

SUMMARY
Human herpesvirus 6 (HHV-6) is a widespread betaherpesvirus which is genetically related to human cytomegalovirus (HCMV) and now encompasses two different species: HHV-6A and HHV-6B. HHV-6 exhibits a wide cell tropism in vivo and, like other herpesviruses, induces a lifelong latent infection in humans. As a noticeable difference with respect to other human herpesviruses, genomic HHV-6 DNA is covalently integrated into the subtelomeric region of cell chromosomes (ciHHV-6) in about 1% of the general population. Although it is infrequent, this may be a confounding factor for the diagnosis of active viral infection. The diagnosis of HHV-6 infection is performed by both serologic and direct methods. The most prominent technique is the quantification of viral DNA in blood, other body fluids, and organs by means of real-time PCR. Many active HHV-6 infections, corresponding to primary infections, reactivations, or exogenous reinfections, are asymptomatic. However, the virus may be the cause of serious diseases, particularly in immunocompromised individuals. As emblematic examples of HHV-6 pathogenicity, exanthema subitum, a benign disease of infancy, is associated with primary infection, whereas further virus reactivations can induce severe encephalitis cases, particularly in hematopoietic stem cell transplant recipients. Generally speaking, the formal demonstration of the causative role of HHV-6 in many acute and chronic human diseases is difficult due to the ubiquitous nature of the virus, chronicity of infection, existence of two distinct species, and limitations of current investigational tools. The antiviral compounds ganciclovir, foscarnet, and cidofovir are effective against active HHV-6 infections, but the indications for treatment, as well as the conditions of drug administration, are not formally approved to date. There are still numerous pending questions about HHV-6 which should stimulate future research works on the pathophysiology, diagnosis, and therapy of this remarkable human virus.
INTRODUCTION
In 1986, the discovery of a novel human herpesvirus was reported in the journal Science (1). The importance of the finding was amplified by the fact that this virus, finally designated human herpesvirus 6 (HHV-6), was initially isolated from patients with lymphoproliferative disorders and AIDS, raising the key question of its pathogenicity. Since that time, the knowledge about this virus and associated diseases has considerably improved, as extensively reported in previously published overviews and books which remain relevant references (2–10). However, many questions on those domains are still pending and constitute true challenges for present and future research. How current tools of medical virology and clinical studies may help researchers to investigate the pathophysiology of HHV-6 infection and consider the means of its control constitutes the core subject of the present review.
(Some of the data and concepts reviewed in the present text have been discussed previously in part in an article referenced herein [11] and in oral communications at the 7th and 8th International Conferences on HHV-6 and HHV-7, which took place in Reston, VA, in 2011 and in Paris, France, in 2013, respectively.)
VIRUS PROPERTIES
Discovery and Classification
HHV-6 was first isolated from the peripheral blood mononuclear cells of patients with lymphoproliferative disorders in attempts to characterize novel lymphotropic human viruses (1). A cytopathic effect made of short-lived large refractile cells was observed in the primary cell cultures and was shown to be transmissible to novel cultures of phytohemagglutinin (PHA)-stimulated human leukocytes. Electron microscopy confirmed virus production and revealed a morphology of virus particles similar to that of herpesviruses. This included a capsid of icosahedral symmetry surrounded by a tegument within an enveloped particle of about 200 nm in diameter (12). The genomic DNA did not cross-hybridize with the genomes of the five other known human herpesviruses, unambiguously demonstrating that the newly isolated virus was different from them (13). HHV-6 was initially characterized as a human B-lymphotropic virus, but it soon appeared that it was essentially a T-lymphotropic virus, and it acquired its definite name (14). Finally, based on both biological properties and genetic analyses, HHV-6 was officially classified as a member of the Herpesvirales order, Herpesviridae family, Betaherpesvirinae subfamily (the type species of which is human cytomegalovirus [HMCV]), and Roseolovirus genus, together with human herpesvirus 7 (HHV-7), a closely related herpesvirus discovered in 1990 (15). Following the description of the initial HHV-6 strain, named GS, other prototypic HHV-6 strains, designated U1102, SIE, LHV, Z29, and HST, were obtained in other laboratories, from HIV-infected patients, mostly of African origin, but also from Japanese patients with exanthema subitum (16–20). Those isolates and the HHV-6 isolates obtained subsequently were stratified into two well-defined, nonoverlapping groups differing by specific genetic changes and phenotypic properties. These two groups were designated variants A and B (HHV-6A and HHV-6B) of the unique HHV-6 species (21). Twenty years later, the differences between the two variants have been considered important enough to recommend the classification of HHV-6A and HHV-6B as two distinct species (22). The term HHV-6 remains in usage and collectively refers to the two species.
Genome and Genetic Variability
The genome of HHV-6 is a linear double-stranded DNA consisting of a unique (U), 143- to 145-kb region flanked by identical terminal direct repeats (DRL and DRR) and having an overall approximate length of 162 to 170 kb (Fig. 1). The U region also contains internal repeat arrays designated R1, R2, and R3. DRL and DRR each contain short unique sequences, the conserved cleavage-packaging motifs pac-1 and pac-2, and two stretches of sequences related to the telomere repeat sequences (TRS) of vertebrate chromosomes (5). The one near the left end of each DR contains reiterations of the hexanucleotide GGGTTA interspersed by related but different sequences, constituting the heterogeneous telomeric-like region het(GGGTTA)n. The right end of each DR contains perfect repeats of GGGTTA. These repeated sequences likely play a major role in the process of chromosomal integration of the HHV-6 genome (ciHHV-6) (see below). This integration has been reported to occur through a junction between host chromosomes and the perfect telomeric repeats at the right end of DRR (23–25). The overall number of protein-encoding open reading frames (ORFs) is about 110 to 120 according to the different published nucleotide sequences (26–28). Most of them are located within the U region, on both genomic strands. This unique region presents strong similarities with that of HHV-7 DNA and the unique long region of the HCMV genome. The core genes coding for the virion proteins and enzymes involved in the virus replication cycle regroup into seven clusters, with each block being composed of two to eight ORFs which are conserved among all herpesviruses and overlay the central part of the U region. In addition, a betaherpesvirus-specific gene cluster and a roseolovirus-specific set of genes are present at the left end of the core genes. Several genes, such as U83 and U94, are unique to HHV-6.
FIG 1.
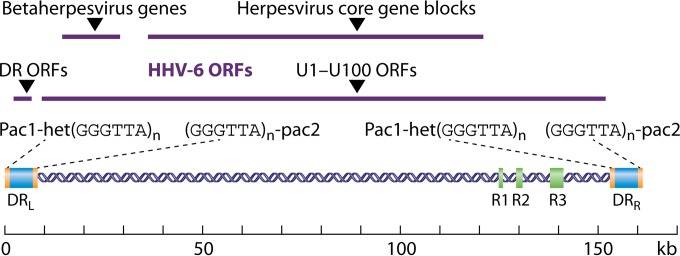
Schematic representation of the HHV-6 genome. The genome is represented as a double-stranded DNA containing specific elements which are described in the text. The repeat elements, shown as green and blue boxes, include the identical terminal repeat sequences DRL and DRR, the internal repeat arrays R1, R2, and R3, and the stretches containing repeat hexanucleotide sequences [(GGGTTA)n] within the DR regions. Purple straight lines indicate the positions of the different sets of ORFs.
The genetic variability of HHV-6 DNA is limited, with the nucleotide identity among all published sequences exceeding 90% as a whole. However, this identity rate differs according to the genes considered, ranging from about 70% for the right part of the genome to about 95% for the central core genes. This genetic polymorphism can be analyzed at three different levels of complexity. As mentioned above, HHV-6A and HHV-6B DNAs exhibit clear specific differences which are scattered throughout the genome and permit their easy recognition, without any ambiguity. These specific signatures concern the rather variable genes of immediate early region 1 (IE1) as well as the highly conserved genes for glycoproteins B (gB) and H (gH) and the U94 product (29–31). Some of the HHV-6A ORFs are thought to have no HHV-6B counterpart and vice versa, although the prediction of functional ORFs often remains debatable in the absence of relevant experimental investigations. Taken as a whole, the differences between HHV-6A and HHV-6B are believed to induce noticeable dissimilarities in the virus replication cycle, in particular regarding splicing patterns and the temporal regulation of gene transcription. Those differences may also affect the binding of viral proteins to their cellular targets, which in turn may alter cell tropism, interactions with the microenvironment, host immune responses, and, ultimately, pathogenesis (4, 22). Note that no recombinant virus originating from a mixed infection with HHV-6A and HHV-6B has ever been identified, although recent results provide some evidence of recombination between ciHHV-6A and ciHHV-6B (32). At an intermediate level of variability, the segregation of strains into distinct subgroups characterized by specific genetic signatures has been reported for HHV-6B isolates but not HHV-6A ones, based on the analysis of IE1, gB, and gH genes (29, 33). However, this segregation is not fully congruent, since it may differ according to the selected gene: the subgroups derived from the phylogenetic analysis of gB gene sequences did not exactly fit those derived from gH gene sequences. Conversely, the study of gB gene polymorphisms provided indirect evidence for genetic recombination among HHV-6B subgroups, suggesting that this genetic process may contribute to creating novel allelic combinations among these putative subgroups. The question remains as to whether the segregation into subgroups is not simply a misinterpretation of basic interstrain polymorphism, which constitutes the lowest level of variability. Overall, this genetic variability is low in terms of nucleotide sequence, and even lower in terms of amino acid sequence (34). However, despite its modest magnitude, the interstrain variability may provide useful markers for differentiating viruses in molecular epidemiology studies (35). A particular aspect concerns the number of TRS in the DR region, which does not depend on the classification as HHV-6A or HHV-6B: it varies widely among the different strains studied but remains stable for a given isolate, even after numerous serial passages in cell culture, and that property can be used to track specific HHV-6 strains in human infections (36, 37). Nevertheless, previous reports mentioned that the length of the DR region changed on viral passage in cell culture in the case of HHV-6B strain Z29 (27, 38).
Cell, Tissue, and Host Tropism
HHV-6 infects a wide range of human cells in vitro, but it preferentially replicates in activated CD4+ T lymphocytes (3, 5). At least one component of the cell receptor permitting virus anchorage to the cell surface differs according to HHV-6 species: HHV-6A uses CD46, a regulator of complement activation expressed on all nucleated cells, while CD134 (also called OX40), a member of the tumor necrosis factor (TNF) receptor superfamily present only on activated T lymphocytes, functions as a specific entry receptor for HHV-6B (39, 40). Note that CD46 is a receptor for other human pathogens, including the vaccine strains of measles virus and Neisseria gonorrhoeae, behaving as a pathogen magnet (41). In addition to CD4+ T lymphocytes, HHV-6 can infect in vitro CD8+ T lymphocytes (only with HHV-6A), human fibroblasts, natural killer cells, liver cells, epithelial cells, endothelial cells, astrocytes, oligodendrocytes, and microglial cells. However, its capacity to infect continuous T cell lines is limited, and in many cases, it can be obtained only through an adaptation process consisting of serial blind passages of a primary isolate on the target cells. The capability to infect different cell lines is generally higher for HHV-6A than for HHV-6B and appears to be a phenotypic character for discriminating both species. As a whole, no continuous cell line can be recommended for isolation of the virus. The primary isolation of HHV-6 from a human specimen usually requires cocultivation with primary highly susceptible cells consisting of peripheral blood mononuclear cells (PBMCs) or umbilical cord blood lymphocytes.
As for HCMV, the host tissue range of HHV-6 in vivo appears to be broader than might be expected from in vitro studies and includes the brain, tonsils, salivary glands, kidneys, liver, lymph nodes, endothelial cells, and monocytes/macrophages (3, 42–48). The latter cell types are suspected to be preferential sites for virus latency, in parallel with bone marrow progenitors and central nervous system (CNS) cells (49–51). Although HHV-6 infection is naturally restricted to human cells and tissues, simian cells and monkeys can be infected experimentally, but the availability of this model is extremely low (52, 53). In transgenic mice expressing human CD46 and infected with HHV-6A, the virus persisted in the brain for months, and a significant inflammatory response developed, opening the possibility of a rodent model for virus-induced neuroinflammatory diseases (54, 55).
Replication Cycle
HHV-6 attaches to its cell receptor by means of a tetrameric viral ligand complex made up of the glycoproteins H (gH), L (gL), Q1 (gQ1), and Q2 (gQ2) (8). Following attachment, HHV-6 entry into cell occurs through a fusion between the viral envelope and the cell membrane by a mechanism which involves gB and gH functions but remains poorly understood. The nucleocapsid is then transported through the cytoplasm to the nucleus, likely using the pathway of the microtubule network. HHV-6 DNA is released into the nucleoplasm. Viral genes are expressed in a temporally ordered manner, starting with immediate early (IE) genes from the IE-A locus, which is constituted of two genetic units, IE1 and IE2 (56–58). Those genes are transcribed in the absence of de novo protein synthesis, and this step is followed by the transcription/expression of early (E) and late (L) genes. The replication of the genome occurs after the synthesis of E proteins, which have enzymatic activities dedicated to nucleotide metabolism and DNA synthesis, i.e., phosphotransferase, ribonucleotide reductase, uracil-DNA glycosylase, origin-binding protein, DNA polymerase, polymerase processivity factor, major DNA-binding protein, and helicase-primase complex activities. Viral DNA is assumed to be replicated through a rolling circle process. Progeny DNA is yielded in the form of concatemeric strands, which are cleaved and packaged into capsid precursors thanks to specific cleavage-packaging signals present in the DRL and DRR regions (8). The capsids exit the nucleus, acquiring an intermediate envelope by budding through the inner part of the nuclear membrane, are deenveloped by fusion with the external part of this membrane, and appear as tegumentary forms in the cytoplasm. The acquisition of the final envelope carrying viral glycoproteins occurs in the trans-Golgi network, and mature virions are released by exocytosis. The occurrence of a complete replication cycle, which lasts about 3 days, has a major impact on host cell functions and morphology. Infected cells engaged in this virus-producing process ultimately die by apoptosis and/or necrosis.
Latency and Reactivation
Like other human herpesviruses, HHV-6 persists indefinitely in its host and is capable of reactivation, meaning the active production of detectable mature virions in some body compartments following a phase of apparently complete clearance. These properties rely on the putative capacity of its genome to be maintained in a nuclear latent form or to drive a low-level productive infection in some cells while inducing a fully lytic infection in other cells. For other human herpesviruses, such as herpes simplex virus, the latent DNA genome has the form of a covalently closed circular episome associated with cellular nuclear proteins. The existence of such a latent nuclear form has not been demonstrated formally for HHV-6, although an episomal state was shown after experimental infection of cervical carcinoma cell lines (59).The viral gene U94, which is expressed during latent infection, is assumed to play a major role in the establishment and maintenance of intracellular latency (60). Other latency-associated transcripts have also been described (61). Reactivation occurs through the transcription of IE genes in the IE1 and IE2 regions following the likely transactivation effect of cellular and/or viral factors whose nature is still unknown. This reactivation process results in the induction of a replication cycle and the possible appearance of a cytopathic effect (48).
Chromosomal Integration
The integration of the HHV-6 genome into human chromosomes (ciHHV-6) was initially described for transformed cell lines (62, 63). This phenomenon was further reported to be present in human cells in vivo, including cells which can be transmitted as germinal cells to offspring and hematopoietic stem cells transferred to a transplant recipient (64, 65). It appears to be a unique feature among human herpesviruses and raises numerous novel questions regarding both pathophysiology and diagnosis (25, 66). The covalent linkage between viral and cellular DNAs occurs within the subtelomeric region of chromosomes, likely by a mechanism of homologous recombination between telomeric repeat sequences of viral and cellular origins. The phenomenon has been described for both HHV-6A and HHV-6B and occurs in 0.2 to 1% of the general population in developed countries. It might be generated in the context of de novo infection and is considered by some authors to be the default pathway of HHV-6 latency, including in non-germ line cells and before persistence of viral DNA as an episome (23). Although there is no in vivo evidence for that assumption, it must be kept in mind knowing that HHV-6 has the ability to infect sperm cells (67, 68). Thus, de novo HHV-6 infection of germinal cells might result in individuals harboring the integrated virus in their germ line and transmitting it to their offspring (25). Moreover, ciHHV-6 might lead directly to reactivation, as reflected by the production of viral transcripts, proteins, and even transmissible virions (23, 35, 69). This emphasizes the tight relationship between ciHHV-6, latency, and reactivation. In that context, it is worth recalling the homology between HHV-6 U94 and the human adeno-associated virus (AAV) type 2 rep gene (70). rep gene products are involved in the site-specific integration of AAV DNA into host cells. Therefore, U94 products might have a pivotal role both in the establishment of latency and in ciHHV-6.
Impacts of Viral Gene Expression on Cell Functions
As previously mentioned, the occurrence of a complete replication cycle has profound effects on cell functions and viability in the context of either de novo infection or reactivation. In addition, independently of any complete virus-producing process, the expression of certain HHV-6 genes might occur from persisting episomal or ciHHV-6 forms of viral DNA. Many publications have reviewed the formally demonstrated or putative effects of virally encoded gene products on the regulation and modification of cell functions (3, 4, 8). As an example, considering the gene products of the IE-A region, IE2 might behave as a general transcriptional activator of many viral and cellular genes, while IE1 interacts with PML bodies (71, 72). The proteins encoded by the IE-B region have also been shown to transactivate heterologous promoters, such as the HIV-1 long terminal repeat (LTR) (30). The products of the DR7 gene appear to demonstrate a cell-transforming activity, presumably through an interaction with p53 (73). Regarding the U94 gene, which is analogous to the AAV rep gene, it can bind to the human TATA-binding protein, and its expression in endothelial cells decreases cell migration and angiogenesis (74, 75). The U95 gene product interacts with the mitochondrial GRIM-19 protein, a component of the oxidative phosphorylation system involved in apoptotic processes (76). As indicated below, several proteins encoded by the HHV-6 genome have immunomodulatory functions. Taken together, all these features provide molecular bases for understanding the pathological processes associated with acute and chronic HHV-6 infections.
HUMAN INFECTION
Epidemiology
HHV-6A and HHV-6B are ubiquitous viruses that are detected in all human populations around the world, as reviewed elsewhere (3, 5, 8). Current serologic assays do not permit discrimination of HHV-6A and HHV-6B infections (77). Consequently, a precise view of their respective seroprevalences in different human populations is not available at present. As a whole, HHV-6 infection is detected in more than 90% of adult populations in developed countries, although the data on seroprevalence may reveal significant differences according to geographic location, age of subjects, and sensitivity and specificity of serologic assays. Standardized species-specific serologic tests will help to clarify the meaning of these differences and to determine whether the circulation of HHV-6A and HHV-6B truly differs in different ethnic groups, countries, or continents.
HHV-6 infection is usually acquired very early in life, between 6 months and 2 years of age, following the loss of protective maternal antibodies (78). At an even earlier period of life, congenital infection following intrauterine transmission has been reported for about 1% of children, a frequency close to that observed with HCMV, and cases of perinatal transmission have been described (79–82). As described below, congenital infection is mainly linked with ciHHV-6 in mothers (78). Primary infection can also happen later, in adults, as reported in a few cases (83). Saliva is assumed to be the main vehicle for virus transmission, as supported by the frequent detection of HHV-6 in saliva and salivary glands. Virus transmission through organ transplantation has been described infrequently, while blood transfusion and breast feeding have never been reported to be origins of primary infections (84, 85). To date, a clear view of the respective temporality of HHV-6A and HHV-6B infections is missing. It is generally believed that in the majority of countries, primary HHV-6B infection occurs first, in many cases associated with clinical symptoms, whereas HHV-6A is acquired later, through asymptomatic infection (see below). Ultimately, the concomitant detection of HHV-6A and HHV-6B in blood or tissues from adults indicates that the two viruses chronically infect many, if not most, individuals (86, 87).
Physiology
No experimental in vivo model of human infection is currently available. Several monkey species are infected with HHV-6-related viruses, but their capability to provide a relevant model regarding HHV-6 pathophysiology is still unknown (88, 89). Monkeys as well as transgenic mice expressing human CD46 could be infected with HHV-6A and developed a specific antiviral immune response; in some cases, neurologic symptoms and neuropathological lesions were observed (54, 55, 90, 91). However, to date, none of these animal models has given a conveniently workable picture of the course of human infection. This course can only be hypothesized from clinical and biological findings observed during the natural process (92). Clinical symptoms as well as virological data reported for primary infection cases suggest that following its entry into the body by the oral route, the virus replicates in the salivary glands and satellite lymphoid tissues of the oropharynx, probably the tonsils and cervical lymph nodes. Systemic diffusion of infection would occur by the blood route, taking advantage of the virus tropism for PBMCs and vascular endothelial cells, as reflected by the isolation of infectious virus from blood during the acute phase of infection. A spreading of virus and infected cells via lymphatic vessels is also possible. This spreading leads to the active, abortive, or latent infection of susceptible cells in other organs, including T lymphocytes and monocytic cells in lymphoid tissue and the liver, kidney and skin epithelial cells, hematopoietic stem cells in bone marrow, and neuroglial cells in the CNS. The entry of HHV-6 into the CNS might occur by crossing of the blood-brain barrier through the olfactory pathway (93). In most cases, this primary infection is self-limiting while a specific immune response develops (see below), and the viremia finally decreases to undetectable levels as measured by conventional diagnostic assays.
The establishment of HHV-6 latency in the body raises numerous questions regarding chronology, cell location, gene expression level, and potential differences between HHV-6A and HHV-6B. HHV-6 DNA is detected, in particular, in saliva, blood monocytes, endothelial cells, and bone marrow progenitors (48, 50, 94). As mentioned previously, latency-associated transcripts from U94 and the IE region have been detected in PBMCs (8). Currently, it is not known whether the transcription of other genes, synthesis of proteins, and even production of virions can occur at low levels in particular cells, tissues, or organs during the latency stage of infection, which would make the putative frontier between latent and active infections even narrower (95). Some authors have suggested that HHV-6A does not exhibit the same capability to establish latency in target cells as that of HHV-6B. However, this hypothesis has to be modulated by the fact that regardless of infection stage, and for still unknown reasons, HHV-6B is detected much more frequently than HHV-6A in PBMCs and cerebrospinal fluid (CSF) (8). As mentioned above, ciHHV-6, present in about 1% of the adult population, is another form of virus quiescence in which each cell in the body contains one copy of the viral genome.
Reactivation can be defined as the reappearance of replication cycle transcripts and yield of infectious virus in peripheral blood or in a specific tissue or organ from an individual who has experienced a primary infection. This may lead to extended reinfection of bodily tissues and cause disease. The initial cellular events can be induced experimentally in cultures of latently infected cells following exposure to a phorbol ester, such as tetradecanoyl-phorbol-13-acetate (TPA), or superinfection with HHV-7 (48, 96). Note that stimulation using TPA or trichostatin A has been shown to promote virus activation from ciHHV-6 in PBMCs as well as in cell lines (23). In vivo, the recognition of reactivation may reveal more complexity due to heterogeneity of patient presentations and limitations of virological methods used to address this issue (see below). In particular, endogenous reactivation has to be distinguished from reinfection of exogenous origin with a novel strain of HHV-6 (11). Exogenous reinfection in an individual who is already seropositive with respect to the same virus species, either HHV-6A or HHV-6B, is assumed to occur, as reflected by the concomitant detection of distinct viral strains by molecular epidemiology approaches (36). However, the frequency of that phenomenon, as well as its pathophysiological consequences, is still unknown.
Interactions with the Immune System
A specific immune response to HHV-6 was recognized very soon after its discovery (8, 97–99). For patients experiencing primary infection, serologic studies have shown the appearance of specific IgM antibodies during the first week and their subsequent disappearance after 1 month, while IgG antibodies are detected later than IgM but persist indefinitely. These antibodies react with a wide range of virally encoded proteins, some of which are considered major antigens by immunoblot studies, such as the U11 gene product. Some of these antibodies have a virus-neutralizing activity, but their activity in the control of active infection is not well understood. Cellular immunity is believed to play the major role in this control, as reflected by the deleterious effects of T cell immune response suppression. Recent publications reported the proliferation of CD4+ and CD8+ T cells in response to HHV-6A and HHV-6B antigens in most healthy adults (100–102). Those studies permitted the fine characterization of a significant number of HHV-6 T cell epitopes but showed the following two important limitations for future investigations: the low frequency of circulating HHV-6-specific T cells, requiring in vitro expansion prior to any functional characterization; and the high degree of cross-reactivity between HHV-6A and HHV-6B epitopes. Prior to the emergence of adaptive immunity, HHV-6 infection has the capacity to stimulate the effectors of innate immunity: an increased secretion of proinflammatory cytokines, such as interleukin-1β (IL-1β), TNF-α, and alpha interferon (IFN-α), is observed in PBMCs, while NK cell activity associated with IL-15 synthesis is elevated in HHV-6A infection (103, 104).
Like other herpesviruses, HHV-6 exhibits a wide range of biological properties which might explain its ability to both stimulate and modulate immune responses (3, 4, 8). This modulation, in turn, would permit evasion of the HHV-6-specific immune response and improve the microenvironmental conditions for promoting virus persistence. As an example, the upregulation of proinflammatory cytokines in PBMCs is associated with a downregulation of IL-2 synthesis and a subsequent decrease of T cell activation. Accordingly, HHV-6 has been shown to promote the shift of the T-helper cell profile from Th1 to Th2 by upregulating IL-10 and downregulating IL-12. HHV-6A infection has been shown to downregulate the expression of HLA class I expression on dendritic cells. In parallel, HHV-6 infection has strong suppressor effects on the growth and differentiation of bone marrow progenitors, which may affect the differentiation of macrophages and the population of thymocyte precursors. Many of these effects are specifically mediated by HHV-6-encoded proteins which act as analogues of cell chemokines and are believed to promote viral growth, viral dissemination, and/or escape from the immune response. The U21 protein has been shown to reduce the expression of HLA class I expression as mentioned above. As other examples, the U83 gene encodes a chemotactic protein which is an agonist for several human CC chemokine receptors (CCRs) and the U12 and U51 genes encode chemokine receptors which presumably activate and recruit host cells (105, 106). The U24 gene product induces the internalization of the T cell receptor/CD3 complex, which may alter the patterns of T cell activation. In vitro studies have often provided conflicting evidence concerning the effects of these proteins, depending on the type of cells and the HHV-6 species investigated. However, it seems clear that the virus has the capacity to perform a fine-tuning regulation of cell functions. This capacity is likely extended to the modulation of other viral infections affecting the same target cells and organs. The role of HHV-6 as a cofactor of HIV in AIDS remains a matter of discussion. It is substantiated by several in vitro findings relying on interactions between both viruses, i.e., the common tropism of HHV-6 and HIV for CD4+ T cells, transactivation of the HIV-1 LTR by HHV-6 proteins, and induction of CD4 expression on CD8+ T cells and NK cells by HHV-6 making these cells susceptible to HIV infection (107). HHV-6 infection has also been shown to stimulate the activation of Epstein-Barr virus (EBV) from latency and, more recently, the expression of the human endogenous retrovirus K-18 (108, 109).
VIROLOGICAL DIAGNOSIS
Objectives and Means
The aim of virological diagnosis is first to provide proof of HHV-6A or HHV-6B infection, i.e., the presence of the virus(es) in an investigated subject. Second, it is necessary to define the status of this infection as latent, active (which can also be termed productive or acute), or ciHHV-6 related. Third, the viral load and, if possible, the expression of viral genes have to be quantified in peripheral blood and specific body compartments in order to determine the possible causative relationship with the concomitant clinical symptoms. Fourth, the question of treating the infection with antiviral drugs active against HHV-6 has to be considered. Consequently, diagnostic procedures are implemented for the monitoring of infection following initial diagnosis and therapy, if started. This includes the serial quantification of virus replication and the detection of putative resistance to antivirals in case of therapeutic failure (110).
Additional objectives refer to the study of interhuman transmission cases and the phylogenic relationships between viral strains and viral subpopulations in the context of a mixed infection within the same individual. These require the implementation of molecular studies based on gene amplification and nucleotide sequencing. Since HHV-6A and HHV-6B are now defined as distinct viral species, it is essential that differentiation between them be obtained at an early step of any HHV-6 infection diagnostic procedure, and this is also required for publication of HHV-6-related scientific articles (22).
Diagnostic procedures performed for either the management of a sole patient or the planned study of a human cohort are basically similar (Table 1). A wide range of human specimens can support these procedures, with the most common ones being whole blood, plasma, and serum. Cerebrospinal fluid is essential for the diagnosis of central nervous system infections, while bronchoalveolar lavage permits investigation of lung infections. The relevance of saliva samples for diagnosis needs to be clarified, as is the case for any cell fraction or extract obtained from a body fluid, cell smear, or tissue biopsy specimen (111). However, the frequent detection of HHV-6 in saliva and the capacity of saliva to contaminate lower respiratory tract specimens have to be taken into account in the interpretation of results.
TABLE 1.
Overview of diagnostic procedures for HHV-6 infection
| Diagnostic approach | Method | Advantages and usefulness | Disadvantages and limitationsb |
|---|---|---|---|
| Indirect (serology) | Assays for IgG and IgM detection (IFA, ELISA)a and avidity assays | Easy collection and storage of serum samples, readily accessible techniques, diagnosis of primary infection, seroprevalence studies | Lack of interpretation for diagnosis of reactivations, no discrimination between HHV-6A and HHV-B, delayed/altered response if immune deficiency is present, cross-reactivity with other betaherpesviruses |
| Direct | Virus isolation in cell culture | Reference method in virology, evidence of infectious virus, precise investigations of virus strains | Labor-intensive method, high cost, limited sensitivity |
| Antigen detection | Uses conventional equipment, gives evidence of virus gene expression, discrimination between HHV-6A and HHV-6B | Need for standardization, limited sensitivity with current reagents, difficulties of readout in some cases | |
| Qualitative viral DNA PCR | High sensitivity and specificity, discrimination between HHV-6A and HHV-6B | No distinction between active infection, latency, and ciHHV-6 | |
| Quantitative viral DNA real-time PCR | High sensitivity and specificity, discrimination between HHV-6A and HHV-6B, longitudinal follow-up studies, comparison of viral loads in blood versus organs | Need for international standardization, need for specific thresholds for active infections and ciHHV-6 | |
| Detection of viral transcripts by RT-PCR | Distinction between active and latent infections, recognition of active infection in ciHHV-6 subjects | Limited sensitivity (to be evaluated), need for standardization | |
| Droplet digital PCR | Precise method for measuring nucleic acid amounts, identification of ciHHV-6 | Limited sensitivity (to be evaluated), adaptation to clinical specimen diversity |
IFA, immunofluorescence assay; ELISA, enzyme-linked immunosorbent assay; RT-PCR, reverse transcriptase PCR; ciHHV-6, chromosomally integrated human herpesvirus 6.
The availability of the mentioned tests may be restricted in some medical centers.
Two general complementary approaches can be used (110). Direct diagnosis is based on the detection and characterization of whole virions or some of their specific components, the most convenient of which currently are nucleic acids. These components may come from either cell-free virions or infected cells. Infected cells contain not only the components of released viral particles but also transcripts and additional virus-encoded proteins, which provides an even larger set of viral targets. The indirect approach, also known as serology, is based on the detection and characterization of virus-specific antibodies in a body fluid, usually serum, using reference viral antigens. Due to the stability of antibodies even after prolonged storage of samples, serologic assays provide reproducible results and are very convenient for retrospective studies. However, the interpretation of serologic results may be equivocal for many reasons that can be summarized as follows. HHV-6A and HHV-6B infections are widespread and lifelong in the general population, making seropositivity highly prevalent and poorly discriminant. Rapid increases in IgG responses and the presence of IgM antibodies are not highly specific for acute primary infections, since these phenomena may also be associated with virus reactivations. The serologic profile may be atypical in the case of immune suppression or ciHHV-6. Cross-reactivity has been reported between antibodies to the four human betaherpesviruses, i.e., HHV-6A, HHV-6B, HHV-7, and HCMV. Lastly, commercially available serologic assays targeting HHV-6 antibodies cannot differentiate HHV-6A from HHV-6B infections to date.
Serology
Serologic methods are mainly founded on indirect immunofluorescence assays (IFA). In such assays, HHV-6-infected cells are fixed on glass slides, incubated with serum, and observed with an optical microscope. The readout of the reaction relies on both the number of fluorescent foci and characteristic patterns of cell staining, also taking into account the intensity of nonspecific background fluorescence signals. The search for neutralizing antibodies is motivated by their specificity and their putative correlation with protective immunity, but this technique is cumbersome and expensive (112). A few enzyme-linked immunosorbent assays (ELISAs) have been developed and commercialized, using either a crude lysate of infected cells or purified virus obtained from cell culture supernatant as the antigen (113), but the question of their specificity has been raised repeatedly. The use of synthetic peptides as antigens is expected to improve their quality in the future, as well as the extension of the use of immunoblot assays or measurements of antibody avidity (114, 115). However, it is not certain that this evolution can circumvent some of the shortcomings mentioned above, in particular the difficulty in interpreting the presence of IgM and the cross-reactivity sometimes observed between distinct betaherpesviruses (116). Clearly, a better knowledge of the humoral immune response against HHV-6 at different stages of infection (primary production, latency, and ciHHV-6) is required to permit significant advances in this domain. The main indications of serologic assays currently remain the diagnosis of primary infection, identification of HHV-6-naive subjects, and use for seroprevalence studies. Consequently, HHV-6 serology has limited usefulness in the management of adult infections (77).
Direct Diagnosis
The isolation of HHV-6A, HHV-6B, and HHV-7 in cell cultures is a reference method and unambiguously demonstrates the presence of infectious viral particles in a sample. However, this method is poorly sensitive, time-consuming, and expensive. It cannot be used for routine diagnosis and is not available in most centers. All HHV-6 isolates theoretically grow on PBMCs or cord blood lymphocytes, leading to a cytopathic effect made of enlarged and refractive cells, which might be missing in some cases. Isolation on other cell types, such as fibroblasts, and growth adaptation on cell lines are possible in some cases but even more difficult than culture on primary permissive cells.
The detection of HHV-6 antigens in PBMCs and tissue biopsy specimens enables observation of viral proteins expressed at different stages of infection and even provides an approximate quantification of this process (117, 118). This is particularly useful for showing active viral infection at the site of tissue lesions by use of immunohistochemistry techniques. However, the panel of available reference antibodies that can be used to develop such assays is limited, and the sensitivity of detection is considered low with current reagents. This explains why current antigen detection studies and histological investigations are performed mainly for research objectives rather than diagnostic procedures.
The detection, quantification, and sequencing of HHV-6 nucleic acids have been combined to become the gold standard of diagnostic procedures applied to HHV-6A and HHV-6B. By means of numerous assays based on real-time PCR, HHV-6 DNA can be detected and reproducibly quantified in a broad range of clinical specimens, including whole blood, cerebrospinal fluid, and any other bodily fluid or tissue (110, 119, 120). The methods are financially accessible, quick, safe, and currently widespread. Most of the previous problems related to the nonspecific inhibition of DNA amplification and carryover inside labs have now been solved. In addition, these approaches also readily permit differentiation of the two species of HHV-6, even in cases of mixed infection (121). However, there is an obvious need for standardization of the various molecular assays in use in order to permit the unambiguous comparison and interpretation of results obtained in different laboratories; in that context, the introduction of a reference international standard would constitute a significant advance, as observed recently for HCMV diagnosis (122).
The detection and quantification of viral transcripts appear to comprise a valuable complementary approach, with the theoretical possibility of recognizing the different steps of the virus cycle. With that purpose, the detection of late gene transcripts would help to identify productive infections, whereas the finding of a remotely detectable amount of U94 transcripts would reveal a predominantly latent phase of infection. The detection of transcripts is facilitated by targeting of spliced mRNAs, with the amplimers obtained from transcripts thus being distinctly shorter than those amplified from genes containing introns. However, the knowledge of the different transcription patterns of HHV-6 is currently far from complete, and the methods of mRNA quantification are not yet properly standardized.
The need for a precise molecular characterization of HHV-6 DNA and transcripts has emerged from recent questions addressing the epidemiology and physiology of HHV-6 infections. These questions concern the classification of HHV-6A and HHV-6B into two different species, as well as the understanding of transplacentally acquired congenital infection and reactivation from the ciHHV-6 stage (29, 35, 69). The general strategy is the combination of gene amplification, nucleotide sequencing, and phylogenic study of selected loci of HHV-6 DNA. This also allows the genetic recognition of resistance to antivirals by targeting the search for specific mutations known to confer a decreased susceptibility to drugs (123, 124). The most recent developments of molecular techniques, namely, droplet digital PCR and next-generation sequencing, will offer novel opportunities for the accurate investigation of infection pathophysiology. In particular, these developments will permit us to address complex molecular phenomena, such as coinfections of HHV-6A and HHV-B, ciHHV-6, and modulation of gene expression (125–127). In addition, they might help to map chromosomal integration sites in ciHHV-6 subjects in parallel with the classical cytogenetic approach, which uses fluorescence in situ hybridization.
INTERPRETING VIROLOGICAL RESULTS
Active Infection
One goal of virological investigations is the distinction between active (or acute) and latent infections, although this distinction is made difficult by the absence of a neat frontier between the two stages of infection. In terms of pathophysiology, active infections correspond to primary infections, endogenous reactivations, and exogenous reinfections (11). They tend to attract a great deal of attention because they are more accessible to current direct diagnosis procedures, are potential targets for specific antiviral therapy, which so far is directed mainly against HHV-6 DNA replication, and can be correlated more convincingly with concomitant disease.
Detectable viremia is generally considered the hallmark of a systemic active infection (4). According to several authors, whole blood is the most valuable specimen for detecting such viremia by means of real-time PCR (120, 128). Alternatively, the use of PBMCs for such detection is justified, since HHV-6 remains mostly cell associated during active infection. In contrast, the use of plasma, albeit more convenient, raises controversy regarding the origin of viral DNA present in this blood compartment, corresponding to either a true virus production from lymphoid tissue or an incidental DNA release from the lysis of circulating cells. The high sensitivity of real-time PCR and ubiquitous presence of latent HHV-6 infection in adults lead to a high frequency of positive qualitative detection of viral DNA. Therefore, for a proper interpretation, the HHV-6 DNA load in blood has to be computed precisely. So far, no threshold has formally been defined as the frontier between latent and active infections. As a preliminary approximation, the threshold of 1,000 genome-equivalent copies per ml of whole blood delineates a fluctuating gray zone separating the two stages of HHV-6 infection (129–131). This broadly corresponds to 1,000 copies per million PBMCs when the white blood cell count is within the normal range. Viral loads corresponding to active infections are thus located within a wide range, from 1,000 up to several hundreds of thousands of copies per ml of whole blood. These values may be confused with those resulting from ciHHV-6. Indeed, in the few individuals who have ciHHV-6, HHV-6 DNA is present in every nucleated cell, in particular cells from hair follicles, the CNS, and peripheral blood. In ciHHV-6 subjects, HHV-6 DNAemia in whole blood usually exceeds 1 million copies per ml, which is far beyond the values observed in most active infections (120, 132). However, when the white blood cell count is significantly decreased, as observed in hematopoietic stem cell transplant (HSCT) recipients, the results for absolute viral loads in blood may be ambiguous. In this case, the expression of results as numbers of copies per cell, with values of 1 or more, may help to distinguish ciHHV-6 from most active infections, in which the average viral load per cell is usually much lower (133). The use of droplet digital PCR may also be valuable for the identification of ciHHV-6 by precisely determining the ratio of HHV-6 DNA to cellular DNA without the use of a standard curve (126, 134).
In the case of active infection limited to a specific body compartment, viral loads in whole blood and in the concerned compartment (bodily fluid, cell fraction, or organ biopsy specimen) have to be compared with each other in order to estimate the respective contributions of blood input and in situ virus multiplication. In this situation, it is admitted that the value of viremia does not need to be significantly high to establish the diagnosis of active infection. Accordingly, due to the sensitivity and specificity of current PCR techniques, the detection of HHV-6 DNA in CSF is considered sufficient for the diagnosis of an active infection of the CNS, regardless of the level of viremia (135, 136). This raises the question of possible restricted local reactivations in the CNS, as suggested in some cases for HHV-6 encephalitis after HSCT (137, 138). In this context, the quantification of HHV-6 DNA in CSF, instead of its simple qualitative detection, may be worthy, particularly in cases of longitudinal follow-up. In ciHHV-6 patients, unusually large numbers of HHV-6 DNA copies are observed in body fluids, which may be interpreted falsely as acute infection in these body compartments: this has been described in particular for CSF, with the risk of misdiagnosing CNS infections (139).
The detection and/or quantification of transcripts specific to a productive viral cycle, for instance, late gene transcripts, would be another way to characterize active infections. Several requirements have to be satisfied for that purpose, namely, the specificity of target mRNAs as markers of active infection in any cell or tissue, the definition of clear cutoff values for quantitative analyses, and the standardization of quantification procedures. For now, these criteria are not fulfilled. The same comment can be made regarding the detection of specific virus-encoded proteins by means of their antigenic properties or by mass spectrometry techniques (140). Moreover, detection procedures based on proteins raise the additional challenge of a reduced sensitivity compared with that of studies of nucleic acids.
Latent Infection
Latent HHV-6 infection corresponds to the widespread chronic infection classically described for all human herpesviruses, although its molecular definition at the cellular level is far from clear and cannot be restricted solely to episomal persistence in the absence of any gene expression (see above). It also corresponds to the existence of ciHHV-6, which is present in only a minority of the general population. In the latter case, the potential pathogenic effects of HHV-6 may affect any tissue and organ, far beyond the usual main locations of community-acquired infection, which particularly involves lymphoid tissue and the CNS. Concerning the clinical syndromes possibly related to latent infection, our current understanding of the disease mechanism is extremely limited, but it hypothetically tends to invoke discrete expression of viral genes, an inflammatory response, and immune dysfunction rather than a modest but present HHV-6 multiplication.
Seropositivity for HHV-6 in the absence of active infection markers thus may appear to be a convenient marker for latent infection. However, there are limitations of serologic assays, as mentioned above. In addition, the production of serum antibodies in some ciHHV-6 individuals is affected, presumably through a phenomenon of immune tolerance, which constitutes an additional challenge for diagnosis (66). As indicated previously, the presence and amount of U94 transcripts might reflect episomal latency and, in some cases, ciHHV-6. Moreover, a recombinant U94 protein has been used to create an ELISA that would be suitable for exploring the antiviral immune response in HHV-6-related chronic diseases (141). However, the relevance of U94-related markers remains to be demonstrated formally.
The spectrum of other virological findings observed during in vivo latent infection is theoretically wide but practically unknown. Except for the detection of HHV-6 DNA in ciHHV-6 subjects, the major hindrance for the detection of direct viral markers is their intrinsically low level of expression. In that context, indirect markers related to HHV-6-related immune responses may reveal interesting features, provided that studies are founded on relevant pathophysiological hypotheses, large human populations, and numerous controls. Accordingly, particular attention has been paid to viral factors potentially involved either directly or indirectly in chronic diseases, in particular those presumably stimulating inflammation, cell transformation, and autoimmunity processes.
The association of HHV-6 with cell transformation and cancer has been a debated question since the early times of virus discovery (142). HHV-6 has direct transforming capacities in vitro, which have been mapped to the DR7 gene, also designated ORF1 (73, 143). As mentioned previously, the virus can transactivate the expression of other viruses, such as EBV and human herpesvirus 8 (HHV-8), which are truly oncogenic viruses (144, 145). In addition, the special context of ciHHV-6 may offer the opportunity to express specific HHV-6 genes in particular cells which do not express virus receptors and usually escape from exogenous infection. However, to date, no report has mentioned an increased frequency of malignancies in ciHHV-6 subject populations. Finally, the ability of HHV-6 to promote oncogenesis even in the absence of direct transforming activity may rely on its inflammatory properties, as discussed above. This provides an incredibly large number of cellular signaling pathways to explore. Similarly, concerning autoimmunity, multiple mechanisms involving HHV-6 either directly or indirectly have been hypothesized (142). A molecular mimicry which might result in cross-reactivity between self and viral epitopes has been found between the sequences of the U24 protein and myelin basic protein (146). The reactivity of HHV-6-specific T cells against infected cells, as investigated in vitro by lymphoproliferative responses to viral antigens, may induce a bystander activation or damage of uninfected cells present within the same microenvironment. The interaction of HHV-6A with its CD46 receptor, which is a regulator of complement activation, may cause an upregulation of this process, including deleterious lytic cell effects and an increase of the soluble form of CD46 (147). The dysfunction of HHV-6-induced immune responses leading to autoimmune processes would involve not only specific T cell responses but also NK cell-mediated activation and killing (148). All these phenomena would converge to promote release of self antigens and polyclonal activation.
HHV-6 Species
It is now highly recommended to identify the causative HHV-6 species as HHV-6A, HHV-6B, or a mixture of both when a diagnosis of HHV-6 infection is made (22). Knowing the causative HHV-6 species precisely has no impact on diagnosis or management at present. Nevertheless, this information will prove valuable in defining the spectrum of diseases associated with each of the viruses and in the event that virus-specific therapeutic options become available. Any HHV-6 isolate can be classified unambiguously as HHV-6A or HHV-6B by using diverse methods based mainly on PCR and/or nucleotide sequencing. Over 2 decades, many publications concerning in vivo studies have neglected this differentiation step, which is still not possible using conventional serologic assays. As a result, our knowledge of the specific epidemiology and pathophysiology of each HHV-6 species is far from complete. Concomitant or sequential infections with the two species can occur, as well as simultaneous infection with two different strains of the same species, which increases the complexity of this analysis. As a whole, HHV-6B is by far the most frequently detected species in peripheral blood, saliva, and CSF, both for asymptomatic infections and for diseases potentially associated with HHV-6 (3, 9, 121, 149). In most populations studied, HHV-6B also appears to be the first species acquired early in life and the quasi-exclusive agent of exanthema subitum, the prototypic disease associated with HHV-6 primary infection (see below) (3, 9). Whether that predominance of HHV-6B detection is due to technical and physiological constraints or reflects a real higher involvement of HHV-6B over HHV-6A within human HHV-6 infections remains an enigma.
Other Favoring and Confounding Factors
Immunosuppression is the major factor promoting endogenous HHV-6 reactivations, so this virus is a true opportunistic pathogen (3, 135, 150). Such pathogenicity has been observed most frequently in situations in which the functions of cell immunity are impaired, in particular in HSCT and solid organ transplant (SOT) recipients as well as HIV-infected individuals. Except for the physiological loss of maternal immunity, which opens the way to primary infection during infancy, no other circumstances, including pregnancy and long-term corticosteroid therapy, have been reported to favor active HHV-6 infection. Conversely, it is worth mentioning that HHV-6 reactivations can be detected in immunocompetent healthy individuals, as evidenced by elevated HHV-6 DNA loads in blood and saliva in the absence of any ciHHV-6 (9, 151, 152).
As mentioned previously, HHV-6 itself is presumably an immunomodulatory virus that is capable of complex interactions with the immune system and other pathogens, particularly HCMV (150). These intricate interactions may induce immune dysfunctions, which may be translated into specific diseases appearing, at least in part, to be indirect effects of HHV-6 infection (Table 2). As examples, HHV-6 reactivations boost the activity of the immune system, which may lead to graft rejection in transplant recipients, hypersensitivity syndrome in the context of exposure to particular drugs, or, in contrast, enhancement of HCMV disease (150).
TABLE 2.
Clinical syndromes consistently (C) or hypothetically (H) associated with HHV-6 infections
| Stage of HHV-6 infection | Disease or symptoma |
|---|---|
| Primary infection (congenital) | Abnormalities at birth and during immediate postnatal period (H) |
| CNS developmental defects (H) | |
| Primary infection (postnatal) | Exanthema subitum (roseola infantum, sixth disease) (C) |
| Fever, seizures (C) | |
| Mild gastrointestinal and respiratory tract symptoms (C) | |
| Thrombocytopenia, infectious mononucleosis-like syndrome (C) | |
| Hepatitis, gastroenteritis, colitis (C) | |
| Meningoencephalitis and encephalitis (C) | |
| Hemophagocytic syndrome (H) | |
| Temporal lobe epilepsy (H) | |
| Reactivation (and possible reinfection) | Fever (C) |
| Rash (C) | |
| Thrombocytopenia, leukopenia, anemia, bone marrow suppression (C) | |
| Hepatitis (C) | |
| Encephalitis, neurocognitive dysfunction (C) | |
| Retinitis (C) | |
| Pneumonitis (C) | |
| Drug-induced hypersensitivity syndrome (C) | |
| Gastroenteritis, colitis (H) | |
| Temporal lobe epilepsy (H) | |
| Allograft rejection (H) | |
| Graft-versus-host disease (H) | |
| Thrombotic microangiopathy (H) | |
| Higher incidence and severity of infections with HCMV, fungi, and other opportunistic pathogens (H) | |
| Chronic latent infection (with possible sporadic reactivations) | Multiple sclerosis (H) |
| Hashimoto's thyroiditis (H) | |
| Myocarditis, cardiomyopathy (H) | |
| Chronic fatigue syndrome (H) | |
| Acceleration of evolution to AIDS in HIV-positive individuals (H) | |
| Hodgkin's disease (H) |
HCMV, human cytomegalovirus; C or H, consistent or hypothetical association with HHV-6 (keeping in mind that robust large-scale studies are lacking in most cases, which makes this classification prone to frequent changes and subject to personal interpretation).
In addition to sequential infections of most subjects by HHV-6B and HHV-6A, the existence of exogenous HHV-6 reinfections can be speculated from reports of intraspecies recombination and dual infection with two distinct strains of the same species (3, 29, 36). However, it is not known whether these exogenous reinfections are characterized by a particular pattern regarding replication dynamics, serologic responses, establishment of latency, and putative interactions with ciHHV-6 compared with endogenous reactivations. In order to clarify these questions, the design of future investigations of active infections may include not only the identification of the causative HHV-6 species but also the precise characterization of the involved strain by means of molecular epidemiology techniques.
CLINICAL IMPACTS OF HHV-6 INFECTIONS
Primary Infections
Since the initial report in 1988, converging data have proven that HHV-6 primary infections cause acute febrile diseases in young children of 6 months to 3 years of age, with the most emblematic one being exanthema subitum (also known as roseola infantum or sixth disease) (20, 78, 153). Other, less typical combinations of fever, seizures, skin rash, and gastrointestinal and respiratory tract symptoms have been reported, with most of them leading to a quick, favorable outcome (Table 2). As a whole, HHV-6 primary infections account for 10 to 20% of febrile illnesses at this age, and the majority of these infections are associated with clinical symptoms, in disagreement with the previous assumption that the majority of these infections are asymptomatic (9, 80). The causative relationship between active HHV-6 infection and disease has been established from the temporal association between the following clinical and virological findings: evidence of viremia, demonstrated first by means of virus isolation and next by PCR, at the acute phase of illness; presence of HHV-6 DNA in the CSF of children with seizures, pointing to direct infection of the CNS; and seroconversion observed after recovery (3). For still unknown reasons, the clinical symptoms might differ slightly according to country: the rate of exanthema subitum associated with primary infection seems to be higher in Japan than in the United States (153). As mentioned above, the responsible variant was found to be HHV-6B in most cases. Primary infections with HHV-6A have been reported much less frequently, and it is generally assumed that HHV-6A is acquired after HHV-6B, through an asymptomatic infection. However, symptomatic primary infections with HHV-6A have been described for children in the United States as well as in sub-Saharan Africa (154–156).
Infrequently, primary infection is associated with a more severe disease, such as hepatitis, including fulminant forms in some cases, thrombocytopenia, infectious mononucleosis-like syndrome, hemophagocytic syndrome, gastroenteritis, colitis, or myocarditis (3, 9, 153).
Neurological complications include meningoencephalitis and encephalitis, which represent ultimate stages of CNS involvement during primary infection and whose outcomes remain uncertain (6, 9, 78). A nationwide survey in Japan showed an unexpectedly poor prognosis for exanthema subitum-associated encephalitis (157). In addition, two ultimately fatal cases of encephalopathy following acute HHV-6 infection were recently reported for two children with underlying genetic mitochondrial disorders (158). As in benign forms of CNS involvement, the recognition of a concomitant active infection permits the etiological diagnosis and is founded on a combination of virological parameters which include the finding of HHV-6 DNA in blood, saliva, and CSF, as well as specific seroconversion and IgM detection (3, 136). The question of active virus replication has been more difficult to address in considering cases of mesial temporal lobe epilepsy which have been related to HHV-6B in children: there was no evidence of systemic viral infection, but high levels of HHV-6 DNA were detected in the hippocampal regions following temporal lobectomy, suggesting an active local multiplication of the virus, possibly focused in astrocytes (6, 42). The role of HHV-6 in febrile status epilepticus, putatively related to encephalitis, was addressed among 169 children in the context of a multicenter prospective study: 54 children (32%) were found to have HHV-6B viremia, while none had HHV-6A infection, and no HHV-6 DNA was detected in any CSF specimen, keeping the question of subsequent development of hippocampal sclerosis and temporal lobe epilepsy under investigation (78, 159). HHV-6 primary infections seem infrequent among adults due to the nearly universal occurrence of this event in early childhood. The question is whether clinical manifestations are usually as benign as those observed in most children. The diagnosis of encephalitis associated with primary infection is less likely to be suspected in this context, whereas at least one case of mononucleosis-like syndrome has been reported for an adult (83, 160).
Congenital HHV-6 infection following primary infection of the embryo or fetus during pregnancy has been found to occur in about 1% of children, a frequency close to that observed for HCMV. In contrast with the case for HCMV, few data are available concerning associated diseases. No symptomatic early infection has been reported, even for children who exhibit detectable active infection at birth (9, 82). However, it was recently reported that HHV-6 congenital infection was associated with lower scores on the Bayley scale of infant development II MDI at 12 months of age, which requires further confirmation (161). Congenital infection results from either the germ line transmission of ciHHV-6 (i.e., congenital ciHHV-6) or transplacental passage of the virus following endogenous reactivation (or putative exogenous reinfection) in the mother. Unexpectedly, in view of its low frequency in adults, ciHHV-6 has been found to be the predominant context associated with congenital HHV-6 infection (162, 163). In addition, HHV-6A was detected significantly more frequently in congenital infection than in postnatally acquired infection, and in a study limited to 6 children with transplacentally acquired infection, all the mothers had ciHHV-6. These data have led to a picture of congenital infection which appears to be far more complex than that of HCMV and in which ciHHV-6 plays a prominent role (162, 163). These results also raise numerous questions, which mainly refer to the capability of ciHHV-6 to feed virus reactivation, tolerate exogenous HHV-6 reinfection, induce protective immunity, and enhance potential developmental deficits, particularly in the CNS.
Reactivations and Reinfections
Active HHV-6 infections often remain totally asymptomatic. Together with the fact that HHV-6 infections are ubiquitous and highly prevalent, this explains the difficulty in unambiguously establishing the causative role of the virus in clinical manifestations concomitant with active infections. However, the following elements may help to demonstrate or at least cause one to suspect the responsibility of HHV-6 in a given disease: a favoring condition, such as a defect of cellular immunity; the temporal convergence between the clinical events and dynamics of viral replication; the correspondence between the nature of symptoms and virus tissue tropism; the absence of any other pathogen known to be a cause of the disease; and the consistency of association between active infection and disease in a similar context.
The clinical symptoms associated with HHV-6 reactivations in transplant recipients appear to occur in a minority of patients but are involved in a wide spectrum of syndromes (164–166). Some symptoms may be considered nonspecific, such as fever, rash, and transiently decreased numbers of circulating blood cells belonging to the granulocyte/macrophage, erythroid, and megakaryocytic lineages (Table 2). In contrast, subacute limbic encephalitis and delayed engraftment are now recognized as typical opportunistic diseases due to HHV-6 reactivation in HSCT recipients (135, 150, 160). These patients, particularly those receiving allogeneic transplants, are at high risk of developing a reactivation within the first 4 weeks after cell transfer and, subsequently, suffering a life-threatening illness concerning the CNS and/or bone marrow, two well-known sites of HHV-6 latency. However, encephalitis develops in only a small proportion of patients experiencing HHV-6 reactivation. HHV-6 encephalitis is usually diagnosed by the detection of viral DNA in CSF, HHV-6 viremia, and abnormal magnetic resonance imaging findings in temporal lobes, with the onset of symptoms occurring at the end of the first month after transplant, on average. The majority of cases are due to HHV-6B. Additional evidence for the direct effect of HHV-6 on the CNS came from a study which showed a strong correlation between HHV-6 reactivation and CNS dysfunction as measured by delirium and neurocognitive decline in HSCT patients (167). Bone marrow suppression in HSCT patients is the other major complication associated with HHV-6 reactivation, which may evolve to secondary graft failure. Other serious illnesses, such as graft-versus-host disease, thrombotic microangiopathy, HCMV reactivation, and gastrointestinal disease, have been found to be associated with HHV-6 reactivation, but the causative relationship is less clear and warrants further investigations.
HHV-6 reactivations have also been detected frequently in SOT patients, but with different rates according to the characteristics of the transplanted organ, the nature of immunosuppressive therapy, and the administration of prophylactic anti-HCMV treatment, which is presumably also active against HHV-6 (3, 150, 168). HHV-6B again remains the main species detected. Concomitant febrile episodes and other nonspecific symptoms, such as leukopenia and thrombocytopenia, have often been observed (164). Specific severe diseases have been reported for SOT patients, but the causative relationship with HHV-6 does not appear to be as convincing as that for HSCT patients. In renal transplant recipients, the spectrum of clinical manifestations seems limited, whereas hepatitis, pneumonitis, bone marrow suppression, and encephalitis have been reported for liver, lung, and heart transplant patients (168, 169). However, a synergistic pathogenic role of HCMV could not be ruled out in all cases. The role of HHV-6 in graft rejection remains a debated question, as is the facilitation of superinfections with fungi and other opportunistic pathogens (7, 164).
For HIV-positive patients, the clinical impact of HHV-6 reactivation was considered an important question before the development of antiretroviral therapy, in parallel with the major pathogenic role observed for HCMV in this context. An increased frequency of active infections was observed in late stages of the course of AIDS but was questioned for earlier stages (170, 171). As a whole, the capability of HHV-6 to accelerate the progression to AIDS remains controversial, while its opportunistic role among AIDS-associated infections was convincingly demonstrated by reports of cases of encephalitis, pneumonitis, and retinitis (172–175).
Drug-induced hypersensitivity syndrome (also known as drug rash with eosinophilia and systemic symptoms [DRESS]) appears to often be associated with active HHV-6 infection (176–178). The disease is constituted of severe adverse drug reactions associated to various degrees with skin rash, fever, lymph node enlargement, liver dysfunction, and blood leukocyte abnormalities. The strength of the association between virus and disease has been recognized by authors from Japan, who have included HHV-6 active infection in the list of criteria used for the diagnosis of this syndrome, a proposal which is not yet universally accepted (179). The point is to understand how active viral infection interacts with drug exposure and immune dysfunction to generate the disease. The possible role of EBV, another human herpesvirus, in this syndrome has also been suggested, which enables the proposal of a scenario founded on herpesvirus reactivation (179, 180). The starting event would be the triggering of virus multiplication by the drug, thus resulting in immune activation by viral antigens and extensive antiviral T cell responses at the origin of the disease. Therefore, it is tempting to speculate that a similar mechanism is possible for HHV-6, a hypothesis supported by the finding that drugs inducing the syndrome, such as amoxicillin and sodium valproate, are capable of directly stimulating HHV-6 replication (4, 181, 182).
Chronic Infections
Numerous challenges arise in considering the chronic clinical manifestations that can be related either directly or indirectly to the persistence of HHV-6 in the body, apart from or independent of clear-cut episodes of active viral infection. This definition makes the demonstration of HHV-6 as the cause of these diseases extremely difficult. Again, the main obstacle comes from the fact that HHV-6 infection occurs early in life, is nearly universal, and is lifelong. Therefore, many of the criteria essential for evaluating causality are lacking, in particular the specificity and temporality of associations between the virus and symptoms. In addition, the current limitations of investigational tools concern the interpretation of results regarding viral loads, transcripts, and immune markers, as well as the low level of viral expression during latent infection, the existence of two HHV-6 species, and the lack of relevant animal models.
The role of HHV-6 as a possible trigger for multiple sclerosis (MS), an inflammatory demyelinating disease of the CNS, has been debated for a long time (6, 183, 184). MS is an autoimmune condition whose pathogenesis seems to be multifactorial, with numerous environmental factors being suspected to act in the genesis, development, and relapses of the disease. Among them, HHV-6 appears to be one of the most serious candidates in the category of infectious triggers, on the basis of immunological, virological, and experimental data (6). These data include the isolation of this virus from diseased CNS tissues of MS patients, serologic studies of HHV-6 antibody reactivity in their sera and CSF, and in situ detection of HHV-6 DNA transcripts (141, 183, 185–188). This hypothesis also results from general considerations founded on the neurotropism of the virus combined with its theoretical capacity to induce both neuroinflammation and autoimmunity. However, to date, no definite proof of this causative role has been provided (184). Such a proof would open the way to novel MS therapies based on specific antiviral effectors.
Hashimoto's thyroiditis, also known as chronic lymphocytic thyroiditis, is another autoimmune disease in which viral infections are suspected to act as environmental triggers (10). A recent study suggested the putative role of HHV-6A infection (148). The virus was detected more frequently in thyroid cells of patients suffering from Hashimoto's thyroiditis than in those of control patients, such as patients with Graves' disease or multinodular goiter. This study also provided evidence that HHV-6-infected thyroid cells were susceptible to NK cell-mediated cell killing, suggesting a possible mechanism for autoimmunity induction.
The association of HHV-6 with cardiac and vascular diseases has been suggested by several authors. HHV-6 has been listed among the viruses which can cause myocarditis and subsequent chronic cardiomyopathy, as reflected by the frequent detection of the virus in heart muscle biopsy specimens (7, 189–191). HHV-6 is capable of infecting vascular endothelium both in vivo and in vitro, which might support a role in the genesis of diseases affecting coronary and peripheral arteries (47, 192). These effects might be strengthened by the inhibition of angiogenesis induced by U94 gene expression (75).
Chronic fatigue syndrome is a chronic illness causing a major functional impairment whose pathogenesis is incompletely understood (193). The underlying biological abnormalities include markers of chronic immune activation as well as neuroendocrine dysfunction. The possible role of HHV-6 in this syndrome is supported by many studies demonstrating an increased rate of viral reactivation events, as shown by PCR studies of plasma, serum, and CSF (194–196). As expected, the data referring to HHV-6 immune responses are more conflicting. Again, the neurotropism of the virus and its capacity to dysregulate inflammatory responses are indirect arguments for considering its role in the disease but do not appear to be sufficient to recommend HHV-6 therapy in this context.
Regarding malignancies, a putative role of HHV-6 has been discussed for Hodgkin's disease in parallel with the accumulation of data which incriminate EBV (5, 7). This particularly concerns the nodular sclerosis form of the disease in young adults (143, 197). Note that recent results found no association between ciHHV-6 and classical Hodgkin's disease (198). As far as other lymphoproliferative diseases are concerned, a hypothetical role of HHV-6 in the pathogenesis and progression of angioimmunoblastic T cell lymphoma is supported by a higher detection frequency and load of HHV-6 DNA in tumor tissue than those in other lymphomas (199, 200).
TREATMENT OF HHV-6 INFECTIONS
Antiviral Agents
Three drugs initially developed to target HCMV infection have been shown to be efficient against HHV-6 infection both in vitro and in vivo: ganciclovir, foscarnet, and cidofovir (Table 3). Acyclovir, the emblematic antiviral drug against herpes simplex virus and varicella-zoster virus infections, is active against HHV-6 in vitro, but only at very high concentrations. These concentrations cannot readily be obtained in body fluids in vivo, which explains why HHV-6 has to be considered naturally resistant to this agent (201). The three efficient anti-HHV-6 compounds exhibit the same inhibition activity against both HHV-6A and HHV-6B in vitro (3, 202–204). The mechanisms of this antiviral activity are similar for both HCMV and HHV-6: the viral DNA polymerase is specifically inhibited by the triphosphorylated form of ganciclovir, the diphosphorylated form of cidofovir, and foscarnet, in the latter case without any chemical modification.
TABLE 3.
Compounds showing effectiveness against HHV-6
| Druga | Prodrug | Chemical structure | HHV-6 target | EC50b (μM) | Clinical use | Reference(s) |
|---|---|---|---|---|---|---|
| Ganciclovir | Valganciclovir | Nucleoside analogue | DNA polymerase | 2.0–68.6 | Yes (approved for HCMV) | 203, 204 |
| Foscarnet | None | Pyrophosphate analogue | DNA polymerase | 6–86 | Yes (approved for HCMV) | 203, 204 |
| Cidofovir | Brincidofovir | Nucleotide analogue | DNA polymerase | 0.3–46 | Yes (approved for HCMV) | 203, 204 |
| H2G | Valomaciclovir | Nucleoside analogue | DNA polymerase | 36.6 | Yes (clinical trials) | 214–216 |
| Cyclopropavir | 6-Deoxycyclopropavir | Nucleoside analogue | DNA synthesis step | 0.5–7 | No (pharmaceutical development) | 203, 204, 217–219 |
| S2242 | None | Nucleoside analogue | DNA polymerase | 0.02–0.03 | No (pharmaceutical development) | 216, 220 |
| 3-Deaza-HPMPA | None | Nucleotide analogue | DNA polymerase | 1.2–1.4 | No (discontinued development) | 220 |
| CMV423 | None | Indolizinecarboxamide | Tyrosine kinasec | 0.017–0.053 | No (discontinued development) | 203, 204, 222 |
| Artesunate | None | Semisynthetic derivative of artemisinin | Sp1/NF-κB activation pathwaysc | 3.8 | Yes (approved for malaria) | 212, 213 |
No drug is officially approved for use on HHV-6.
EC50, 50% effective dose (range or unique value, according to the data in referenced studies).
Cellular component target.
The first phosphorylation step of ganciclovir is catalyzed by a protein kinase, encoded by the HCMV UL97 or HHV-6 U69 gene, while the further phosphorylation steps of ganciclovir and the two phosphorylation steps of cidofovir are dependent upon the activity of cellular kinases. In agreement with this mechanism of action, acquired virus resistance to these drugs has been related to mutations of target viral genes involved in drug susceptibility for both HCMV and HHV-6: protein kinase and DNA polymerase genes in the case of resistance to ganciclovir and the DNA polymerase gene only in the case of resistance to foscarnet and cidofovir (123, 205–208). Accordingly, the risk of simultaneous resistance of different herpesvirus species to the same drug from the concomitant selection of mutations in homologous genes has to be considered. This was illustrated by a report of HHV-6 resistance to ganciclovir following prolonged treatment of HCMV infection with this drug (205). HHV-6 and HCMV do not express any thymidine kinase, the enzyme which performs the first phosphorylation step for other antiherpetic drugs, such as acyclovir, penciclovir, and brivudin. This explains in part the low activities of those drugs against HCMV and HHV-6.
In addition to their cost and the possibility of selecting drug-resistant viruses, efficient anti-HHV-6 drugs exhibit another drawback which may restrict their use in patients: despite their selectivity with respect to viral enzymes, they exhibit a certain degree of toxicity for human cells and organs, i.e., bone marrow in the case of ganciclovir and the kidneys in the case of foscarnet and cidofovir. The usefulness of these antivirals against diseases thought to be caused by HHV-6 has been reported for humans, but only in the context of uncontrolled studies (5). Therefore, the assumed efficacy of these drugs in vivo is not formally demonstrated, and none is officially approved for treatment of HHV-6 infection. Similarly, the possible synergistic activity of these compounds administered together, for instance, ganciclovir and foscarnet, as previously demonstrated for HCMV, has not yet been proven for HHV-6.
Other nucleoside or nucleotide analogues are potentially efficient drugs, particularly some phosphonate derivatives which are currently under preclinical development or in early clinical trials (203, 209, 210). Brincidofovir, also known as CMX001, is an orally administered lipid-ester derivative of cidofovir which is active against numerous DNA viruses, including HHV-6, is detected at significant levels in CSF from treated patients, and is less toxic for the kidney than cidofovir (137, 204, 211). Artesunate is a derivative of artemisinin, a drug used for the treatment of malaria. It has been proven to inhibit the replication of several human herpesviruses in vitro, in particular HCMV and HHV-6, and has been given to treat HCMV infections in vivo as a compassionate use. Its mechanism of antiviral activity likely involves the modulation of cellular activation pathways involving Sp1 and NF-κB (212, 213). Valomaciclovir, a drug developed initially against varicella-zoster virus, has also shown significant activity against HHV-6 in vitro (214–216). Other antiviral compounds have demonstrated promising experimental anti-HHV-6 activity, including the nucleoside/nucleotide analogues SS2242, A-5021, cyclopropavir, and 3-deaza-HPMPA and the nonnucleoside inhibitor CMV423 (4, 217–222). These are all still at a step of preclinical development.
Recently, the complementary use of immunotherapy, particularly in the context of HSCT, has been considered through the generation of polyclonal cytotoxic T lymphocytes targeted to several opportunistic viruses, including HHV-6 (100, 203). The concept of an adoptive therapy based on these T cells is very attractive, due to the possibility of circumventing drug cytotoxicity and resistance, but requires validation in ongoing clinical trials.
Indications and Management of Clinical HHV-6 Treatments
Independently of any antiviral drug administration, a key factor for the control of active HHV-6 infection is the reversal of immunosuppression when this favoring factor is present and accessible to medical modulation. Thus, reducing the dose of immunosuppressive drugs is a valuable approach to consider when this option is available (11, 223).
The general strategy of antiviral therapy against HHV-6 may be inspired by the current practices of anti-HCMV treatment, thus leading to a choice between prophylactic, preemptive, and curative approaches (Fig. 2). The essential target is active infection, since the available drugs are not efficient against the potential deleterious effects of latent infection. Hence, virological findings provide valuable information in order to define the most convenient strategy. Prophylactic treatment has the advantage of totally protecting at-risk individuals from the deleterious effects of active HHV-6 replication by preventing either primary infection (mainly for children) or reactivation (for most adults). However, this strategy requires the treatment of a large number of individuals, for instance, virtually all immunocompromised patients; induces high costs; and exposes individuals to numerous side effects as well as to emergence of resistance. Conversely, curative therapy is initiated once HHV-6-associated disease has been diagnosed, which concerns fewer people but exposes them to the risk of therapeutic failure due to late intervention. Preemptive therapy is the median solution, with the detection of viral replication at a significant level being the starting signal for therapy prior to any declared disease. Its cost-effectiveness is presumably favorable, but its indications require a precise analysis of HHV-6 replication dynamics. Note that regarding HCMV infection, the early detection of virus reactivation by use of PCR-based molecular tools has permitted successful preemptive therapy, particularly in transplant recipients (122).
FIG 2.

Overview of the three general options for the treatment of HHV-6 infection. The “+” and “−” symbols indicate the presence, absence, and/or level of the indicated parameter.
It is tempting to speculate on the following items as possible additive criteria for initiating therapy: (i) demonstrated active infection with a significantly elevated viral load in the absence of ciHHV-6; (ii) immune suppression context; (iii) concomitant clinical symptoms relating to virus replication on a pathophysiological basis; and (iv) absence of any other causative agent, in particular HCMV, but keeping in mind that ganciclovir, foscarnet, and cidofovir are also active against HCMV infection. As indicated previously, to date, the nature of the HHV-6 species, either HHV-6A or HHV-6B, has not been considered a key feature for treatment decisions. Taking all available data together, it currently seems premature to propose a prophylactic therapy in order to prevent HHV-6 reactivations or a preemptive therapy of virologically proven active infection in order to prevent clinical manifestations. The spontaneous evolution of HHV-6 reactivations and their association with disease are not sufficiently understood to enable such therapeutic strategies. It is obvious that active HHV-6 infections are spontaneously controlled in many circumstances, which emphasizes the risk of giving a treatment without any need. Accordingly, the International Herpesvirus Management Forum and the American Society of Transplantation do not recommend antiviral prophylaxis for HHV-6 infection but promote the initiation of antiviral therapy in case of HHV-6 encephalitis. Intravenous ganciclovir and foscarnet are proposed as first-line drugs (164, 223, 224). However, no other recommendation has been approved internationally for the therapy of active HHV-6 infections while numerous questions are pending. Those questions refer to the precise timing for therapy initiation, the preferred drug to be used, and treatment duration. An additional question is that of maintenance therapy administered in order to prevent relapses after the acute disease has been controlled.
Indeed, the limbic encephalitis of HSCT patients fulfils most of the criteria listed above and obviously is a potential target for antiviral drugs. The severe CNS diseases associated with primary infection in children might be another priority indication, but the data supporting this assumption need to be strengthened. As a general comment, in the case of CNS disease associated with HHV-6 infection, ganciclovir and foscarnet, either alone or in combination, might be preferred over cidofovir, as the ability of the latter drug to penetrate the blood-brain barrier in humans is controversial (225). No study has yet been performed to define the specific dosing and duration of antiviral therapy. Therefore, the patterns of CMV therapy are used in most cases, with an approximate duration of 3 to 4 weeks (165).
Whatever the indication, the effectiveness of antiviral therapy must be monitored by the concomitant regression of clinical symptoms and decrease of viral load, as performed to assess the response to anti-HCMV treatments (122). A divergence between the evolution of disease and that of viral parameters would lead one to suspect a causative agent other than HHV-6 or a pathogenic mechanism distinct from direct effects of active infection. In addition, a therapeutic failure following the prolonged administration of an efficient antiviral drug may lead to suspicion of the emergence of a resistant virus and should encourage performance of a drug susceptibility test based on a phenotypic assay or a genetic analysis (226). The resistance of HHV-6 to antiviral drugs is not currently as crucial a question as it is for HCMV (122). This weak concern about resistance is correlated with the present limited use of antivirals against this virus. However, the risk of HHV-6 resistance resulting from a previous exposure to drugs in the context of other concomitant herpesvirus infections, notably HCMV infection, does exist and must be kept in mind (205).
For any indication, the validation of therapeutic options against HHV-6 infections will require randomized controlled therapeutic trials, the only valuable strategy for establishing formal treatment guidelines (203).
PENDING QUESTIONS AND PERSPECTIVES
Deciphering the Clinical Impact of Chromosomal Integration
In addition to the difficulties in interpreting results for nucleic acid testing diagnosis, ciHHV-6 raises numerous theoretical clinical concerns related to virus reactivation, dysregulation of immune responses, and instability of the cell genome. If proven, these phenomena might significantly alter the frequencies of cancer and inflammatory, autoimmune, or drug-induced hypersensitivity diseases in ciHHV-6 subjects. They also might modify the rate of side effects and the overall prognosis of transplantation and transfusion for patients receiving ciHHV-6 blood cells or organs (66). Therefore, further research is needed to clarify these points and to provide clear information for persons carrying ciHHV-6. This can be done by means of retrospective studies in order to determine whether ciHHV-6 is significantly more frequent in some diseases. Alternatively, prospective studies of cohorts of ciHHV-6 individuals will analyze the occurrences of particular illnesses compared to those for cohorts of control subjects.
Differential Pathogenic Roles of HHV-6A and HHV-6B
HHV-6A and HHV-6B are now defined as distinct virus species (22). Their epidemiologies are also suspected to differ from each other regarding the time of virus acquisition in life, their role in HHV-6 primary infection according to geographic region, their distribution in human tissues, and their modes of human-to-human transmission. Major questions refer to disease associations. Some reports have suggested an increased severity of HHV-6A over HHV-6B, particularly in acute and chronic CNS diseases suspected to be linked with HHV-6 infection. However, only HHV-6B has been found to be associated with mesial temporal lobe epilepsy and status epilepticus, while only HHV-6A has been associated with Hashimoto's thyroiditis (148, 159, 227). It is now essential to make a clear distinction between both species in all research projects regarding HHV-6 epidemiology and pathogenicity. This distinction might provide dramatic insights into our understanding of the various infectious events that are collectively designated HHV-6 infection. This, in turn, would enable us to define specific diagnostic and treatment strategies on a more relevant basis.
Causative Role of HHV-6 in Chronic Diseases
The association of HHV-6 with chronic diseases has been supported by in vivo immunological and molecular findings as well as experimental data from cell models. However, the convincing demonstration of a causative role of HHV-6 in diseases such as MS, Hashimoto's thyroiditis, and chronic cardiomyopathy, either as an initial inducer or as a further trigger, is a huge challenge. The ubiquitous distribution and lifelong duration of HHV-6 infection create major obstacles for case-control and/or prospective studies. Clinical studies based on the potential beneficial effects of anti-HHV-6 drugs or immune effectors in these diseases have very limited options. These limitations are due to the small number of available virus inhibitors, the huge number of parameters to be investigated, and the long duration of experimental therapy, which may expose treated patients to numerous side effects. In addition, complex chronic illnesses may rely on multiple pathogens which are also sensitive to the administered antiviral treatments, such as HCMV and other herpesviruses. This makes the interpretation of therapy effects difficult regarding the possible etiological role of HHV-6.
Algorithms and Priorities for HHV-6 Treatments
Despite the uncertainties regarding HHV-6 pathogenicity and the limitations of therapeutic options, it would be worth setting up algorithms with the double purpose of improving the knowledge about HHV-6 infection and its overall prognosis in patients. The situation which currently appears to be the most favorable for the application of such algorithms is that of active severe HHV-6 infections (Fig. 3). There are many reasons for this. Active HHV-6 replication can be diagnosed confidently and quantified by means of current molecular tools, its dynamics evaluated in sequential samples can be paralleled with clinical symptoms, it might induce highly serious diseases, particularly in immunocompromised patients, and it is a convenient target for inhibitors of viral DNA polymerase. Although no extensive study has been done regarding the kinetics of viral replication in the human host, it can be speculated that the dynamics of quantitative viremia, at least to some extent, may predict HHV-6 disease risk, particularly in immunocompromised patients, as is the case for HCMV (122). However, one must keep in mind that many active HHV-6 infections are asymptomatic, even in the context of immune deficiency, and can revert to latency without any therapy. Moreover, if an active HHV-6 infection is suspected, it is essential to rule out the possibility of ciHHV-6. If the latter case is true, then virus reactivation may still have occurred and may be a potential source of disease. However, the patterns of transcription revealing active virus replication are not completely known, and the quantification of transcripts is not yet standardized, which makes the interpretation of virological findings ambiguous in many cases.
FIG 3.

Hypothetical algorithm for diagnosis and therapy decisions in a situation of severe disease potentially associated with active HHV-6 infection.
The development of extensive laboratory support, including the standardized quantification of viral nucleic acids and in situ detection of viral antigen expression in bodily tissues, is needed. Combined with the initiation of well-designed therapeutic trials, the improvement of laboratory approaches will open wide research perspectives on this pathogenic, albeit ubiquitous, human herpesvirus.
ACKNOWLEDGMENTS
We thank the editors and conference organizers with respect to the previously presented work mentioned in the introduction for giving us the opportunity to share our thoughts with the scientific community. We also express our gratitude to the HHV-6 Foundation, in particular to Dharam Ablashi, Kristin S. Loomis, and all the members of the scientific advisory board, for constant support, constructive comments, and stimulating discussions in the field of HHV-6 research.
This work was supported in part by the Université Pierre et Marie Curie (UPMC) and the Institut National de la Santé et de la Recherche Médicale (INSERM).
Biographies

Henri Agut, M.D., Ph.D., is Head of the Virology Department at La Pitié-Salpêtrière Hospital, Professor of Virology at the University Pierre et Marie Curie, and Leader of the research team PVI, dedicated to the study of persistent viral infections, at the CIMI-Paris INSERM U1135 Center. He serves as an expert of medical virology on several boards and committees dedicated to the study and control of viral diseases. His major interests in research are the genetics and pathogenicity of human viruses, particularly HIV and herpesviruses, with a specific current orientation toward the improvement of diagnostic methods and antiviral therapies. Specifically, in the field of HHV-6 and HHV-7, he has investigated the genetic variability of these viruses, including the differentiation between HHV-6A and HHV-6B, their pathogenic role in immunocompromised patients, the implementation of PCR-based detection and quantification methods, and the mechanisms of resistance to antiherpetic drugs.

Pascale Bonnafous received a Ph.D. in Microbiology-Virology and is now a Research Engineer of the University Pierre et Marie Curie, working on the PVI team of the CIMI-Paris INSERM U1135 Center. She has a long-standing interest in the mechanisms of activity of antibiotics and antiviral agents. In particular, she has developed phenotypic and genetic assays for the study of HHV-6 susceptibility to antivirals and has applied those techniques for the characterization of resistant mutants. She is currently enrolled in a research program studying the resistance of HHV-6 to DNA polymerase inhibitors and a project aiming at a better understanding of the clinical impact of ciHHV-6. She also has extended training experience in the field of health and safety in research laboratories.

Agnès Gautheret-Dejean, Pharm.D., Ph.D., is Associate Professor at the Pharmacy School of the University Paris Descartes, Senior Clinical Virologist in the Virology Department at La Pitié-Salpêtrière Hospital, and Principal Investigator for the PVI team of the CIMI-Paris INSERM U1135 Center. She serves as an expert for the editorial boards of several international virology journals. She is also an expert on the evaluation of viral infection risk concerning the biosafety of laboratory operations and medical products of biological origin. Her current research interests concern both the development of rapid diagnostic assays, particularly in the field of HIV and herpesviruses, and the pathogenesis of HHV-6 infection, especially in the context of ciHHV-6.
REFERENCES
- 1.Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, Halligan G, Biberfeld P, Wong-Staal F, Kramarsky B. 1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234:596–601. doi: 10.1126/science.2876520. [DOI] [PubMed] [Google Scholar]
- 2.Pellett PE, Black JB, Yamamoto M. 1992. Human herpesvirus 6: the virus and the search for its role as a human pathogen. Adv Virus Res 41:1–52. doi: 10.1016/S0065-3527(08)60034-2. [DOI] [PubMed] [Google Scholar]
- 3.De Bolle L, Naesens L, De Clercq E. 2005. Update on human herpesvirus 6 biology, clinical features, and therapy. Clin Microbiol Rev 18:217–245. doi: 10.1128/CMR.18.1.217-245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flamand L, Komaroff AL, Arbuckle JH, Medveczky PG, Ablashi DV. 2010. Human herpesvirus-6—basic biology, diagnostic testing, and antiviral efficacy. J Med Virol 82:1560–1568. doi: 10.1002/jmv.21839. [DOI] [PubMed] [Google Scholar]
- 5.Braun DK, Dominguez G, Pellett PE. 1997. Human herpesvirus 6. Clin Microbiol Rev 10:521–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao K, Crawford JR, Komaroff AL, Ablashi DV, Jacobson S. 2010. Human herpesvirus-6 in central nervous system diseases. J Med Virol 82:1669–1678. doi: 10.1002/jmv.21861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ablashi DV, Devin CL, Yoshikawa T, Lautenschlager I, Luppi M, Kuhl U, Komaroff AL. 2010. Human herpesvirus-6 in multiple non-neurological diseases. J Med Virol 82:1903–1910. doi: 10.1002/jmv.21860. [DOI] [PubMed] [Google Scholar]
- 8.Yamanishi K, Mori Y, Pellett PE. 2013. Human herpesviruses 6 and 7, p 2058–2079. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 9.Zerr DM. 2006. Human herpesvirus 6: a clinical update. Herpes 13:20–24. [PubMed] [Google Scholar]
- 10.Broccolo F. 2014. HHV-6A and HHV-6B in autoimmune disease, p 167–178. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 11.Agut H. 2011. Deciphering the clinical impact of acute human herpesvirus 6 (HHV-6) infections. J Clin Virol 52:164–171. doi: 10.1016/j.jcv.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 12.Biberfeld P, Kramarsky B, Salahuddin SZ, Gallo RC. 1987. Ultrastructural characterization of a new human B lymphotropic DNA virus (human herpesvirus 6) isolated from patients with lymphoproliferative disease. J Natl Cancer Inst 79:933–941. [PubMed] [Google Scholar]
- 13.Josephs SF, Salahuddin SZ, Ablashi DV, Schachter F, Wong-Staal F, Gallo RC. 1986. Genomic analysis of the human B-lymphotropic virus (HBLV). Science 234:601–603. doi: 10.1126/science.3020691. [DOI] [PubMed] [Google Scholar]
- 14.Ablashi DV, Salahuddin SZ, Josephs SF, Imam F, Lusso P, Gallo RC, Hung C, Lemp J, Markham PD. 1987. HBLV (or HHV-6) in human cell lines. Nature 329:207. doi: 10.1038/329207a0. [DOI] [PubMed] [Google Scholar]
- 15.Frenkel N, Schirmer EC, Wyatt LS, Katsafanas G, Roffman E, Danovich RM, June CH. 1990. Isolation of a new herpesvirus from human CD4+ T cells. Proc Natl Acad Sci U S A 87:748–752. doi: 10.1073/pnas.87.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Downing RG, Sewankambo N, Serwadda D, Honess R, Crawford D, Jarrett R, Griffin BE. 1987. Isolation of human lymphotropic herpesviruses from Uganda. Lancet ii:390. [DOI] [PubMed] [Google Scholar]
- 17.Tedder RS, Briggs M, Cameron CH, Honess R, Robertson D, Whittle H. 1987. A novel lymphotropic herpesvirus. Lancet ii:390–392. [DOI] [PubMed] [Google Scholar]
- 18.Agut H, Guetard D, Collandre H, Dauguet C, Montagnier L, Miclea JM, Baurmann H, Gessain A. 1988. Concomitant infection by human herpesvirus 6, HTLV-I, and HIV-2. Lancet i:712. [DOI] [PubMed] [Google Scholar]
- 19.Lopez C, Pellett P, Stewart J, Goldsmith C, Sanderlin K, Black J, Warfield D, Feorino P. 1988. Characteristics of human herpesvirus-6. J Infect Dis 157:1271–1273. doi: 10.1093/infdis/157.6.1271. [DOI] [PubMed] [Google Scholar]
- 20.Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, Kurata T. 1988. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet i:1065–1067. [DOI] [PubMed] [Google Scholar]
- 21.Ablashi D, Agut H, Berneman Z, Campadelli-Fiume G, Carrigan D, Ceccerini-Nelli L, Chandran B, Chou S, Collandre H, Cone R, Dambaugh T, Dewhurst S, DiLuca D, Foa-Tomasi L, Fleckenstein B, Frenkel N, Gallo RC, Gompels UA, Hall C, Jones M, Lawrence G, Martin M, Montagnier L, Neipel F, Nicholas J, Pellett PE, Razzaque A, Torrelli G, Thomson B, Salahuddin SZ, Wyatt L, Yamanishi K. 1993. Human herpesvirus-6 strain groups: a nomenclature. Arch Virol 129:363–366. doi: 10.1007/BF01316913. [DOI] [PubMed] [Google Scholar]
- 22.Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, Diluca D, Flamand L, Frenkel N, Gallo R, Gompels UA, Hollsberg P, Jacobson S, Luppi M, Lusso P, Malnati M, Medveczky P, Mori Y, Pellett PE, Pritchett JC, Yamanishi K, Yoshikawa T. 2014. Classification of HHV-6A and HHV-6B as distinct viruses. Arch Virol 159:863–870. doi: 10.1007/s00705-013-1902-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arbuckle JH, Medveczky MM, Luka J, Hadley SH, Luegmayr A, Ablashi D, Lund TC, Tolar J, De Meirleir K, Montoya JG, Komaroff AL, Ambros PF, Medveczky PG. 2010. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc Natl Acad Sci U S A 107:5563–5568. doi: 10.1073/pnas.0913586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arbuckle JH, Pantry SN, Medveczky MM, Prichett J, Loomis KS, Ablashi D, Medveczky PG. 2013. Mapping the telomere integrated genome of human herpesvirus 6A and 6B. Virology 442:3–11. doi: 10.1016/j.virol.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufer BB, Flamand L. 2014. Chromosomally integrated HHV-6: impact on virus, cell and organismal biology. Curr Opin Virol 9:111–118. doi: 10.1016/j.coviro.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Gompels UA, Nicholas J, Lawrence G, Jones M, Thomson BJ, Martin ME, Efstathiou S, Craxton M, Macaulay HA. 1995. The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology 209:29–51. doi: 10.1006/viro.1995.1228. [DOI] [PubMed] [Google Scholar]
- 27.Dominguez G, Dambaugh TR, Stamey FR, Dewhurst S, Inoue N, Pellett PE. 1999. Human herpesvirus 6B genome sequence: coding content and comparison with human herpesvirus 6A. J Virol 73:8040–8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isegawa Y, Mukai T, Nakano K, Kagawa M, Chen J, Mori Y, Sunagawa T, Kawanishi K, Sashihara J, Hata A, Zou P, Kosuge H, Yamanishi K. 1999. Comparison of the complete DNA sequences of human herpesvirus 6 variants A and B. J Virol 73:8053–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achour A, Malet I, Le Gal F, Dehee A, Gautheret-Dejean A, Bonnafous P, Agut H. 2008. Variability of gB and gH genes of human herpesvirus-6 among clinical specimens. J Med Virol 80:1211–1221. doi: 10.1002/jmv.21205. [DOI] [PubMed] [Google Scholar]
- 30.Gravel A, Gosselin J, Flamand L. 2002. Human herpesvirus 6 immediate-early 1 protein is a sumoylated nuclear phosphoprotein colocalizing with promyelocytic leukemia protein-associated nuclear bodies. J Biol Chem 277:19679–19687. doi: 10.1074/jbc.M200836200. [DOI] [PubMed] [Google Scholar]
- 31.Rapp JC, Krug LT, Inoue N, Dambaugh TR, Pellett PE. 2000. U94, the human herpesvirus 6 homolog of the parvovirus nonstructural gene, is highly conserved among isolates and is expressed at low mRNA levels as a spliced transcript. Virology 268:504–516. doi: 10.1006/viro.1999.0163. [DOI] [PubMed] [Google Scholar]
- 32.Tweedy J, Spyrou MA, Hubacek P, Kuhl U, Lassner D, Gompels UA. 2015. Analyses of germline, chromosomally integrated human herpesvirus 6A and B genomes indicate emergent infection and new inflammatory mediators. J Gen Virol 96:370–389. doi: 10.1099/vir.0.068536-0. [DOI] [PubMed] [Google Scholar]
- 33.Stanton R, Wilkinson GW, Fox JD. 2003. Analysis of human herpesvirus-6 IE1 sequence variation in clinical samples. J Med Virol 71:578–584. doi: 10.1002/jmv.10508. [DOI] [PubMed] [Google Scholar]
- 34.Yamanishi K, Mori Y, Pellett PE. 2007. Human herpesviruses 6 and 7, p 2819–2845. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 35.Gravel A, Hall CB, Flamand L. 2013. Sequence analysis of transplacentally acquired human herpesvirus 6 DNA is consistent with transmission of a chromosomally integrated reactivated virus. J Infect Dis 207:1585–1589. doi: 10.1093/infdis/jit060. [DOI] [PubMed] [Google Scholar]
- 36.Achour A, Malet I, Deback C, Bonnafous P, Boutolleau D, Gautheret-Dejean A, Agut H. 2009. Length variability of telomeric repeat sequences of human herpesvirus 6 DNA. J Virol Methods 159:127–130. doi: 10.1016/j.jviromet.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Kato Y, Ihira M, Umeda M, Higashimoto Y, Kawamura Y, Ohashi M, Ishi J, Yoshikawa T. 2014. Copy numbers of telomeric repeat sequences of human herpesvirus 6B in clinical isolates: possibility of mixed infections. J Clin Microbiol 52:419–424. doi: 10.1128/JCM.02192-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindquester GJ, Pellett PE. 1991. Properties of the human herpesvirus 6 strain Z29 genome: G+C content, length, and presence of variable-length directly repeated terminal sequence elements. Virology 182:102–110. doi: 10.1016/0042-6822(91)90653-S. [DOI] [PubMed] [Google Scholar]
- 39.Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P. 1999. CD46 is a cellular receptor for human herpesvirus 6. Cell 99:817–827. doi: 10.1016/S0092-8674(00)81678-5. [DOI] [PubMed] [Google Scholar]
- 40.Tang H, Serada S, Kawabata A, Ota M, Hayashi E, Naka T, Yamanishi K, Mori Y. 2013. CD134 is a cellular receptor specific for human herpesvirus-6B entry. Proc Natl Acad Sci U S A 110:9096–9099. doi: 10.1073/pnas.1305187110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cattaneo R. 2004. Four viruses, two bacteria, and one receptor: membrane cofactor protein (CD46) as pathogens' magnet. J Virol 78:4385–4388. doi: 10.1128/JVI.78.9.4385-4388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donati D, Akhyani N, Fogdell-Hahn A, Cermelli C, Cassiani-Ingoni R, Vortmeyer A, Heiss JD, Cogen P, Gaillard WD, Sato S, Theodore WH, Jacobson S. 2003. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology 61:1405–1411. doi: 10.1212/01.WNL.0000094357.10782.F9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roush KS, Domiati-Saad RK, Margraf LR, Krisher K, Scheuermann RH, Rogers BB, Dawson DB. 2001. Prevalence and cellular reservoir of latent human herpesvirus 6 in tonsillar lymphoid tissue. Am J Clin Pathol 116:648–654. doi: 10.1309/Y2HH-B1CK-0F5L-U7B8. [DOI] [PubMed] [Google Scholar]
- 44.Fox JD, Briggs M, Ward PA, Tedder RS. 1990. Human herpesvirus 6 in salivary glands. Lancet 336:590–593. doi: 10.1016/0140-6736(90)93392-3. [DOI] [PubMed] [Google Scholar]
- 45.Okuno T, Higashi K, Shiraki K, Yamanishi K, Takahashi M, Kokado Y, Ishibashi M, Takahara S, Sonoda T, Tanaka K. 1990. Human herpesvirus 6 infection in renal transplantation. Transplantation 49:519–522. doi: 10.1097/00007890-199003000-00009. [DOI] [PubMed] [Google Scholar]
- 46.Harma M, Hockerstedt K, Lautenschlager I. 2003. Human herpesvirus-6 and acute liver failure. Transplantation 76:536–539. doi: 10.1097/01.TP.0000069233.13409.DF. [DOI] [PubMed] [Google Scholar]
- 47.Caruso A, Rotola A, Comar M, Favilli F, Galvan M, Tosetti M, Campello C, Caselli E, Alessandri G, Grassi M, Garrafa E, Cassai E, Di Luca D. 2002. HHV-6 infects human aortic and heart microvascular endothelial cells, increasing their ability to secrete proinflammatory chemokines. J Med Virol 67:528–533. doi: 10.1002/jmv.10133. [DOI] [PubMed] [Google Scholar]
- 48.Kondo K, Kondo T, Okuno T, Takahashi M, Yamanishi K. 1991. Latent human herpesvirus 6 infection of human monocytes/macrophages. J Gen Virol 72:1401–1408. doi: 10.1099/0022-1317-72-6-1401. [DOI] [PubMed] [Google Scholar]
- 49.Luppi M, Barozzi P, Maiorana A, Marasca R, Torelli G. 1994. Human herpesvirus 6 infection in normal human brain tissue. J Infect Dis 169:943–944. doi: 10.1093/infdis/169.4.943. [DOI] [PubMed] [Google Scholar]
- 50.Luppi M, Barozzi P, Morris C, Maiorana A, Garber R, Bonacorsi G, Donelli A, Marasca R, Tabilio A, Torelli G. 1999. Human herpesvirus 6 latently infects early bone marrow progenitors in vivo. J Virol 73:754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andre-Garnier E, Milpied N, Boutolleau D, Saiagh S, Billaudel S, Imbert-Marcille BM. 2004. Reactivation of human herpesvirus 6 during ex vivo expansion of circulating CD34+ haematopoietic stem cells. J Gen Virol 85:3333–3336. doi: 10.1099/vir.0.80319-0. [DOI] [PubMed] [Google Scholar]
- 52.Lusso P, Secchiero P, Crowley RW. 1994. In vitro susceptibility of Macaca nemestrina to human herpesvirus 6: a potential animal model of coinfection with primate immunodeficiency viruses. AIDS Res Hum Retroviruses 10:181–187. doi: 10.1089/aid.1994.10.181. [DOI] [PubMed] [Google Scholar]
- 53.Yalcin S, Mukai T, Kondo K, Ami Y, Okawa T, Kojima A, Kurata T, Yamanishi K. 1992. Experimental infection of cynomolgus and African green monkeys with human herpesvirus 6. J Gen Virol 73:1673–1677. doi: 10.1099/0022-1317-73-7-1673. [DOI] [PubMed] [Google Scholar]
- 54.Reynaud JM, Jegou JF, Welsch JC, Horvat B. 2014. Human herpesvirus 6A infection in CD46 transgenic mice: viral persistence in the brain and increased production of proinflammatory chemokines via Toll-like receptor 9. J Virol 88:5421–5436. doi: 10.1128/JVI.03763-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horvat B, Berges BK, Lusso P. 2014. Recent developments in animal models for human herpesvirus 6A and 6B. Curr Opin Virol 9:97–103. doi: 10.1016/j.coviro.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schiewe U, Neipel F, Schreiner D, Fleckenstein B. 1994. Structure and transcription of an immediate-early region in the human herpesvirus 6 genome. J Virol 68:2978–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsao EH, Kellam P, Sin CS, Rasaiyaah J, Griffiths PD, Clark DA. 2009. Microarray-based determination of the lytic cascade of human herpesvirus 6B. J Gen Virol 90:2581–2591. doi: 10.1099/vir.0.012815-0. [DOI] [PubMed] [Google Scholar]
- 58.Tomoiu A, Gravel A, Flamand L. 2006. Mapping of human herpesvirus 6 immediate-early 2 protein transactivation domains. Virology 354:91–102. doi: 10.1016/j.virol.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 59.Chen M, Popescu N, Woodworth C, Berneman Z, Corbellino M, Lusso P, Ablashi DV, DiPaolo JA. 1994. Human herpesvirus 6 infects cervical epithelial cells and transactivates human papillomavirus gene expression. J Virol 68:1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rotola A, Ravaioli T, Gonelli A, Dewhurst S, Cassai E, Di Luca D. 1998. U94 of human herpesvirus 6 is expressed in latently infected peripheral blood mononuclear cells and blocks viral gene expression in transformed lymphocytes in culture. Proc Natl Acad Sci U S A 95:13911–13916. doi: 10.1073/pnas.95.23.13911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kondo K, Shimada K, Sashihara J, Tanaka-Taya K, Yamanishi K. 2002. Identification of human herpesvirus 6 latency-associated transcripts. J Virol 76:4145–4151. doi: 10.1128/JVI.76.8.4145-4151.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daibata M, Taguchi T, Taguchi H, Miyoshi I. 1998. Integration of human herpesvirus 6 in a Burkitt's lymphoma cell line. Br J Haematol 102:1307–1313. doi: 10.1046/j.1365-2141.1998.00903.x. [DOI] [PubMed] [Google Scholar]
- 63.Luppi M, Barozzi P, Marasca R, Torelli G. 1994. Integration of human herpesvirus-6 (HHV-6) genome in chromosome 17 in two lymphoma patients. Leukemia 8(Suppl 1):S41–S45. [PubMed] [Google Scholar]
- 64.Daibata M, Taguchi T, Nemoto Y, Taguchi H, Miyoshi I. 1999. Inheritance of chromosomally integrated human herpesvirus 6 DNA. Blood 94:1545–1549. [PubMed] [Google Scholar]
- 65.Clark DA, Nacheva EP, Leong HN, Brazma D, Li YT, Tsao EH, Buyck HC, Atkinson CE, Lawson HM, Potter MN, Griffiths PD. 2006. Transmission of integrated human herpesvirus 6 through stem cell transplantation: implications for laboratory diagnosis. J Infect Dis 193:912–916. doi: 10.1086/500838. [DOI] [PubMed] [Google Scholar]
- 66.Pellett PE, Ablashi DV, Ambros PF, Agut H, Caserta MT, Descamps V, Flamand L, Gautheret-Dejean A, Hall CB, Kamble RT, Kuehl U, Lassner D, Lautenschlager I, Loomis KS, Luppi M, Lusso P, Medveczky PG, Montoya JG, Mori Y, Ogata M, Pritchett JC, Rogez S, Seto E, Ward KN, Yoshikawa T, Razonable RR. 2012. Chromosomally integrated human herpesvirus 6: questions and answers. Rev Med Virol 22:144–155. doi: 10.1002/rmv.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Godet AN, Soignon G, Koubi H, Bonnafous P, Agut H, Poirot C, Gautheret-Dejean A. 20 May 2014. Presence of HHV-6 genome in spermatozoa in a context of couples with low fertility: what type of infection? Andrologia doi: 10.1111/and.12299. [DOI] [PubMed] [Google Scholar]
- 68.Michou V, Liarmakopoulou S, Thomas D, Tsimaratou K, Makarounis K, Constantoulakis P, Angelopoulou R, Tsilivakos V. 2012. Herpes virus infected spermatozoa following density gradient centrifugation for IVF purposes. Andrologia 44:174–180. doi: 10.1111/j.1439-0272.2010.01121.x. [DOI] [PubMed] [Google Scholar]
- 69.Endo A, Watanabe K, Ohye T, Suzuki K, Matsubara T, Shimizu N, Kurahashi H, Yoshikawa T, Katano H, Inoue N, Imai K, Takagi M, Morio T, Mizutani S. 2014. Molecular and virological evidence of viral activation from chromosomally integrated human herpesvirus 6A in a patient with X-linked severe combined immunodeficiency. Clin Infect Dis 59:545–548. doi: 10.1093/cid/ciu323. [DOI] [PubMed] [Google Scholar]
- 70.Thomson BJ, Efstathiou S, Honess RW. 1991. Acquisition of the human adeno-associated virus type-2 rep gene by human herpesvirus type-6. Nature 351:78–80. doi: 10.1038/351078a0. [DOI] [PubMed] [Google Scholar]
- 71.Flamand L, Romerio F, Reitz MS, Gallo RC. 1998. CD4 promoter transactivation by human herpesvirus 6. J Virol 72:8797–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gravel A, Tomoiu A, Cloutier N, Gosselin J, Flamand L. 2003. Characterization of the immediate-early 2 protein of human herpesvirus 6, a promiscuous transcriptional activator. Virology 308:340–353. doi: 10.1016/S0042-6822(03)00007-2. [DOI] [PubMed] [Google Scholar]
- 73.Kashanchi F, Araujo J, Doniger J, Muralidhar S, Hoch R, Khleif S, Mendelson E, Thompson J, Azumi N, Brady JN, Luppi M, Torelli G, Rosenthal LJ. 1997. Human herpesvirus 6 (HHV-6) ORF-1 transactivating gene exhibits malignant transforming activity and its protein binds to p53. Oncogene 14:359–367. doi: 10.1038/sj.onc.1200840. [DOI] [PubMed] [Google Scholar]
- 74.Mori Y, Dhepakson P, Shimamoto T, Ueda K, Gomi Y, Tani H, Matsuura Y, Yamanishi K. 2000. Expression of human herpesvirus 6B rep within infected cells and binding of its gene product to the TATA-binding protein in vitro and in vivo. J Virol 74:6096–6104. doi: 10.1128/JVI.74.13.6096-6104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caruso A, Caselli E, Fiorentini S, Rotola A, Prandini A, Garrafa E, Saba E, Alessandri G, Cassai E, Di Luca D. 2009. U94 of human herpesvirus 6 inhibits in vitro angiogenesis and lymphangiogenesis. Proc Natl Acad Sci U S A 106:20446–20451. doi: 10.1073/pnas.0905535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yeo WM, Isegawa Y, Chow VT. 2008. The U95 protein of human herpesvirus 6B interacts with human GRIM-19: silencing of U95 expression reduces viral load and abrogates loss of mitochondrial membrane potential. J Virol 82:1011–1020. doi: 10.1128/JVI.01156-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gautheret-Dejean A, Agut H. 2014. Practical diagnostic procedures for HHV-6A, HHV-6B, and HHV-7, p 9–34. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 78.Tesini BL, Epstein LG, Caserta MT. 2014. Clinical impact of primary infection with roseoloviruses. Curr Opin Virol 9:91–96. doi: 10.1016/j.coviro.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aubin JT, Poirel L, Agut H, Huraux JM, Bignozzi C, Brossard Y, Mulliez N, Roume J, Lecuru F, Taurelle R. 1992. Intrauterine transmission of human herpesvirus 6. Lancet 340:482–483. doi: 10.1016/0140-6736(92)91801-E. [DOI] [PubMed] [Google Scholar]
- 80.Hall CB, Long CE, Schnabel KC, Caserta MT, McIntyre KM, Costanzo MA, Knott A, Dewhurst S, Insel RA, Epstein LG. 1994. Human herpesvirus-6 infection in children. A prospective study of complications and reactivation. N Engl J Med 331:432–438. [DOI] [PubMed] [Google Scholar]
- 81.Mendel I, de Matteis M, Bertin C, Delaporte B, Maguer D, Collandre H, Buffet-Janvresse C. 1995. Fulminant hepatitis in neonates with human herpesvirus 6 infection. Pediatr Infect Dis J 14:993–997. doi: 10.1097/00006454-199511000-00013. [DOI] [PubMed] [Google Scholar]
- 82.Hall CB, Caserta MT, Schnabel KC, Boettrich C, McDermott MP, Lofthus GK, Carnahan JA, Dewhurst S. 2004. Congenital infections with human herpesvirus 6 (HHV6) and human herpesvirus 7 (HHV7). J Pediatr 145:472–477. doi: 10.1016/j.jpeds.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 83.Akashi K, Eizuru Y, Sumiyoshi Y, Minematsu T, Hara S, Harada M, Kikuchi M, Niho Y, Minamishima Y. 1993. Brief report: severe infectious mononucleosis-like syndrome and primary human herpesvirus 6 infection in an adult. N Engl J Med 329:168–171. doi: 10.1056/NEJM199307153290304. [DOI] [PubMed] [Google Scholar]
- 84.Ward KN, Gray JJ, Efstathiou S. 1989. Brief report: primary human herpesvirus 6 infection in a patient following liver transplantation from a seropositive donor. J Med Virol 28:69–72. doi: 10.1002/jmv.1890280203. [DOI] [PubMed] [Google Scholar]
- 85.Kusuhara K, Takabayashi A, Ueda K, Hidaka Y, Minamishima I, Take H, Fujioka K, Imai S, Osato T. 1997. Breast milk is not a significant source for early Epstein-Barr virus or human herpesvirus 6 infection in infants: a seroepidemiologic study in 2 endemic areas of human T-cell lymphotropic virus type I in Japan. Microbiol Immunol 41:309–312. doi: 10.1111/j.1348-0421.1997.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 86.Fillet AM, Raphael M, Visse B, Audouin J, Poirel L, Agut H. 1995. Controlled study of human herpesvirus 6 detection in acquired immunodeficiency syndrome-associated non-Hodgkin's lymphoma. The French Study Group for HIV-Associated Tumors. J Med Virol 45:106–112. [DOI] [PubMed] [Google Scholar]
- 87.Cone RW, Huang ML, Hackman RC, Corey L. 1996. Coinfection with human herpesvirus 6 variants A and B in lung tissue. J Clin Microbiol 34:877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lacoste V, Verschoor EJ, Nerrienet E, Gessain A. 2005. A novel homologue of human herpesvirus 6 in chimpanzees. J Gen Virol 86:2135–2140. doi: 10.1099/vir.0.81034-0. [DOI] [PubMed] [Google Scholar]
- 89.Staheli JP, Dyen MR, Lewis P, Barcy S. 2014. Discovery and biological characterization of two novel pig-tailed macaque homologs of HHV-6 and HHV-7. Virology 471–473:126–140. doi: 10.1016/j.virol.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reynaud JM, Horvat B. 2013. Animal models for human herpesvirus 6 infection. Front Microbiol 4:174. doi: 10.3389/fmicb.2013.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leibovitch E, Wohler JE, Cummings Macri SM, Motanic K, Harberts E, Gaitan MI, Maggi P, Ellis M, Westmoreland S, Silva A, Reich DS, Jacobson S. 2013. Novel marmoset (Callithrix jacchus) model of human herpesvirus 6A and 6B infections: immunologic, virologic and radiologic characterization. PLoS Pathog 9:e1003138. doi: 10.1371/journal.ppat.1003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krueger G, Lautenschlager I. 2014. Pathologic features of HHV-6A, HHV-6B, and HHV-7 infection (light and electron microscopy), p 35–67. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 93.Harberts E, Yao K, Wohler JE, Maric D, Ohayon J, Henkin R, Jacobson S. 2011. Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci U S A 108:13734–13739. doi: 10.1073/pnas.1105143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gautheret-Dejean A, Dejean O, Vastel L, Kerboull M, Aubin JT, Franti M, Agut H. 2000. Human herpesvirus-6 and human herpesvirus-7 in the bone marrow from healthy subjects. Transplantation 69:1722–1723. [DOI] [PubMed] [Google Scholar]
- 95.Kondo K, Sashihara J, Shimada K, Takemoto M, Amo K, Miyagawa H, Yamanishi K. 2003. Recognition of a novel stage of betaherpesvirus latency in human herpesvirus 6. J Virol 77:2258–2264. doi: 10.1128/JVI.77.3.2258-2264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Katsafanas GC, Schirmer EC, Wyatt LS, Frenkel N. 1996. In vitro activation of human herpesviruses 6 and 7 from latency. Proc Natl Acad Sci U S A 93:9788–9792. doi: 10.1073/pnas.93.18.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saxinger C, Polesky H, Eby N, Grufferman S, Murphy R, Tegtmeir G, Parekh V, Memon S, Hung C. 1988. Antibody reactivity with HBLV (HHV-6) in U.S. populations. J Virol Methods 21:199–208. doi: 10.1016/0166-0934(88)90066-3. [DOI] [PubMed] [Google Scholar]
- 98.Calvo-Calle JM, Stern LJ. 2014. The immune response to HHV-6, p 235–249. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 99.Becerra A, Gibson L, Stern LJ, Calvo-Calle JM. 2014. Immune response to HHV-6 and implications for immunotherapy. Curr Opin Virol 9:154–161. doi: 10.1016/j.coviro.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gerdemann U, Keukens L, Keirnan JM, Katari UL, Nguyen CT, de Pagter AP, Ramos CA, Kennedy-Nasser A, Gottschalk SM, Heslop HE, Brenner MK, Rooney CM, Leen AM. 2013. Immunotherapeutic strategies to prevent and treat human herpesvirus 6 reactivation after allogeneic stem cell transplantation. Blood 121:207–218. doi: 10.1182/blood-2012-05-430413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nastke MD, Becerra A, Yin L, Dominguez-Amorocho O, Gibson L, Stern LJ, Calvo-Calle JM. 2012. Human CD4+ T cell response to human herpesvirus 6. J Virol 86:4776–4792. doi: 10.1128/JVI.06573-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martin LK, Schub A, Dillinger S, Moosmann A. 2012. Specific CD8(+) T cells recognize human herpesvirus 6B. Eur J Immunol 42:2901–2912. doi: 10.1002/eji.201242439. [DOI] [PubMed] [Google Scholar]
- 103.Flamand L, Stefanescu I, Menezes J. 1996. Human herpesvirus-6 enhances natural killer cell cytotoxicity via IL-15. J Clin Invest 97:1373–1381. doi: 10.1172/JCI118557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Flamand L, Gosselin J, D'Addario M, Hiscott J, Ablashi DV, Gallo RC, Menezes J. 1991. Human herpesvirus 6 induces interleukin-1 beta and tumor necrosis factor alpha, but not interleukin-6, in peripheral blood mononuclear cell cultures. J Virol 65:5105–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dewin DR, Catusse J, Gompels UA. 2006. Identification and characterization of U83A viral chemokine, a broad and potent beta-chemokine agonist for human CCRs with unique selectivity and inhibition by spliced isoform. J Immunol 176:544–556. doi: 10.4049/jimmunol.176.1.544. [DOI] [PubMed] [Google Scholar]
- 106.Catusse J, Spinks J, Mattick C, Dyer A, Laing K, Fitzsimons C, Smit MJ, Gompels UA. 2008. Immunomodulation by herpesvirus U51A chemokine receptor via CCL5 and FOG-2 down-regulation plus XCR1 and CCR7 mimicry in human leukocytes. Eur J Immunol 38:763–777. doi: 10.1002/eji.200737618. [DOI] [PubMed] [Google Scholar]
- 107.Lusso P, Gallo RC. 1995. Human herpesvirus 6 in AIDS. Immunol Today 16:67–71. doi: 10.1016/0167-5699(95)80090-5. [DOI] [PubMed] [Google Scholar]
- 108.Flamand L, Menezes J. 1996. Cyclic AMP-responsive element-dependent activation of Epstein-Barr virus zebra promoter by human herpesvirus 6. J Virol 70:1784–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tai AK, Luka J, Ablashi D, Huber BT. 2009. HHV-6A infection induces expression of HERV-K18-encoded superantigen. J Clin Virol 46:47–48. doi: 10.1016/j.jcv.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 110.Gautheret-Dejean A, Agut H. 2014. Practical diagnostic procedures for HHV-A, HHV-6B, and HHV-7, p 9–34. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 111.Descamps V, Avenel-Audran M, Valeyrie-Allanore L, Bensaid B, Barbaud A, Al Jawhari M, Ranger-Rogez S. 2013. Saliva polymerase chain reaction assay for detection and follow-up of herpesvirus reactivation in patients with drug reaction with eosinophilia and systemic symptoms (DRESS). JAMA Dermatol 149:565–569. doi: 10.1001/jamadermatol.2013.2018. [DOI] [PubMed] [Google Scholar]
- 112.Asada H, Yalcin S, Balachandra K, Higashi K, Yamanishi K. 1989. Establishment of titration system for human herpesvirus 6 and evaluation of neutralizing antibody response to the virus. J Clin Microbiol 27:2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chou SW, Scott KM. 1990. Rises in antibody to human herpesvirus 6 detected by enzyme immunoassay in transplant recipients with primary cytomegalovirus infection. J Clin Microbiol 28:851–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.LaCroix S, Stewart JA, Thouless ME, Black JB. 2000. An immunoblot assay for detection of immunoglobulin M antibody to human herpesvirus 6. Clin Diagn Lab Immunol 7:823–827. doi: 10.1128/CDLI.7.5.823-827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ward KN, Gray JJ, Joslin ME, Sheldon MJ. 1993. Avidity of IgG antibodies to human herpesvirus-6 distinguishes primary from recurrent infection in organ transplant recipients and excludes cross-reactivity with other herpesviruses. J Med Virol 39:44–49. doi: 10.1002/jmv.1890390109. [DOI] [PubMed] [Google Scholar]
- 116.Sutherland S, Christofinis G, O'Grady J, Williams R. 1991. A serological investigation of human herpesvirus 6 infections in liver transplant recipients and the detection of cross-reacting antibodies to cytomegalovirus. J Med Virol 33:172–176. doi: 10.1002/jmv.1890330306. [DOI] [PubMed] [Google Scholar]
- 117.Lautenschlager I, Hockerstedt K, Linnavuori K, Taskinen E. 1998. Human herpesvirus-6 infection after liver transplantation. Clin Infect Dis 26:702–707. doi: 10.1086/514592. [DOI] [PubMed] [Google Scholar]
- 118.Loginov R, Karlsson T, Hockerstedt K, Ablashi D, Lautenschlager I. 2010. Quantitative HHV-6B antigenemia test for the monitoring of transplant patients. Eur J Clin Microbiol Infect Dis 29:881–886. doi: 10.1007/s10096-010-0923-1. [DOI] [PubMed] [Google Scholar]
- 119.Flamand L, Gravel A, Boutolleau D, Alvarez-Lafuente R, Jacobson S, Malnati MS, Kohn D, Tang YW, Yoshikawa T, Ablashi D. 2008. Multicenter comparison of PCR assays for detection of human herpesvirus 6 DNA in serum. J Clin Microbiol 46:2700–2706. doi: 10.1128/JCM.00370-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Caserta MT, Hall CB, Schnabel K, Lofthus G, Marino A, Shelley L, Yoo C, Carnahan J, Anderson L, Wang H. 2010. Diagnostic assays for active infection with human herpesvirus 6 (HHV-6). J Clin Virol 48:55–57. doi: 10.1016/j.jcv.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Boutolleau D, Duros C, Bonnafous P, Caiola D, Karras A, Castro ND, Ouachee M, Narcy P, Gueudin M, Agut H, Gautheret-Dejean A. 2006. Identification of human herpesvirus 6 variants A and B by primer-specific real-time PCR may help to revisit their respective role in pathology. J Clin Virol 35:257–263. doi: 10.1016/j.jcv.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 122.Razonable RR, Hayden RT. 2013. Clinical utility of viral load in management of cytomegalovirus infection after solid organ transplantation. Clin Microbiol Rev 26:703–727. doi: 10.1128/CMR.00015-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bonnafous P, Naesens L, Petrella S, Gautheret-Dejean A, Boutolleau D, Sougakoff W, Agut H. 2007. Different mutations in the HHV-6 DNA polymerase gene accounting for resistance to foscarnet. Antivir Ther 12:877–888. [PubMed] [Google Scholar]
- 124.Isegawa Y, Matsumoto C, Nishinaka K, Nakano K, Tanaka T, Sugimoto N, Ohshima A. 2010. PCR with quenching probes enables the rapid detection and identification of ganciclovir-resistance-causing U69 gene mutations in human herpesvirus 6. Mol Cell Probes 24:167–177. doi: 10.1016/j.mcp.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 125.Leibovitch EC, Brunetto GS, Caruso B, Fenton K, Ohayon J, Reich DS, Jacobson S. 2014. Coinfection of human herpesviruses 6A (HHV-6A) and HHV-6B as demonstrated by novel digital droplet PCR assay. PLoS One 9:e92328. doi: 10.1371/journal.pone.0092328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sedlak RH, Cook L, Huang ML, Magaret A, Zerr DM, Boeckh M, Jerome KR. 2014. Identification of chromosomally integrated human herpesvirus 6 by droplet digital PCR. Clin Chem 60:765–772. doi: 10.1373/clinchem.2013.217240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tuddenham L, Jung JS, Chane-Woon-Ming B, Dolken L, Pfeffer S. 2012. Small RNA deep sequencing identifies microRNAs and other small noncoding RNAs from human herpesvirus 6B. J Virol 86:1638–1649. doi: 10.1128/JVI.05911-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Achour A, Boutolleau D, Gautheret-Dejean A, Agut H. 2006. Is human herpesvirus-6 DNA in plasma the right marker for active infection? J Infect Dis 194:1795–1796. doi: 10.1086/509624. [DOI] [PubMed] [Google Scholar]
- 129.Boutolleau D, Fernandez C, Andre E, Imbert-Marcille BM, Milpied N, Agut H, Gautheret-Dejean A. 2003. Human herpesvirus (HHV)-6 and HHV-7: two closely related viruses with different infection profiles in stem cell transplantation recipients. J Infect Dis 187:179–186. doi: 10.1086/367677. [DOI] [PubMed] [Google Scholar]
- 130.Ljungman P, Wang FZ, Clark DA, Emery VC, Remberger M, Ringden O, Linde A. 2000. High levels of human herpesvirus 6 DNA in peripheral blood leucocytes are correlated to platelet engraftment and disease in allogeneic stem cell transplant patients. Br J Haematol 111:774–781. doi: 10.1111/j.1365-2141.2000.02422.x. [DOI] [PubMed] [Google Scholar]
- 131.Gautheret-Dejean A, Manichanh C, Thien-Ah-Koon F, Fillet AM, Mangeney N, Vidaud M, Dhedin N, Vernant JP, Agut H. 2002. Development of a real-time polymerase chain reaction assay for the diagnosis of human herpesvirus-6 infection and application to bone marrow transplant patients. J Virol Methods 100:27–35. doi: 10.1016/S0166-0934(01)00390-1. [DOI] [PubMed] [Google Scholar]
- 132.Ward KN, Leong HN, Nacheva EP, Howard J, Atkinson CE, Davies NW, Griffiths PD, Clark DA. 2006. Human herpesvirus 6 chromosomal integration in immunocompetent patients results in high levels of viral DNA in blood, sera, and hair follicles. J Clin Microbiol 44:1571–1574. doi: 10.1128/JCM.44.4.1571-1574.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gautheret-Dejean A, Henquell C, Mousnier F, Boutolleau D, Bonnafous P, Dhedin N, Settegrana C, Agut H. 2009. Different expression of human herpesvirus-6 (HHV-6) load in whole blood may have a significant impact on the diagnosis of active infection. J Clin Virol 46:33–36. doi: 10.1016/j.jcv.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 134.Hill JA, Sedlak RH, Jerome KR. 2014. Past, present, and future perspectives on the diagnosis of Roseolovirus infections. Curr Opin Virol 9C:84–90. doi: 10.1016/j.coviro.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ogata M. 2009. Human herpesvirus 6 in hematological malignancies. J Clin Exp Hematop 49:57–67. doi: 10.3960/jslrt.49.57. [DOI] [PubMed] [Google Scholar]
- 136.Yao K, Honarmand S, Espinosa A, Akhyani N, Glaser C, Jacobson S. 2009. Detection of human herpesvirus-6 in cerebrospinal fluid of patients with encephalitis. Ann Neurol 65:257–267. doi: 10.1002/ana.21611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Caserta MT, Dewhurst S. 2011. Dazed and confused by HHV-6. Blood 117:5016–5018. doi: 10.1182/blood-2011-03-342741. [DOI] [PubMed] [Google Scholar]
- 138.Kawamura Y, Sugata K, Ihira M, Mihara T, Mutoh T, Asano Y, Yoshikawa T. 2011. Different characteristics of human herpesvirus 6 encephalitis between primary infection and viral reactivation. J Clin Virol 51:12–19. doi: 10.1016/j.jcv.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 139.Ward KN, Leong HN, Thiruchelvam AD, Atkinson CE, Clark DA. 2007. Human herpesvirus 6 DNA levels in cerebrospinal fluid due to primary infection differ from those due to chromosomal viral integration and have implications for diagnosis of encephalitis. J Clin Microbiol 45:1298–1304. doi: 10.1128/JCM.02115-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Alenda R, Alvarez-Lafuente R, Costa-Frossard L, Arroyo R, Mirete S, Alvarez-Cermeno JC, Villar LM. 2014. Identification of the major HHV-6 antigen recognized by cerebrospinal fluid IgG in multiple sclerosis. Eur J Neurol 21:1096–1101. doi: 10.1111/ene.12435. [DOI] [PubMed] [Google Scholar]
- 141.Caselli E, Boni M, Bracci A, Rotola A, Cermelli C, Castellazzi M, Di Luca D, Cassai E. 2002. Detection of antibodies directed against human herpesvirus 6 U94/REP in sera of patients affected by multiple sclerosis. J Clin Microbiol 40:4131–4137. doi: 10.1128/JCM.40.11.4131-4137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Crawford JR. 2014. HHV-6A and HHV-6B in cancer, p 297–310. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 143.Lacroix A, Collot-Teixeira S, Mardivirin L, Jaccard A, Petit B, Piguet C, Sturtz F, Preux PM, Bordessoule D, Ranger-Rogez S. 2010. Involvement of human herpesvirus-6 variant B in classic Hodgkin's lymphoma via DR7 oncoprotein. Clin Cancer Res 16:4711–4721. doi: 10.1158/1078-0432.CCR-10-0470. [DOI] [PubMed] [Google Scholar]
- 144.Cuomo L, Trivedi P, de Grazia U, Calogero A, D'Onofrio M, Yang W, Frati L, Faggioni A, Rymo L, Ragona G. 1998. Upregulation of Epstein-Barr virus-encoded latent membrane protein by human herpesvirus 6 superinfection of EBV-carrying Burkitt lymphoma cells. J Med Virol 55:219–226. doi:. [DOI] [PubMed] [Google Scholar]
- 145.Lu C, Zeng Y, Huang Z, Huang L, Qian C, Tang G, Qin D. 2005. Human herpesvirus 6 activates lytic cycle replication of Kaposi's sarcoma-associated herpesvirus. Am J Pathol 166:173–183. doi: 10.1016/S0002-9440(10)62242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ. 2003. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol 53:189–197. doi: 10.1002/ana.10425. [DOI] [PubMed] [Google Scholar]
- 147.Soldan SS, Fogdell-Hahn A, Brennan MB, Mittleman BB, Ballerini C, Massacesi L, Seya T, McFarland HF, Jacobson S. 2001. Elevated serum and cerebrospinal fluid levels of soluble human herpesvirus type 6 cellular receptor, membrane cofactor protein, in patients with multiple sclerosis. Ann Neurol 50:486–493. doi: 10.1002/ana.1135. [DOI] [PubMed] [Google Scholar]
- 148.Caselli E, Zatelli MC, Rizzo R, Benedetti S, Martorelli D, Trasforini G, Cassai E, degli Uberti EC, Di Luca D, Dolcetti R. 2012. Virologic and immunologic evidence supporting an association between HHV-6 and Hashimoto's thyroiditis. PLoS Pathog 8:e1002951. doi: 10.1371/journal.ppat.1002951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Reddy S, Manna P. 2005. Quantitative detection and differentiation of human herpesvirus 6 subtypes in bone marrow transplant patients by using a single real-time polymerase chain reaction assay. Biol Blood Marrow Transplant 11:530–541. doi: 10.1016/j.bbmt.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 150.Clark DA, Griffiths PD. 2003. Human herpesvirus 6: relevance of infection in the immunocompromised host. Br J Haematol 120:384–395. doi: 10.1046/j.1365-2141.2003.04048.x. [DOI] [PubMed] [Google Scholar]
- 151.Caserta MT, McDermott MP, Dewhurst S, Schnabel K, Carnahan JA, Gilbert L, Lathan G, Lofthus GK, Hall CB. 2004. Human herpesvirus 6 (HHV6) DNA persistence and reactivation in healthy children. J Pediatr 145:478–484. doi: 10.1016/j.jpeds.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 152.Collot S, Petit B, Bordessoule D, Alain S, Touati M, Denis F, Ranger-Rogez S. 2002. Real-time PCR for quantification of human herpesvirus 6 DNA from lymph nodes and saliva. J Clin Microbiol 40:2445–2451. doi: 10.1128/JCM.40.7.2445-2451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Descamps V, Tohyama M, Shiohara T. 2014. HHV-6A and HHV-6B in drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms, p 179–200. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 154.Hall CB, Caserta MT, Schnabel KC, McDermott MP, Lofthus GK, Carnahan JA, Gilbert LM, Dewhurst S. 2006. Characteristics and acquisition of human herpesvirus (HHV) 7 infections in relation to infection with HHV-6. J Infect Dis 193:1063–1069. doi: 10.1086/503434. [DOI] [PubMed] [Google Scholar]
- 155.Bates M, Monze M, Bima H, Kapambwe M, Clark D, Kasolo FC, Gompels UA. 2009. Predominant human herpesvirus 6 variant A infant infections in an HIV-1 endemic region of sub-Saharan Africa. J Med Virol 81:779–789. doi: 10.1002/jmv.21455. [DOI] [PubMed] [Google Scholar]
- 156.Tembo J, Kabwe M, Chilukutu L, Chilufya M, Mwaanza N, Chabala C, Zumla A, Bates M. 2015. Prevalence and risk factors for betaherpesvirus DNAemia in children >3 weeks and <2 years of age admitted to a large referral hospital in sub-Saharan Africa. Clin Infect Dis 60:423–431. doi: 10.1093/cid/ciu853. [DOI] [PubMed] [Google Scholar]
- 157.Yoshikawa T, Ohashi M, Miyake F, Fujita A, Usui C, Sugata K, Suga S, Hashimoto S, Asano Y. 2009. Exanthem subitum-associated encephalitis: nationwide survey in Japan. Pediatr Neurol 41:353–358. doi: 10.1016/j.pediatrneurol.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 158.Al-Zubeidi D, Thangarajh M, Pathak S, Cai C, Schlaggar BL, Storch GA, Grange DK, Watson ME Jr. 2014. Fatal human herpesvirus 6-associated encephalitis in two boys with underlying POLG mitochondrial disorders. Pediatr Neurol 51:448–452. doi: 10.1016/j.pediatrneurol.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 159.Epstein LG, Shinnar S, Hesdorffer DC, Nordli DR, Hamidullah A, Benn EK, Pellock JM, Frank LM, Lewis DV, Moshe SL, Shinnar RC, Sun S. 2012. Human herpesvirus 6 and 7 in febrile status epilepticus: the FEBSTAT study. Epilepsia 53:1481–1488. doi: 10.1111/j.1528-1167.2012.03542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Flamand L, Lautenschlager I, Krueger G, Ablashi D. 2014. Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 161.Caserta MT, Hall CB, Canfield RL, Davidson P, Lofthus G, Schnabel K, Carnahan J, Shelley L, Wang H. 2014. Early developmental outcomes of children with congenital HHV-6 infection. Pediatrics 134:1111–1118. doi: 10.1542/peds.2014-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hall CB, Caserta MT, Schnabel K, Shelley LM, Marino AS, Carnahan JA, Yoo C, Lofthus GK, McDermott MP. 2008. Chromosomal integration of human herpesvirus 6 is the major mode of congenital human herpesvirus 6 infection. Pediatrics 122:513–520. doi: 10.1542/peds.2007-2838. [DOI] [PubMed] [Google Scholar]
- 163.Hall CB, Caserta MT, Schnabel KC, Shelley LM, Carnahan JA, Marino AS, Yoo C, Lofthus GK. 2010. Transplacental congenital human herpesvirus 6 infection caused by maternal chromosomally integrated virus. J Infect Dis 201:505–507. doi: 10.1086/650495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Razonable RR. 2010. Infections due to human herpesvirus 6 in solid organ transplant recipients. Curr Opin Organ Transplant 15:671–675. doi: 10.1097/MOT.0b013e3283404325. [DOI] [PubMed] [Google Scholar]
- 165.Razonable RR, Lautenschlager I. 2010. Impact of human herpesvirus 6 in liver transplantation. World J Hepatol 2:345–353. doi: 10.4254/wjh.v2.i9.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jeulin H, Agrinier N, Guery M, Salmon A, Clement L, Bordigoni P, Venard V. 2013. Human herpesvirus 6 infection after allogeneic stem cell transplantation: incidence, outcome, and factors associated with HHV-6 reactivation. Transplantation 95:1292–1298. doi: 10.1097/TP.0b013e318289958b. [DOI] [PubMed] [Google Scholar]
- 167.Zerr DM, Fann JR, Breiger D, Boeckh M, Adler AL, Xie H, Delaney C, Huang ML, Corey L, Leisenring WM. 2011. HHV-6 reactivation and its effect on delirium and cognitive functioning in hematopoietic cell transplantation recipients. Blood 117:5243–5249. doi: 10.1182/blood-2010-10-316083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Caiola D, Karras A, Flandre P, Boutolleau D, Scieux C, Agut H, Legendre C, Gautheret-Dejean A. 2012. Confirmation of the low clinical effect of human herpesvirus-6 and -7 infections after renal transplantation. J Med Virol 84:450–456. doi: 10.1002/jmv.23206. [DOI] [PubMed] [Google Scholar]
- 169.Humar A, Malkan G, Moussa G, Greig P, Levy G, Mazzulli T. 2000. Human herpesvirus-6 is associated with cytomegalovirus reactivation in liver transplant recipients. J Infect Dis 181:1450–1453. doi: 10.1086/315391. [DOI] [PubMed] [Google Scholar]
- 170.Corbellino M, Lusso P, Gallo RC, Parravicini C, Galli M, Moroni M. 1993. Disseminated human herpesvirus 6 infection in AIDS. Lancet 342:1242. [DOI] [PubMed] [Google Scholar]
- 171.Gautheret A, Aubin JT, Fauveau V, Rozenbaum W, Huraux JM, Agut H. 1995. Rate of detection of human herpesvirus-6 at different stages of HIV infection. Eur J Clin Microbiol Infect Dis 14:820–824. doi: 10.1007/BF01691003. [DOI] [PubMed] [Google Scholar]
- 172.Lusso P, Gallo RC. 1994. Human herpesvirus 6 in AIDS. Lancet 343:555–556. doi: 10.1016/S0140-6736(94)91515-6. [DOI] [PubMed] [Google Scholar]
- 173.Spira TJ, Bozeman LH, Sanderlin KC, Warfield DT, Feorino PM, Holman RC, Kaplan JE, Fishbein DB, Lopez C. 1990. Lack of correlation between human herpesvirus-6 infection and the course of human immunodeficiency virus infection. J Infect Dis 161:567–570. doi: 10.1093/infdis/161.3.567. [DOI] [PubMed] [Google Scholar]
- 174.Knox KK, Harrington DP, Carrigan DR. 1995. Fulminant human herpesvirus six encephalitis in a human immunodeficiency virus-infected infant. J Med Virol 45:288–292. doi: 10.1002/jmv.1890450309. [DOI] [PubMed] [Google Scholar]
- 175.Fillet AM, Reux I, Joberty C, Fournier JG, Hauw JJ, Le Hoang P, Bricaire F, Huraux JM, Agut H. 1996. Detection of human herpes virus 6 in AIDS-associated retinitis by means of in situ hybridization, polymerase chain reaction and immunohistochemistry. J Med Virol 49:289–295. [DOI] [PubMed] [Google Scholar]
- 176.Descamps V, Bouscarat F, Laglenne S, Aslangul E, Veber B, Descamps D, Saraux JL, Grange MJ, Grossin M, Navratil E, Crickx B, Belaich S. 1997. Human herpesvirus 6 infection associated with anticonvulsant hypersensitivity syndrome and reactive haemophagocytic syndrome. Br J Dermatol 137:605–608. doi: 10.1111/j.1365-2133.1997.tb03795.x. [DOI] [PubMed] [Google Scholar]
- 177.Tohyama M, Yahata Y, Yasukawa M, Inagi R, Urano Y, Yamanishi K, Hashimoto K. 1998. Severe hypersensitivity syndrome due to sulfasalazine associated with reactivation of human herpesvirus 6. Arch Dermatol 134:1113–1117. [DOI] [PubMed] [Google Scholar]
- 178.Shiohara T, Iijima M, Ikezawa Z, Hashimoto K. 2007. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations. Br J Dermatol 156:1083–1084. doi: 10.1111/j.1365-2133.2007.07807.x. [DOI] [PubMed] [Google Scholar]
- 179.Yoshikawa K. 2014. HHV-6B and HHV-7 in exanthema subitum and related skin diseases, p 153–166. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 180.Picard D, Janela B, Descamps V, D'Incan M, Courville P, Jacquot S, Rogez S, Mardivirin L, Moins-Teisserenc H, Toubert A, Benichou J, Joly P, Musette P. 2010. Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med 2:46ra62. doi: 10.1126/scitranslmed.3001116. [DOI] [PubMed] [Google Scholar]
- 181.Mardivirin L, Descamps V, Lacroix A, Delebassee S, Ranger-Rogez S. 2009. Early effects of drugs responsible for DRESS on HHV-6 replication in vitro. J Clin Virol 46:300–302. doi: 10.1016/j.jcv.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 182.Mardivirin L, Valeyrie-Allanore L, Branlant-Redon E, Beneton N, Jidar K, Barbaud A, Crickx B, Ranger-Rogez S, Descamps V. 2010. Amoxicillin-induced flare in patients with DRESS (drug reaction with eosinophilia and systemic symptoms): report of seven cases and demonstration of a direct effect of amoxicillin on human herpesvirus 6 replication in vitro. Eur J Dermatol 20:68–73. doi: 10.1684/ejd.2010.0821. [DOI] [PubMed] [Google Scholar]
- 183.Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, Schultz ER, Bennett JL, Garber RL, Chang M, Schad PA, Stewart PM, Nowinski RC, Brown JP, Burmer GC. 1995. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A 92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Leibovitch EC, Jacobson S. 2014. Evidence linking HHV-6 with multiple sclerosis: an update. Curr Opin Virol 9:127–133. doi: 10.1016/j.coviro.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Merelli E, Bedin R, Sola P, Barozzi P, Mancardi GL, Ficarra G, Franchini G. 1997. Human herpes virus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J Neurol 244:450–454. doi: 10.1007/s004150050121. [DOI] [PubMed] [Google Scholar]
- 186.Liedtke W, Malessa R, Faustmann PM, Eis-Hubinger AM. 1995. Human herpesvirus 6 polymerase chain reaction findings in human immunodeficiency virus associated neurological disease and multiple sclerosis. J Neurovirol 1:253–258. doi: 10.3109/13550289509114021. [DOI] [PubMed] [Google Scholar]
- 187.Cermelli C, Berti R, Soldan SS, Mayne M, D'Ambrosia MJ, Ludwin SK, Jacobson S. 2003. High frequency of human herpesvirus 6 DNA in multiple sclerosis plaques isolated by laser microdissection. J Infect Dis 187:1377–1387. doi: 10.1086/368166. [DOI] [PubMed] [Google Scholar]
- 188.Virtanen JO, Pietilainen-Nicklen J, Uotila L, Farkkila M, Vaheri A, Koskiniemi M. 2011. Intrathecal human herpesvirus 6 antibodies in multiple sclerosis and other demyelinating diseases presenting as oligoclonal bands in cerebrospinal fluid. J Neuroimmunol 237:93–97. doi: 10.1016/j.jneuroim.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 189.Mahrholdt H, Wagner A, Deluigi CC, Kispert E, Hager S, Meinhardt G, Vogelsberg H, Fritz P, Dippon J, Bock CT, Klingel K, Kandolf R, Sechtem U. 2006. Presentation, patterns of myocardial damage, and clinical course of viral myocarditis. Circulation 114:1581–1590. doi: 10.1161/CIRCULATIONAHA.105.606509. [DOI] [PubMed] [Google Scholar]
- 190.Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Schultheiss HP. 2005. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 112:1965–1970. doi: 10.1161/CIRCULATIONAHA.105.548156. [DOI] [PubMed] [Google Scholar]
- 191.Leveque N, Boulagnon C, Brasselet C, Lesaffre F, Boutolleau D, Metz D, Fornes P, Andreoletti L. 2011. A fatal case of human herpesvirus 6 chronic myocarditis in an immunocompetent adult. J Clin Virol 52:142–145. doi: 10.1016/j.jcv.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 192.Takatsuka H, Wakae T, Mori A, Okada M, Fujimori Y, Takemoto Y, Okamoto T, Kanamaru A, Kakishita E. 2003. Endothelial damage caused by cytomegalovirus and human herpesvirus-6. Bone Marrow Transplant 31:475–479. doi: 10.1038/sj.bmt.1703879. [DOI] [PubMed] [Google Scholar]
- 193.Zerr DM, Komaroff AL. 2014. Cognitive dysfunction from HHV-6A and HHV-B, p 99–122. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 194.Buchwald D, Cheney PR, Peterson DL, Henry B, Wormsley SB, Geiger A, Ablashi DV, Salahuddin SZ, Saxinger C, Biddle R, Kikinis R, Jolesz FA, Folks T, Balachandran N, Peter JB, Gallo RC, Komaroff AL. 1992. A chronic illness characterized by fatigue, neurologic and immunologic disorders, and active human herpesvirus type 6 infection. Ann Intern Med 116:103–113. doi: 10.7326/0003-4819-116-2-103. [DOI] [PubMed] [Google Scholar]
- 195.Patnaik M, Komaroff AL, Conley E, Ojo-Amaize EA, Peter JB. 1995. Prevalence of IgM antibodies to human herpesvirus 6 early antigen (p41/38) in patients with chronic fatigue syndrome. J Infect Dis 172:1364–1367. doi: 10.1093/infdis/172.5.1364. [DOI] [PubMed] [Google Scholar]
- 196.Ablashi DV, Zompetta C, Lease C, Josephs SF, Balachandra N, Komaroff AL, Krueger GR, Henry B, Lukau J, Salahuddin SZ. 1991. Human herpesvirus 6 (HHV6) and chronic fatigue syndrome (CFS). Can Dis Wkly Rep 17(Suppl 1E):33–40. [PubMed] [Google Scholar]
- 197.Lacroix A, Jaccard A, Rouzioux C, Piguet C, Petit B, Bordessoule D, Ranger-Rogez S. 2007. HHV-6 and EBV DNA quantitation in lymph nodes of 86 patients with Hodgkin's lymphoma. J Med Virol 79:1349–1356. doi: 10.1002/jmv.20868. [DOI] [PubMed] [Google Scholar]
- 198.Bell AJ, Gallagher A, Mottram T, Lake A, Kane EV, Lightfoot T, Roman E, Jarrett RF. 2014. Germ-line transmitted, chromosomally integrated HHV-6 and classical Hodgkin lymphoma. PLoS One 9:e112642. doi: 10.1371/journal.pone.0112642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Luppi M, Marasca R, Barozzi P, Artusi T, Torelli G. 1993. Frequent detection of human herpesvirus-6 sequences by polymerase chain reaction in paraffin-embedded lymph nodes from patients with angioimmunoblastic lymphadenopathy and angioimmunoblastic lymphadenopathy-like lymphoma. Leuk Res 17:1003–1011. doi: 10.1016/0145-2126(93)90049-Q. [DOI] [PubMed] [Google Scholar]
- 200.Zhou Y, Attygalle AD, Chuang SS, Diss T, Ye H, Liu H, Hamoudi RA, Munson P, Bacon CM, Dogan A, Du MQ. 2007. Angioimmunoblastic T-cell lymphoma: histological progression associates with EBV and HHV6B viral load. Br J Haematol 138:44–53. doi: 10.1111/j.1365-2141.2007.06620.x. [DOI] [PubMed] [Google Scholar]
- 201.Agut H, Collandre H, Aubin JT, Guetard D, Favier V, Ingrand D, Montagnier L, Huraux JM. 1989. In vitro sensitivity of human herpesvirus-6 to antiviral drugs. Res Virol 140:219–228. doi: 10.1016/S0923-2516(89)80099-8. [DOI] [PubMed] [Google Scholar]
- 202.Agut H, Aubin JT, Huraux JM. 1991. Homogeneous susceptibility of distinct human herpesvirus 6 strains to antivirals in vitro. J Infect Dis 163:1382–1383. doi: 10.1093/infdis/163.6.1382. [DOI] [PubMed] [Google Scholar]
- 203.Pritchett JC, Naesens L, Montoya JG. 2014. Treating HHV-6 infections—the laboratory efficacy and clinical use of anti-HHV-6 agents, p 311–331. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 204.Prichard MN, Whitley RJ. 2014. The development of new therapies for human herpesvirus 6. Curr Opin Virol 9:148–153. doi: 10.1016/j.coviro.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Manichanh C, Olivier-Aubron C, Lagarde JP, Aubin JT, Bossi P, Gautheret-Dejean A, Huraux JM, Agut H. 2001. Selection of the same mutation in the U69 protein kinase gene of human herpesvirus-6 after prolonged exposure to ganciclovir in vitro and in vivo. J Gen Virol 82:2767–2776. [DOI] [PubMed] [Google Scholar]
- 206.Bonnafous P, Boutolleau D, Naesens L, Deback C, Gautheret-Dejean A, Agut H. 2008. Characterization of a cidofovir-resistant HHV-6 mutant obtained by in vitro selection. Antiviral Res 77:237–240. doi: 10.1016/j.antiviral.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 207.Safronetz D, Petric M, Tellier R, Parvez B, Tipples GA. 2003. Mapping ganciclovir resistance in the human herpesvirus-6 U69 protein kinase. J Med Virol 71:434–439. doi: 10.1002/jmv.10510. [DOI] [PubMed] [Google Scholar]
- 208.Isegawa Y, Hara J, Amo K, Osugi Y, Takemoto M, Yamanishi K, Fukunaga R, Shibata M, Ohshima A, Horiguchi Y, Sugimoto N. 2009. Human herpesvirus 6 ganciclovir-resistant strain with amino acid substitutions associated with the death of an allogeneic stem cell transplant recipient. J Clin Virol 44:15–19. doi: 10.1016/j.jcv.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 209.Naesens L, Bonnafous P, Agut H, De Clercq E. 2006. Antiviral activity of diverse classes of broad-acting agents and natural compounds in HHV-6-infected lymphoblasts. J Clin Virol 37(Suppl 1):S69–S75. doi: 10.1016/S1386-6532(06)70015-4. [DOI] [PubMed] [Google Scholar]
- 210.Naesens L, Stephens CE, Andrei G, Loregian A, De Bolle L, Snoeck R, Sowell JW, De Clercq E. 2006. Antiviral properties of new arylsulfone derivatives with activity against human betaherpesviruses. Antiviral Res 72:60–67. doi: 10.1016/j.antiviral.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 211.Bonnafous P, Bogaert S, Godet AN, Agut H. 2013. HDP-CDV as an alternative for treatment of human herpesvirus-6 infections. J Clin Virol 56:175–176. doi: 10.1016/j.jcv.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 212.Milbradt J, Auerochs S, Korn K, Marschall M. 2009. Sensitivity of human herpesvirus 6 and other human herpesviruses to the broad-spectrum antiinfective drug artesunate. J Clin Virol 46:24–28. doi: 10.1016/j.jcv.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 213.Hakacova N, Klingel K, Kandolf R, Engdahl E, Fogdell-Hahn A, Higgins T. 2013. First therapeutic use of artesunate in treatment of human herpesvirus 6B myocarditis in a child. J Clin Virol 57:157–160. doi: 10.1016/j.jcv.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 214.Yao K, Hoest C, Rashti F, Schott TC, Jacobson S. 2009. Effect of (r)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine (H2G) and AZT-lipid-PFA on human herpesvirus-6B infected cells. J Clin Virol 46:10–14. doi: 10.1016/j.jcv.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bonnafous P, Agut H. 2010. Activity of H2G on human herpesvirus-6B strains either sensitive or resistant to ganciclovir. J Clin Virol 48:153–154. doi: 10.1016/j.jcv.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 216.De Clercq E, Naesens L, De Bolle L, Schols D, Zhang Y, Neyts J. 2001. Antiviral agents active against human herpesviruses HHV-6, HHV-7 and HHV-8. Rev Med Virol 11:381–395. doi: 10.1002/rmv.336. [DOI] [PubMed] [Google Scholar]
- 217.Prichard MN, Williams JD, Komazin-Meredith G, Khan AR, Price NB, Jefferson GM, Harden EA, Hartline CB, Peet NP, Bowlin TL. 2013. Synthesis and antiviral activities of methylenecyclopropane analogs with 6-alkoxy and 6-alkylthio substitutions that exhibit broad-spectrum antiviral activity against human herpesviruses. Antimicrob Agents Chemother 57:3518–3527. doi: 10.1128/AAC.00429-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.De Clercq E, Naesens L. 2006. In search of effective anti-HHV-6 agents. J Clin Virol 37(Suppl 1):S82–S86. doi: 10.1016/S1386-6532(06)70017-8. [DOI] [PubMed] [Google Scholar]
- 219.Komazin-Meredith G, Cardinale SC, Williams JD, Peet NP, Prichard MN, Bowlin TL. 2013. Human herpesvirus 6 U69 kinase phosphorylates the methylenecyclopropane nucleosides cyclopropavir, MBX 2168, and MBX 1616 to their monophosphates. Antimicrob Agents Chemother 57:5760–5762. doi: 10.1128/AAC.00978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Reymen D, Naesens L, Balzarini J, Holy A, Dvorakova H, De Clercq E. 1995. Antiviral activity of selected acyclic nucleoside analogues against human herpesvirus 6. Antiviral Res 28:343–357. doi: 10.1016/0166-3542(95)00058-5. [DOI] [PubMed] [Google Scholar]
- 221.Neyts J, Naesens L, Ying C, De Bolle L, De Clercq E. 2001. Anti-herpesvirus activity of (1′S,2′R)-9-[[1′,2′-bis(hydroxymethyl)-cycloprop-1′-yl]methyl] × guanine (A-5021) in vitro and in vivo. Antiviral Res 49:115–120. doi: 10.1016/S0166-3542(00)00144-3. [DOI] [PubMed] [Google Scholar]
- 222.De Bolle L, Andrei G, Snoeck R, Zhang Y, Van Lommel A, Otto M, Bousseau A, Roy C, De Clercq E, Naesens L. 2004. Potent, selective and cell-mediated inhibition of human herpesvirus 6 at an early stage of viral replication by the non-nucleoside compound CMV423. Biochem Pharmacol 67:325–336. doi: 10.1016/j.bcp.2003.08.042. [DOI] [PubMed] [Google Scholar]
- 223.Lautenschlager I, Razonable RR. 2014. HHV-6A and HHV-6B in solid organ transplantation, p 201–215. In Flamand L, Lautenschlager I, Krueger G, Ablashi D (ed), Human herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed Elsevier, Amsterdam, Netherlands. [Google Scholar]
- 224.Razonable RR, Zerr DM. 2009. HHV-6, HHV-7 and HHV-8 in solid organ transplant recipients. Am J Transplant 9(Suppl 4):S97–S100. doi: 10.1111/j.1600-6143.2009.02899_1.x. [DOI] [PubMed] [Google Scholar]
- 225.Rozenberg F, Deback C, Agut H. 2011. Herpes simplex encephalitis: from virus to therapy. Infect Disord Drug Targets 11:235–250. doi: 10.2174/187152611795768088. [DOI] [PubMed] [Google Scholar]
- 226.Agut H, Boutolleau D, Deback C, Bonnafous P, Gautheret-Dejean A. 2009. Testing the susceptibility of human herpesviruses to antivirals. Future Microbiol 4:1111–1123. doi: 10.2217/fmb.09.83. [DOI] [PubMed] [Google Scholar]
- 227.Theodore WH, Epstein L, Gaillard WD, Shinnar S, Wainwright MS, Jacobson S. 2008. Human herpes virus 6B: a possible role in epilepsy? Epilepsia 49:1828–1837. doi: 10.1111/j.1528-1167.2008.01699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]