

Abstract
Hepatic cancer is a class of cancer that is relatively insensitive to chemotherapy, and cancers that harbor EGFR active mutations are more sensitive to EGFR-TK inhibitor such as gefitinib, which becomes the first-line treatment of this subtype of cancer. However, almost all patients treated with gefitinib will develop drug resistance. Here we show that a protein called integrin beta-8 (ITGB8) when over-expressed, is correlated with the gefitinib resistance of hepatic cancer cell line HepG2/G. After ITGB8 silencing, the drug resistance is reversed as the cell proliferation decreases and apoptosis rate increases significantly by gefitinib treatment when compared to HepG2/G. We demonstrated that multi-drug resistant proteins ABCB1, ABCC2 and ABCG2, anti-apoptosis proteins like survivin and Bcl-2, and cycle promoting protein CDK1 are involved in drug resistance of HepG2/G. Other drug-resistance relative proteins like SOD, GST, TS and HIF-1 are also modulated by ITGB8 silencing, but their role in this gefitinib resistance might be indirect. TGF beta pathway could be a critical pathway by which ITGB8 modulates the sensitivity of HepG2/G to gefitinib.
Keywords: Hepatic cancer, integrin beta-8, gefitinib resistance
Introduction
Hepatic cancer is the most common cancer in both men and women worldwide. It is responsible for 1.38 million deaths annually, as of 2008 [1]. Typically hepatic cancer are less sensitive to chemotherapy, so surgery is the best treatment for early/non-metastatic cases [2]. However, a significant number of patients with advanced hepatic cancer should be treated with chemotherapies. Chemotherapies like platinum drugs and taxol are widely used singly and more often in combined regime [3]. However, the 5 year overall survival rate for these patients are still quite limited [2]. Moreover, the side effects with the chemotherapies decrease the life qualities [3]. In the past decade, targeted therapeutics has a considerable progress in hepatic cancer. A typical example is gefitinib (Trade name IRESSA), which is an EGFR (Epithelial Growth Factor Receptor) tyrosine kinase inhibitor (TKI) and the first one to reach market in class. Gefitinib was first approved for hepatic cancer in Japan in 2002 and then by U.S. FDA under accelerated approval regulation in 2003. In June 2005, the U.S. FDA limited the use of gefitinib in new patients due to lack of evidence that it extended life [4]. Then a detailed investigation into the existed data found that Asian women patients who have never smoked and with pulmonary adenocarcinomas are more likely to better benefit from gefitinib [5]. Also, some pilot studies indicated that a special kind of mutations in EGFR instead of the expression level of EGFR was a predictor of gefitinib response [6,7]. The above mentioned sub-population has a relatively high incidence of such EGFR mutations. The IPASS study is the first phase 3 trials to confirmed gefitinib superiority in this patient population [8]. Nowadays, there are two EGFR-TKI named gefitinib and erlotinib in the market, and both of them have been proven to significantly benefit special sub-population of hepatic cancer [9]. Meanwhile, almost all patients responsive to EGFR-TKIs acquired resistance as treatment going on. The resistant mechanisms include secondary mutation of the EGFR gene, amplification of the MET gene, and overexpression of HGF and so on [10,11].
Integrins are transmembrane receptors that mediate the attachment of cells. They also play roles in signal transduction that involved in the regulation of cell cycle and motility [12,13]. Ju et al. showed that integrin beta 1 over-expression associated with resistance to gefitinib in hepatic cancer [14]. Here we showed that another integrin subtype beta-8 (ITGB8) is associated with gefitinib resistance in hepatic cancer cell line HepG2/G. Several multi-drug resistant proteins and cell cycle and apoptosis related proteins are involved in such modulation.
Materials and methods
Cell culture and establishment of gefitinib-resistant HepG2/G cell line
The human hepatic cancer cell line HepG2 was purchased from RIKEN Biosource Center (Japan) and maintained at 37°C in a humidified atmosphere with 5% CO2. The culture medium was RPMI-1640 supplemented with 10% fetal bovine serum (FBS). The gefitinib resistant cell line HepG2/G was established by continuing exposure of the parental cells to gradient increasing concentration of gefitinib.
Silencing of ITGB8 by shRNA
The lentiviral-mediated gene silencing of ITGB8 was employed. The ITGB8 shRNA lentiviral particles (Santa Cruz biotechnology) were transfected into the HepG2/G and the transfected cells could be selected out by puromycin resistance as indicated by the instruction. Control shRNA lentiviral particles were also purchased from Santa Cruz biotechnology.
MTS assay to determine cell proliferation and gefitinib sensitivity
0.1 ml of 4 × 104/ml cells were seeded into each well of the 96-well microplate, and incubated overnight to get the cells to adherence. For cell proliferation determination, every 24 h, CellTiter 96 Aqueous One Solution Reagent (Promega) was added to the respective wells and 4 hrs later the cell viability was determined by measuring the absorbance at 490nm using a microplate reader. For gefitinib sensitivity determination, different concentrations of gefitinib were added to each well and the cells were cultured for another 72 hrs. Then cell mass viability was determined as before.
Flow cytometry to analyze cell cycle, apoptosis, and intracellular accumulation of Rhodamine-123 and expression of p-glycoprotein
About 1 × 106 the cells were harvested and re-suspended in 0.1 mL culture medium. For cell cycle analysis, PI (BD Biosciences) was used to stain DNA. For apoptosis analysis, Annexin V-FITC/PI dual stain (BD Biosciences) was employed. Also, cells were stained with 10 μM rhodamine-123 (Sigma) for 1 hr and the intracellular concentration was determined by flow cytometry (BD, FACSCalibur). The expression of p-glycoprotein on the cells surface was determined by using a direct immunoflurescence staining kit purchased from BD Biosciences.
Real-time PCR analysis
About 2 × 106 of cells were harvested for RT-PCR analysis. The total mRNA was extracted from the cells by the Dynabeads mRNA Direct Kit (Life Science) according to the manual instruction. Total mRNA was then reverse transcribed for 1 hr at 42°C in incubation buffer containing 250 μM of each deoxynucleotide triphosphate, 5 μM oligo (dT) 20, 25 units of RNase inhibitor, and 20 units of avian myeloblastosis virus reverse transcriptase (Roche Diagnostics). The transcription level of ABCB1, ABCC2, ABCG2, survivin, Bcl-2 and so on were detected by semi-quantitative real-time PCR using the icycler iQ detection system (Bio-Rad). The PCR condition was as following: decontamination at 50°C for 2 min, denaturation at 95°C for 2 min, followed by n cycles at 95°C for 20 sec. and at hybridization T°C for 40 sec. The full details were shown in Table 1.
Table 1.
Oligonucleotide sequences used for real-time PCR
| Gene | Sequences (5’-3’) | Hybridization T°C | Cycles |
|---|---|---|---|
| ABCB1 | Forward: AAAAAGATCAACTCGTACCACTC | 65 | 37 |
| Reverse: GCACAAAATACACCAACAA | |||
| ABCC2 | Forward: CTGGGAACATGATTAGGAAGC | 65 | 42 |
| Reverse: GAGGATTTCCCAGAGCCGAC | |||
| ABCG2 | Forward: GGGTTCTCTTCTTCCTGACGACC | 65 | 38 |
| Reverse: CCAGACAACCAGTTAGAGTGTTGGT | |||
| Survivin | Forward: GCATGGGTGCCCCGACGTTG | 60 | 35 |
| Reverse: GCT CCG GCC AGA GGC CTC AA | |||
| BCL-2 | Forward: ACGGGGTGAACTGGGGGAGGA | 60 | 35 |
| Reverse: TGTTTGGGGCAGGCATGTTGACTT | |||
| HIF-1 | Forward: TGCTCATCAGTTGCCACTTCC | 60 | 45 |
| Reverse: CGCTGTGTGTTTTGTTCTTTACCC | |||
| TGF-β | Forward: TGTGTGACGAGCCCAAGGA | 65 | 45 |
| Reverse: TAGTTGGGTCTGGGCCAAAC | |||
| CDK1 | Forward: CAG TCT TCA GGA TGT GCT TAT GC | 65 | 45 |
| Reverse: GAGGTTTTAAGTCTCTGTGAAGAA CTC | |||
| GST | Forward: ACGTGGCAGGAGGGCTCACTC | 60 | 40 |
| Reverse: TACTCAGGGGAGGCCAGCAA | |||
| β-actin | Forward: TGAGCGCGGCTACAGCTT | 60 | 40 |
| Reverse: TCCTTAATGTCACGCACGATTT |
Western blotting
Cells were eliminated by trypsinization and the whole proteins were obtained by RAPI lysis buffer (Millipore) extraction and centrifugation at 12,000 g for 10 min. Total protein concentrations of the supernatants were measured by the BCA kit (Sigma Aldrich). Proteins (20 μg) were separated on 12% SDS-PAGE and transferred onto PVDF membranes. Membranes were blocked for 2 hrs at room temperature in TNT buffer (10 mM Tris-HCl and 150 mM NaCl pH 7.4, 0.1% Tween-20) with 5% non-fat dried milk, followed by incubation overnight at 4°C with rabbit anti-ABCB1, ABCC2, ABCG2, Bcl-2, Survivin and so on (Santa Cruz). Phosphorylation of Smad 2/3 and Akt were also determined by WB. All primary antibodies were diluted according to the instruction manual. Membranes were washed and incubated for 1 hr with peroxidase-labelled anti-rabbit IgG (Santa Cruz, diluted at 1:2000). Finally, membranes were washed three times in TNT and exposed to the ImmobilonTM Western chemiluminescent HRP substrate (Millipore) for 1 min, and then exposed autoradiography film for 2~3 min in the dark.
Statistical analyses
Data were expressed as means ± SD., statistical analysis was carried out using Student’s t-test (two tailed) and P < 0.05 indicates statistical significance.
Results
The gefitinib resistant HepG2/G cells over-expressed ITGB8 and shRNA downregulated expression of ITGB8
The parental HepG2 cell line was a gefitinib sensitive cell. However, after continuing exposure to gefitinib, drug resistance was developed. ITGB8 over-expression was found in HepG2/G (gefitinib resistance) cells. The resistance to gefitinib was indicated as the IC50 of this cell line was 17 folds of that of parental cells (Figure 1A).
Figure 1.
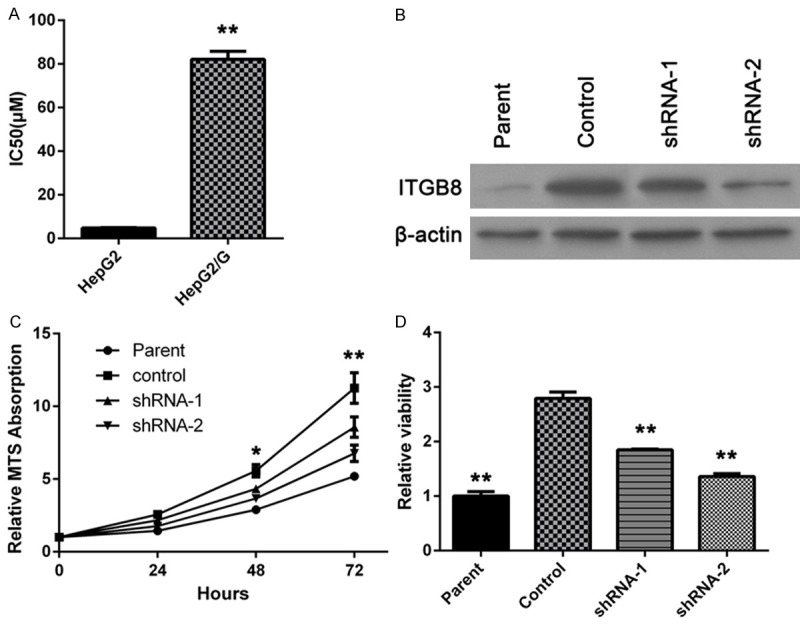
ITGB8 up-regulates in HepG2/G cells and participates in gefitinib resistance. A: Gefitinib IC50 of HepG2 and HepG2/G cells was determined. The data are shown as the means ± SD (n = 3). B: HepG2/G cells were infected with two different ITGB8 shRNAs (shRNA-1 or shRNA-2). Then ITGB8 expression was determined by western blot assay. C: Growth curve of HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA-1 (shRNA-1) or shRNA-2 (shRNA-2) was determined by MTS assay. D: Cells treated with gefitinib for 72 hours, then cell viability was examined by MTS assay. The data are shown as the means ± SD (n = 3). *P < 0.05, **P < 0.01.
After transfecting HepG2/G cells with ITGB8 shRNA lentiviral particles, several clones were selected out by puromyicin. Downregulation of ITGB8 was confirmed by western blotting and a high (about 70~80%) and a low (about 30~40%) silencing clones were selected for further studies. Control lentiviral particles had no effect on ITGB8 expression (Figure 1B).
ITGB8 silencing decreased cell proliferation and increased gefitinib sensitivity
As shown in Figure 1C, the cell proliferation rate was decreased after 24 hours of culture when compare to the parental cells. Hence, in the following studies, we performed the assay within 24 hours after cell seeding. Moreover, gefitinib sensitivity was restored by ITGB8 silencing as shown in Figure 1D, This reverse effect was correlated with ITGB8 silencing level.
Restore of drug sensitivity was accompanied with an intracellular accumulation of Rhodamine-123, cell cycle arrest and increased apoptosis rate
Rhodamine-123 is a fluorescent pigment which is transported by some membrane transporter protein that involved in multi-drug resistance. The flow cytometry results showed that the ITGB8 silencing cells have a greater intracellular fluorescent activity, which mean more Rh-123 was retained in the cell. Meanwhile, FCM showed that cell cycle was halted by G0/G1 phase and apoptosis rate increased significantly after ITGB8 silencing, as shown in Figure 2.
Figure 2.
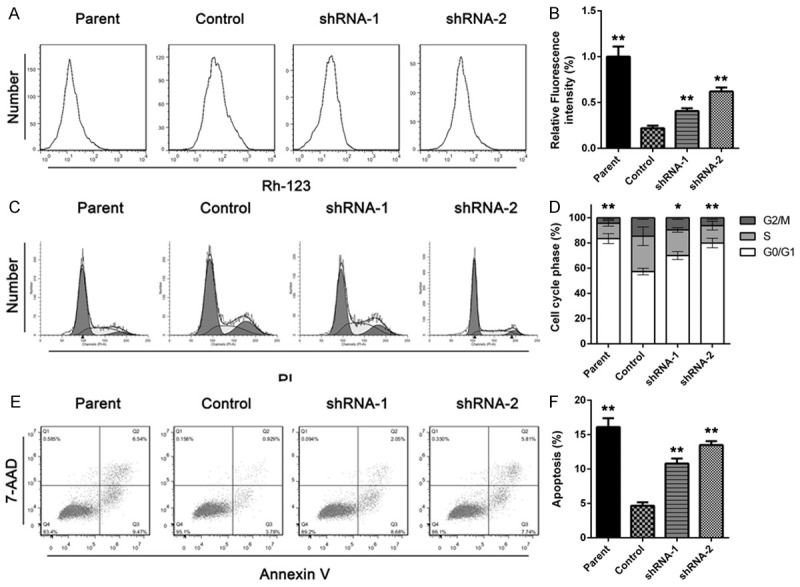
ITGB8 silencing increases accumulation Rhodamine-123 accumulation, cell cycle arrest and apoptosis rate. A and B: Rhodamin-123 accumulation assay was performed on HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA-1 (shRNA-1) or shRNA-2 (shRNA-2). The data are shown as the means ± SD (n = 3). C and D: HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA-1 (shRNA-1) or shRNA-2 (shRNA-2) was treated with gefitinib for 24 hours, then Cell cycle phase was analyzed by flow cytometry. The data are shown as the means ± SD (n = 3). E and F: HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA-1 (shRNA-1) or shRNA-2 (shRNA-2) was treated with gefitinib for 24 hours, then apoptosis rate was determined. The data are shown as the means ± SD. *P < 0.05, **P < 0.01.
The accumulation of Rh-123 was accompanied with downregulation of ABCB1, ABCC2 and ABCG2
Since intracellular Rh-123 increased after ITGB8 silencing, we were interested in whether transporters from ABC family, such as ABCB1, ABCC2 and ABCG2 were involved in the drug resistance. Western blotting analysis shown that HepG2/G cells expressed a high level of ABCB1, ABCC2 and ABCG2; then RT-PCR and western blotting results showed that, when compared to the high expression level observed in the parental cells, the expression of these proteins decreased in the ITGB8 silencing cells (Figure 3).
Figure 3.
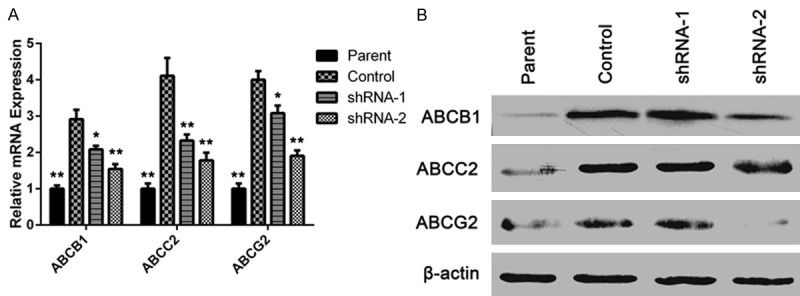
ITGB8 silencing induces downregulation of ABCB1, ABCC2 and ABCG2. A and B: qPCR and Western blot analysis of RASAL2 levels in HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA- 1 (shRNA-1) or shRNA-2 (shRNA-2). The data are shown as the means ± SD. *P < 0.05, **P < 0.01.
SOD, GST, TS (Thymidylate synthase), HIF-1 and TGF-beta
Another subset of proteins like SOD, GST and TS, which could detoxify anti-cancer drugs, were showed to involve in ITGB8 mediated gefitinib resistance in HepG2/G. HIF-1 and TGF-beta was also found to increased in HepG2/G and decrease after ITGB8 silencing (Figure 4). We had a further investigation into TGF-beta pathway which would be described below.
Figure 4.
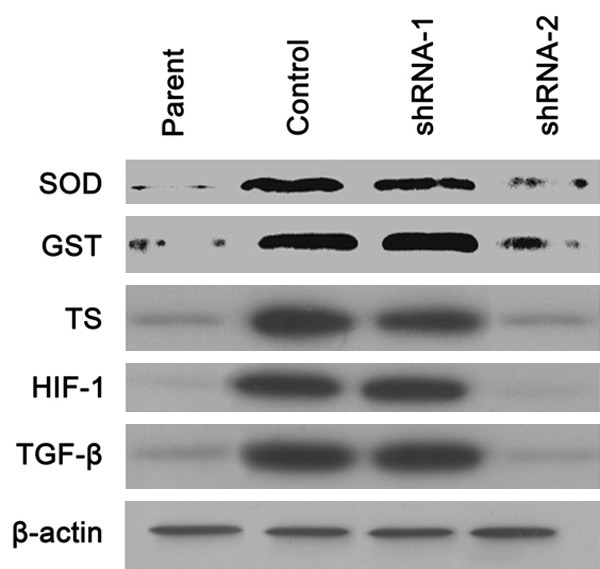
ITGB8 silencing down-regulates SOD, GST, TS (Thymidylate synthase), HIF-1 and TGF-beta. Western blot analysis of indicated protein expression in HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA-1 (shRNA-1) or shRNA-2 (shRNA-2).
ITGB8 modulated the expression of Survivin, Bcl-2, and CDK1
Survivin and Bcl-2 have been implicated in anti-apoptosis, and is also thought to be involved in resistance to conventional cancer treatment. The expression of survivin and Bcl-2 were upregulated in HepG2/G cells, when compare to the parental cells and ITGB8 silencing downregulated the survivin and Bcl-2. The RT-PCR results were consistent to those from western blotting, meanwhile, CDK1, a protein which promotes the cell proliferation, was downregulated by ITGB8 silencing as well (Figure 5).
Figure 5.
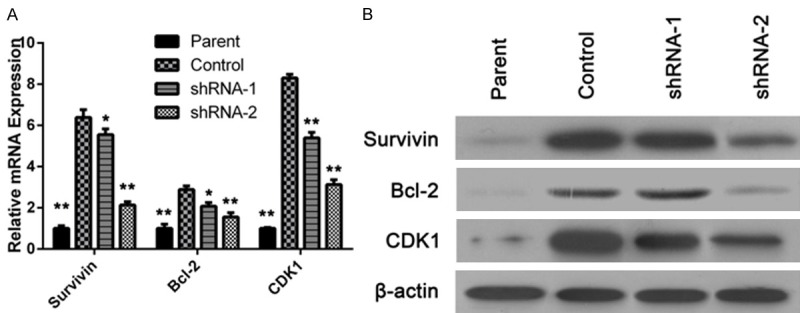
ITGB8 modulated the expression of Survivin, Bcl-2, and CDK1. A and B: qPCR and Western blot analysis of Survivin, Bcl-2, and CDK1 levels in HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA-1 (shRNA-1) or shRNA-2 (shRNA-2). The data are shown as the means ± SD. *P < 0.05, **P < 0.01.
TGF-beta signaling pathway was involved in ITGB8 mediated gefitinib resistance
Since integrin and TGF pathway have some close interaction, we investigated whether TGF pathway was involved in this ITGB8 mediated gefitinib resistance. Above mentioned data showed that TGF-beta expression level changed according to the drug sensitivity. Further investigations showed that signaling downstream of integrin and ITGB8, such as phosphorylation of Smad 2/3 and Akt, was inhibited by ITGB8 silencing (Figure 6).
Figure 6.
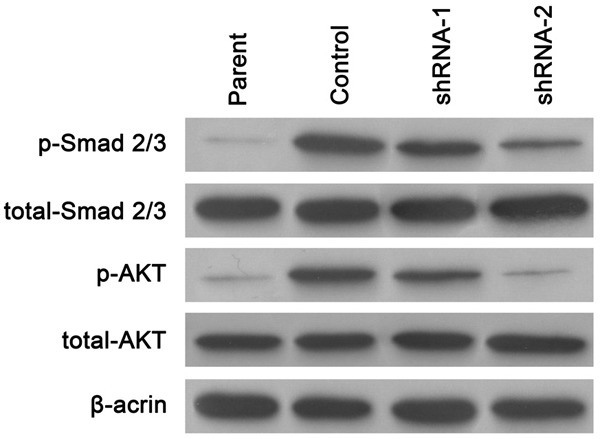
TGF-beta signaling pathway was involved in ITGB8 mediated gefitinib resistance. Western blot analysis of Smad 2/3 and Akt levels in HepG2 cells (Parent), HepG2/G cells (Control) and HepG2/G cells infected with shRNA-1 (shRNA-1) or shRNA-2 (shRNA-2).
Discussion
Currently, gefitinib is highly recommended as a first-line therapeutic for patient who harbored EGFR active mutations. Several large clinical trials have demonstrated that in this sup-population of patients, gefitinib could significantly improve the progress free survival (PFS) when compare to standard chemotherapy. However, almost all patients will develop resistance to gefitinib [4]. Several mechanisms including the common secondary mutation T790M have been found and respective strategies are under developed. Recently integrins are also found to involve in gefetinib and erlotinib (another EGFR-TKI) resistance [14,15]. Here we showed that another integrin called beta 8 could contribute to this resistance.
Previous study has confirmed that HepG2 is a gefitinib sensitive cell with active EGFR mutation [16]. We generated gefitinib resistant HepG2 and several clones were obtained. Interestingly, we found that one clone named HepG2/G overexpressed ITGB8 without T790M mutation. To confirm the role of ITGB8 in gefitinib resistance, we silenced ITGB8 by shRNA and found that drug sensitivity could be restored. Further studies showed that after ITGB8 silencing, intracellular concentration of Rh-123 increased, cell cycle was halted by G0/G1 phase, and apoptosis rate increased as well. So the mechanism behind ITGB8 mediated resistance reverse was explored following these clues.
The increase of intracellular concentration of Rh-123 means that drug efflux by the cancer cells is inhibited. P-gp, ABCB1, ABCG2 and ABCC2 are several most common membrane transporters that efflux drugs out of cells and frequently found to be overexpressed in the drug resistance environment. It has been reported that the polymorphisms of ABCB1, ABCC2 and ABCG2 is associated with irinotecan-pharmacokinetics and clinical outcome in patients with advanced hepatic cancer [17]. There some evidences indicate that gefitinib is a possible substrate to ABCB1 and ABCG2, as Agarwal et al [18] showed that distribution of gefitinib to brain is limited by these two proteins and Elkine et al [19] showed that ABCG2 attenuated gefitinib induced tumor cell death. We demonstrated that ITGB8 silencing could downregulate the expression of these transporter proteins. Hence this effect could improve intracellular concentration of gefitinib.
Another mechanisms involved in chemotherapy resistance is that hepatic cancer cells have elevated detoxification activity. SOD, GST and TS are such kind of proteins. SOD and GST are two antioxidant enzymes that are effectively protected cells, including cancer cells against exogenous free radicals. This effect could protect cancer cells from radiation and chemotherapy. TS plays a central role in DNA biosynthesis and is the target of many chemotherapeutic agents. Cancer cells resistant to cisplatin or doxorubicin show increased levels of this enzyme [20]. Although our studies showed that these proteins were modulated by ITGB8, their roles in gefitinib resistance are unclear since little evidence has been found to indicate that free radicals involve in gefitinib inhibition of cancer. HIF-1 is a transcriptional factor that responds to changes in available oxygen in the cellular environment [21]. For a long time it has been considered that HIF-1 is a possible anti-cancer targets [22,23]. It has also been showed that gefitinib could decrease VEGF expression of hepatic cancer cells by both HIF-1-independent and HIF-1-dependent manner [24]. In our study, HIF-1 was upregulated in HepG2/G and downregulated by ITGB8 silencing. However, according to its biological functions, HIF-1 might not play a direct role in this in vitro gefitinib resistance.
CDK1 is a highly conserved protein that functions as a serine/threonine kinase and is a key player in cell cycle regulation. CDK1 functions abnormally in many cancers and promotes the cancer cells through the cell cycle [25]. Since ITGB8 silencing inhibits cell cycle of HepG2/G, we determine the expression level of CDK1 and found that it was downregulated after ITGB8 silencing. It’s not very clear how ITGB8 modified CDK1. One possible mechanism is through TGF-beta pathway as described below.
Survivin is an anti-apoptosis protein which solely expressed in tumor and embryonic tissue. By inhibiting cancer cell apoptosis and promoting proliferation and angiogenesis, it is a potential anti-cancer target [26,27]. Bcl-2 is another anti-apoptosis protein and in many caners its overexpression is correlated with drug resistance [28]. We demonstrated that survivin and Bcl-2 were involved in gefitinib resistance in HepG2/G as they were both downregulated by ITGB8 silencing.
Most interesting finding in our study is that TGF-beta decreases after ITGB8 silencing. Since it has been found that integrins usually interact with TGF-beta pathway [13,29], we speculated that TGF beta signaling pathway was involved in ITGB8 mediated HepG2/G gefitinib resistance. Indeed, integrins and TGF-beta receptor could coordinate to form a complicated intracellular signaling network which involves in many cellular processes including cell growth, cell differentiation, and apoptosis and so on. Here we showed that the phosphorylation of smad 2/3, two responsive factors of TGF-beta receptors, and Akt was inhibited by ITGB8 silencing, which proved our speculation.
In summary, we found that ITGB8 overexpression could be an inducer of gefitinib resistance of hepatic cancer. ITGB8 overexpression could lead to drug efflux and promote cell cycle and anti-apoptosis. ITGB8 might interact with TGF-beta pathway to achieve its anti-gefitinib effects.
Disclosure of conflict of interest
None.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Zhu Y, Zhu L, Lu L, Zhang L, Zhang G, Wang Q, Yang P. Role and mechanism of the alkylglycerone phosphate synthase in suppressing the invasion potential of human glioma and hepatic carcinoma cells in vitro. Oncol Rep. 2014;32:431–436. doi: 10.3892/or.2014.3189. [DOI] [PubMed] [Google Scholar]
- 3.Murakami T, Tsurusaki M. Hypervascular benign and malignant liver tumors that require differentiation from hepatocellular carcinoma: key points of imaging diagnosis. Liver Cancer. 2014;3:85–96. doi: 10.1159/000343864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen MJ, Zhong W, Zhang L, Zhao J, Li LY, Wang MZ. Recurrence patterns of advanced non-small cell lung cancer treated with gefitinib. Chin Med. 2013;126:2235–2241. [PubMed] [Google Scholar]
- 5.Karroum O, Kengen J, Gregoire V, Gallez B, Jordan BF. Tumor reoxygenation following administration of the EGFR inhibitor, gefitinib, in experimental tumors. Adv Exp Med Biol. 2013;789:265–271. doi: 10.1007/978-1-4614-7411-1_36. [DOI] [PubMed] [Google Scholar]
- 6.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2014;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 7.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 8.Mok TS, Wu YL, Thongprasert S. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 9.Fratto ME, Santini D, Vincenzi B, Silvestris N, Azzariti A, Tommasi S, Zoccoli A, Galluzzo S, Maiello E, Colucci G, Tonini G. Targeting EGFR in bilio-pancreatic and liver carcinoma. Front Biosci. 2011;3:16–22. doi: 10.2741/s128. [DOI] [PubMed] [Google Scholar]
- 10.Chen WC, Chen W, Tseng GC, Lai HC, Shih CM, Hsia TC. Gefitinib as an effective therapy for advanced hepatocellular carcinoma with lung metastasis? Liver Int. 2010;30:1548–1549. doi: 10.1111/j.1478-3231.2010.02239.x. [DOI] [PubMed] [Google Scholar]
- 11.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabodi S, Di Stefano P, Leal Mdel P, Tinnirello A, Bisaro B, Morello V, Damiano L, Aramu S, Repetto D, Tornillo G, Defilippi P. Integrins and signal transduction. Adv Exp Med Biol. 2010;674:43–54. doi: 10.1007/978-1-4419-6066-5_5. [DOI] [PubMed] [Google Scholar]
- 13.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 14.Ju L, Zhou C, Li W, Yan L. Integrin beta1 over-expression associates with resistance to tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. J Cell Biochem. 2010;111:1565–1574. doi: 10.1002/jcb.22888. [DOI] [PubMed] [Google Scholar]
- 15.Dong Y, Xie X, Wang Z, Hu C, Zheng Q, Wang Y, Chen R, Xue T, Chen J, Gao D, Wu W, Ren Z, Cui J. Increasing matrix stiffness upregulates vascular endothelial growth factor expression in hepatocellular carcinoma cells mediated by integrin beta1. Biochem Biophy Res Commun. 2014;444:427–432. doi: 10.1016/j.bbrc.2014.01.079. [DOI] [PubMed] [Google Scholar]
- 16.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, Gerald W, Huang SF, Yang PC, Miller V, Ladanyi M, Yang CH, Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Nat Acad of Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang PP, Xu DJ, Huang C, Wang WP, Xu WK. Astragaloside reduces the expression level of P-glycoprotein in multidrug-resistant human hepatic cancer cell lines. Mol Med Rep. 2014;9:2131–2137. doi: 10.3892/mmr.2014.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334:147–155. doi: 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Várady G, Ujhelly O, Szabó K, Homolya L, Váradi A, Buday L, Kéri G, Német K, Sarkadi B. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–1777. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- 20.Prasad B, Evers R, Gupta A, Hop CE, Salphati L, Shukla S, Ambudkar SV, Unadkat JD. Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab Disposit. 2014;42:78–88. doi: 10.1124/dmd.113.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith TG, Robbins PA, Ratcliffe PJ. The human side of hypoxia-inducible factor. Br J Haematol. 2008;141:325–334. doi: 10.1111/j.1365-2141.2008.07029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12:853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Melillo G. Inhibiting hypoxia-inducible factor 1 for cancer therapy. Mol Cancer Res. 2006;4:601–605. doi: 10.1158/1541-7786.MCR-06-0235. [DOI] [PubMed] [Google Scholar]
- 24.Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006;66:3197–3204. doi: 10.1158/0008-5472.CAN-05-3090. [DOI] [PubMed] [Google Scholar]
- 25.Collins I, Garrett MD. Targeting the cell division cycle in cancer: CDK and cell cycle checkpoint kinase inhibitors. Curr Opin Pharmacol. 2005;5:366–373. doi: 10.1016/j.coph.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Ryan BM, O’Donovan N, Duffy MJ. Survivin: a new target for anti-cancer therapy. Cancer Treat Rev. 2009;35:553–562. doi: 10.1016/j.ctrv.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J. 2010;430:199–205. doi: 10.1042/BJ20100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 29.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]