, 48, or 72 h respectively, and the decrease could be prevented by NAC at 24, 48, and 72 h post-treatment (Figure 1).
Figure 1.
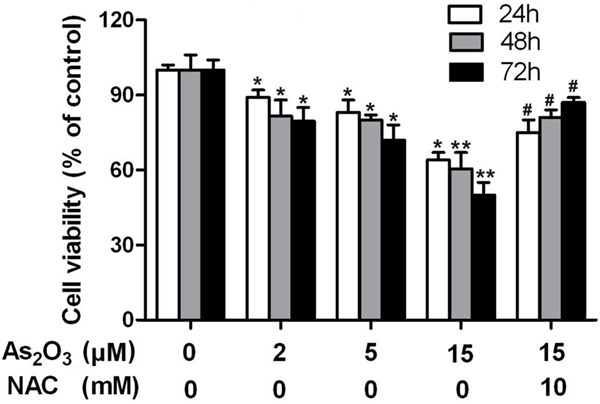
Effect of As2O3 on HepG2 cell viability. Cell viability were significant decrease with concentration of 2, 5, 15 μM As2O3 treated for 24 h, 48 h, 72 h, and the decrease can be prevented by NAC at 24 h, 48 h, 72 h post-treatment, respectively. Data represent means ± SE of three independent experiments *P<0.05 and **P<0.01 compared with control; #P<0.05 compared with 15 μM As2O3 alone.
As2O3-induced ROS generation in HepG2 cells
To further investigate the role of As2O3 in ROS production, we measured production of intracellular ROS in As2O3-treated HepG2 cells by flow cytometric using DCFH-DA. Incubation of HepG2 cell with 2 μM As2O3 induced a significant increase (124.7%) in intracellular ROS content. At As2O3-concentration lead to a ROS burst (282.5% compared to untreated cells, P<0.01), however, the increase was completely inhibited by ROS scavenger NAC (102.3%) (Supplementary Figure 1).
As2O3-induced apoptosis in HepG2 cells
To evaluate whether As2O3 induced apoptosis is related to ROS production in HepG2 cells, annexin V-FITC/PI double staining was performed. After treatment of HepG2 cells with As2O3 at concentration of 2, 5, and 15 μM, respectively, for 24 h with or without pretreatment with 10 mM NAC for 1 h, the percentage of apoptotic cells increased in an arsenic-dose-dependent manner (Figure 2B-D). However, this increase was significantly decreased by pretreatment with the antioxidant NAC (Figure 2E). Characteristic apoptotic morphological changes such as condensation of chromatin and nuclear fragmentation were found in arsenic-treated cells by Hoechst33258 staining (Figure 3B-D). A decreased amount of apoptotic cells were seen by Hoechst33258 staining after pretreatment with NAC (Figure 3E).
Figure 2.
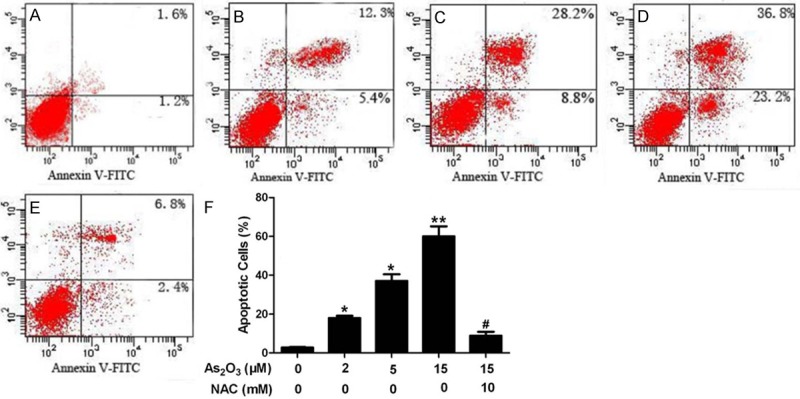
Cell apoptosis and necrosis analyzed by flow cytometry. HepG2 cells were incubated in drug-free medium (A) or medium containing 2 μM As2O3 (B), 5 μM As2O3 (C) and 15 μM As2O3 (D) for 24 h, or cells were pretreated with 10 mM (E) NAC for 1 h and then incubated in 15 μM As2O3 for an additional 24 h. The results show in (F) are the mean and SE for three independent experiments.
Figure 3.

Fluorescence images show the nucleic changes of HepG2 cells incubated in As2O3 with or without NAC. Cells were stained with the DNA-binding fluorochrome Hoechst 33258. (A) shows normal culture medium nucleic morphology, (B-D) respectively show cells cultured in 2 μM, 5 μM and 15 μM As2O3 for 24 h. In addition, cells were pretreated with 10 mM NAC (E) for 1 h and then incubated in As2O3 (15 μM) for an additional 24 h. (F) Histograms showing ratio of condensed nuclei to total nuclei. White arrows represent location of apoptosis cell. Scale bars represent 50 μm. *P<0.05 and **P<0.01 compared with control; #P<0.05 compared with 15 μM As2O3 alone.
As2O3 induces apoptosis via a ROS-mediated mitochondrial pathway
To demonstrate whether As2O3-treatment leads to a collapse of the mitochondrial membrane potential (Δψm) in HepG2 cells, the change of Δψm was evaluated by flow cytometry using rhodamin-123, which can accumulate rapidly and selectively within the mitochondria depending on mitochondrial membrane potential. As2O3 induced a disruption of the Δψm as evidenced by a decline in the proportion of cells with high fluorescence intensity (Supplementary Figure 2). The mean rhodamin-123 fluorescence intensity of HepG2 cells decreased from about 114.8 (control) to 66.2 and 43.4 after 24 h treatment with 5 μM and 15 μM As2O3, respectively (P<0.05). Moreover, the disruption of Δψm was partially prevented by pretreatment with NAC (mean fluorescence intensity 86.6, P<0.05).
Since cytochrome c release is associated with the loss of Δψm, we analyzed cytosolic cytochrome c (cyt c) levels by Western blot analysis. Cyt c gradually accumulated in the cytosol, and these changes were almost completely prevented by NAC pretreatment. Since the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax play an important role in the regulation of the mitochondrial pathway of apoptosis, the ratio of Bax/Bcl-2 protein levels were detected using Western blot analysis (Figure 4). As2O3 caused up-regulation of Bax/Bcl-2 in HepG2 cells. These changes could be inhibited by pretreatment with NAC.
Figure 4.
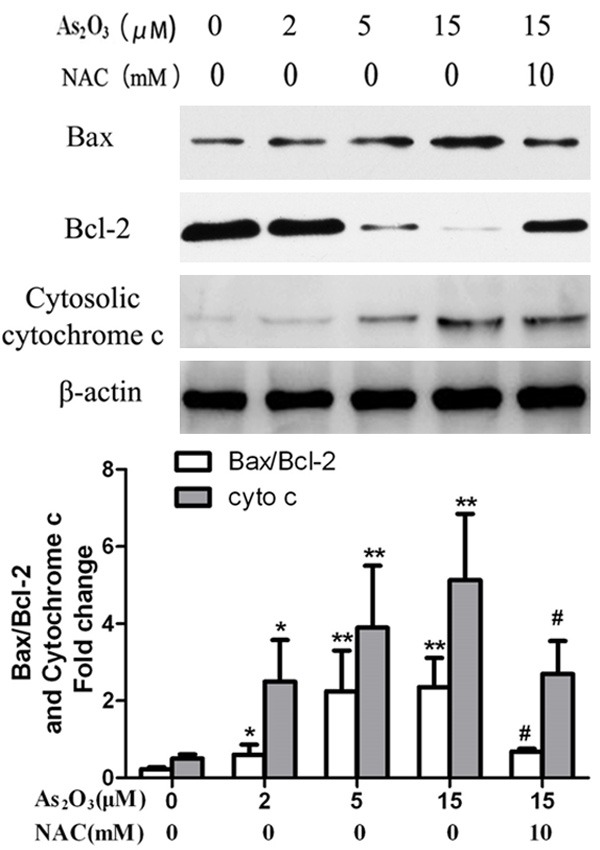
Cytochrome c release and Bax/Bcl-2 expression. Bax/Bcl-2 and Cytochrome c levels in cytosolic were determined by Western blotting analysis. The quantitative analysis of Bax/Bcl-2 and Cytochrome c protein levels. β-actin used as internal control. Data represent as means ± SE of three independent experiments. *P<0.05 and **P<0.01 compared with control; #P<0.05 compared with 15 μM As2O3 alone.
Role of caspases in As2O3-induced apoptosis in HepG2 cells
The caspase cascade plays a key role in apoptosis induction. Therefore, we determined the levels of activated caspase-9, caspase-6, and caspase-3 in As2O3-treated HepG2 cells by Western blot analysis. The levels of activated caspase-3, caspase-6 and caspase-9 increased in a dose-depended manner (Supplementary Figure 3).
Discussion
In the present study, we found that As2O3 induced apoptosis of HepG2 cells in a dose- and time- dependent manner and caused a massive production of ROS. As2O3 induced apoptosis in HepG2 cells by inhibiting Bcl-2 expression, a collapse of MMP, release of cytochrome c, and activation of the caspase cascade. These findings strongly suggest that the As2O3 induced apoptosis is mediated, at least in part, by controlling the ROS-mediated mitochondrial pathway and activation of caspases.
Compelling evidence suggests that most cancer cells are under oxidative stress associated with increased metabolic activity and production of ROS [14-17]. These biochemical characteristics make the cancer cells vulnerable to damage by additional oxidative stress, either through inhibiting ROS elimination, increasing their intracellular production or by adding exogenous ROS [18,19]. The cell-damaging properties of ROS and the increased ROS production in cancer cells may be a potential therapeutic target, if it is possible to exploit the cell killing potential of ROS by using exogenous agents to increase intracellular ROS to a toxic level, or above the threshold that triggers cell death [20]. In the present study, a dramatic ROS burst was observed in As2O3-treated cells, and As2O3-induced cells apoptosis was prevented by pretreatment of the cells with the antioxidant NAC, suggesting that ROS generation is involved in As2O3-induced apoptosis in HepG2 cells.
Mitochondria play a key role in the regulation of apoptosis [21,22]. Mitochondrial dysfunctions during apoptosis induction include the loss of mitochondrial membrane potential (Δψm), permeability transition, and release of cytochrome c from mitochondria into the cytosol [23-25]. Here, we showed that As2O3 induced loss of mitochondrial membrane potential and release of cytochrome c into the cytosol in a dose-dependent manner in HepG2 cells. In addition, As2O3 significantly inhibited the expression of Bcl-2 and up-regulated the expression of Bax. However, pretreatment of the cells with NAC prevented these changes, suggesting a ROS-mediated mitochondrial apoptotic pathway in the induction of cell death by As2O3 in HepG2 cells.
Caspases are intracellular cysteine proteases that are key components in classic apoptosis. Caspase activation occurs in response to various types of mitochondrial damage and pro-apoptotic stimuli, which cause cytochrome c to be released into the cytosol, where it binds and activates Apaf-1, which in turn activates procaspase-9, caspase-9 cleaves procaspase-3, and downstream of caspase-3, caspase-6, and eventually lead to cell apoptosis [26-29]. Here, we showed that As2O3 treatment caused activation of caspase-9, -3, -6, suggesting that As2O3 induced cell death through a caspase-dependent mechanism.
In conclusion, As2O3 induced apoptosis in HepG2 cells at least in part through a ROS-mediated mitochondrial pathway and activation of caspases. These results also provide important new insights into the possible molecular mechanism of As2O3 treatment for HCC.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Laing ST, Lemoy MJ, Sammak RL, Tarara RP. Metastatic hepatocellular carcinoma in a juvenile rhesus macaque (Macaca mulatta) Comp Med. 2013;63:448–53. [PMC free article] [PubMed] [Google Scholar]
- 2.Lau WY, Lai EC. Hepatocellular carcinoma: current management and recent advances. Hepatobiliary Pancreat Dis Int. 2008;7:237–57. [PubMed] [Google Scholar]
- 3.Liu Y, Jiang L, Mu Y. Somatostatin receptor subtypes 2 and 5 are associated with better survival in operable hepatitis B-related hepatocellular carcinoma following octreotide long-acting release treatment. Oncol Lett. 2013;6:821–828. doi: 10.3892/ol.2013.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Memon K, Kulik L, Lewandowski RJ, Gupta R, Ryu RK, Miller FH, Vouche M, Atassi R, Ganger D, Mulcahy MF, Salem R. Prospective evaluation of patients with early-/intermediate-stage hepatocellular carcinoma with disease progression following arterial locoregional therapy: candidacy for systemic treatment or clinical trials. J Vasc Interv Radiol. 2013;24:1189–97.e2. doi: 10.1016/j.jvir.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 5.Chan JY, Siu KP, Fung KP. Effect of arsenic trioxide on multidrug resistant hepatocellular carcinoma cells. Cancer Lett. 2006;236:250–8. doi: 10.1016/j.canlet.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 6.Park MJ, Kim YK, Park HJ, Hwang J, Lee WJ. Scirrhous hepatocellular carcinoma on gadoxetic Acid-enhanced magnetic resonance imaging and diffusion-weighted imaging: emphasis on the differentiation of intrahepatic cholangiocarcinoma. J Comput Assist Tomogr. 2013;37:872–81. doi: 10.1097/RCT.0b013e31829d44c1. [DOI] [PubMed] [Google Scholar]
- 7.Fu ZC, Zhang B, Fu ZJ. As2O3 may be a treatment option for adenoid cystic carcinoma of salivary gland. Med Hypotheses. 2010;75:490–1. doi: 10.1016/j.mehy.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Novick SC, Warrell RP Jr. Arsenicals in hematologic cancers. Semin Oncol. 2000;27:495–501. [PubMed] [Google Scholar]
- 9.Siu KP, Chan JY, Fung KP. Effect of arsenic trioxide on human hepatocellular carcinoma HepG2 cells: inhibition of proliferation and induction of apoptosis. Life Sci. 2002;71:275–85. doi: 10.1016/s0024-3205(02)01622-3. [DOI] [PubMed] [Google Scholar]
- 10.Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, Jin XL, Tang W, Li XS, Xong SM, Shen ZX, Sun GL, Ma J, Zhang P, Zhang TD, Gazin C, Naoe T, Chen SJ, Wang ZY, Chen Z. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996;88:1052–61. [PubMed] [Google Scholar]
- 11.Zhang Y, Zhang Z, Li J, Li L, Han X, Han L, Hu L, Wang S, Zhao Y, Li X, Zhang Y, Fan S, Lv C, Li Y, Su Y, Zhao H, Zhang X, Zhou J. Long-term efficacy and safety of arsenic trioxide for first-line treatment of elderly patients with newly diagnosed acute promyelocytic leukemia. Cancer. 2013;119:115–25. doi: 10.1002/cncr.27650. [DOI] [PubMed] [Google Scholar]
- 12.Alarifi S, Ali D, Alkahtani S, Siddiqui MA, Ali BA. Arsenic trioxide-mediated oxidative stress and genotoxicity in human hepatocellular carcinoma cells. Onco Targets Ther. 2013;6:75–84. doi: 10.2147/OTT.S38227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–7. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 14.Aune TM, Pogue SL. Inhibition of tumor cell growth by interferon-gamma is mediated by two distinct mechanisms dependent upon oxygen tension: induction of tryptophan degradation and depletion of intracellular nicotinamide adenine dinucleotide. J Clin Invest. 1989;84:863–75. doi: 10.1172/JCI114247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K, Habermehl GG. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem J. 1989;263:539–45. doi: 10.1042/bj2630539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaughnessy SG, Buchanan MR, Turple S, Richardson M, Orr FW. Walker carcinosarcoma cells damage endothelial cells by the generation of reactive oxygen species. Am J Pathol. 1989;134:787–96. [PMC free article] [PubMed] [Google Scholar]
- 17.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- 18.Fang J, Sawa T, Akaike T, Maeda H. Tumor-targeted delivery of polyethylene glycol-conjugated D-amino acid oxidase for antitumor therapy via enzymatic generation of hydrogen peroxide. Cancer Res. 2002;62:3138–43. [PubMed] [Google Scholar]
- 19.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–5. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- 20.Sen CK. Redox signaling and the emerging therapeutic potential of thiol antioxidants. Biochem Pharmacol. 1998;55:1747–58. doi: 10.1016/s0006-2952(97)00672-2. [DOI] [PubMed] [Google Scholar]
- 21.Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84:143–52. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- 22.Ma Y, Ogino T, Kawabata T, Li J, Eguchi K, Okada S. Cupric nitrilotriacetate-induced apoptosis in HL-60 cells association with lipid peroxidation, release of cytochrome C from mitochondria, and activation of caspase-3. Free Radic Biol Med. 1999;27:227–33. doi: 10.1016/s0891-5849(99)00083-0. [DOI] [PubMed] [Google Scholar]
- 23.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345 Pt 2:271–8. [PMC free article] [PubMed] [Google Scholar]
- 24.Atlante A, Calissano P, Bobba A, Azzariti A, Marra E, Passarella S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J Biol Chem. 2000;275:37159–66. doi: 10.1074/jbc.M002361200. [DOI] [PubMed] [Google Scholar]
- 25.Bossy-Wetzel E, Green DR. Assays for cytochrome c release from mitochondria during apoptosis. Methods Enzymol. 2000;322:235–42. doi: 10.1016/s0076-6879(00)22024-7. [DOI] [PubMed] [Google Scholar]
- 26.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denault JB, Boatright K. Apoptosis in Biochemistry and Structural Biology. 3-8 February 2004, Keystone, CO, USA. IDrugs. 2004;7:315–7. [PubMed] [Google Scholar]
- 28.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 29.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem. 1999;274:17941–5. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.